
Abstract
Photosynthesis is tightly regulated in order to withstand dynamic light environments. Under high light intensities, a mechanism known as non-photochemical quenching (NPQ) dissipates excess excitation energy, protecting the photosynthetic machinery from damage. An obstacle that lies in the way of understanding the molecular mechanism of NPQ is the large gap between in vitro and in vivo studies. On the one hand, the complexity of the photosynthetic membrane makes it challenging to obtain molecular information from in vivo experiments. On the other hand, a suitable in vitro system for the study of quenching is not available. Here we have developed a minimal NPQ system using proteoliposomes. With this, we demonstrate that the combination of low pH and PsbS is both necessary and sufficient to induce quenching in LHCII, the main antenna complex of plants. This proteoliposome system can be further exploited to gain more insight into how PsbS and other factors (e.g. zeaxanthin) influence the quenching mechanism observed in LHCII.
Similar content being viewed by others

Introduction
In plants, light absorption is maximized with specialized light-harvesting complexes (LHCs) that bind a high density of chlorophylls (Chls) and carotenoids. The specific organization of pigments and proteins enables efficient transfer of excitation energy to the Photosystem II (PSII) reaction centre where photosynthesis is initiated with photochemical charge separation.1 However, the intensity of natural light can fluctuate rapidly, and the amount of light absorbed often exceeds the capacity of the photosynthetic reactions. A feedback mechanism known as non-photochemical quenching (NPQ) acts as a safety valve, dissipating the excess excitation energy as heat, preventing the formation of Chl triplets and the inevitable production of reactive oxygen species2.
NPQ is triggered by an increase in lumen proton concentration and the subsequent protonation and activation of the integral membrane protein, PsbS3,4,5. Although PsbS is a member of the LHC superfamily, it has some unusual characteristics. It is not an integral part of the PSII-LHC supercomplex6,7,8, it is substoichiometric with respect to the PSII core9, and its compact four-transmembrane helix fold precludes the binding of pigments10. The absence of stably bound pigments suggests PsbS is not the site of quenching; therefore, the current view is that PsbS promotes the formation of quenching sites in the LHCs11 or at the interface between LHCs and PsbS12. This is supported by the substantial reduction of NPQ capacity in the absence of LHCs in vivo13.
The first hypothesis proposes that PsbS is responsible for quenching in LHCs by promoting LHC oligomerization and/or increasing the probability of the quenched conformation. It has long been observed that LHC oligomers are highly quenched in vitro14,15. The oligomers (sometimes referred to as aggregates) can be formed via detergent removal16,17 or spontaneous clustering in the lipid bilayer of proteoliposomes18. Oligomerization is not necessary for LHCs to access the quenched state per se. LHCs can access a variety of spectral and kinetic states via conformational changes in the protein scaffold modifying pigment–pigment interactions19,20,21,22,23. An oligomer consists of a heterogeneous mixture of these conformational states, and quenching arises from a small number of complexes in a strongly quenched conformation being energetically connected to the bulk of the unquenched complexes24,25. Parallels have been drawn between aggregation-related quenching in vitro and what occurs during NPQ in vivo. For example, increased far-red fluorescence emission, a spectroscopic signature of LHC oligomerization, has been observed in leaves following NPQ induction26,27. Furthermore, freeze-fracture electron microscopy of intact plant chloroplasts shows that LHCII has a tendency to cluster during NPQ28. This clustering is inhibited in the PsbS knockout mutant and enhanced in the PsbS overexpressor17,29.
The second hypothesis proposes a more direct role of PsbS in quenching, via the formation of a quenching site at a PsbS–LHC interface12,30,31. The idea is that a pH-dependent conformational change of PsbS, such a monomerization, could allow the transient binding of a pigment present in the thylakoid membrane. The PsbS-pigment complex could then strongly interact with an LHC, positioning the carotenoid in close contact with one of the peripheral Chls, thus creating a quenching site. While the pigment in question has been suggested to be zeaxanthin12, it could equally be another pigment with the capacity to quench Chl excited singlet states such as lutein23,32.
To further investigate the interaction between PsbS and LHCs, several studies have utilized a proteoliposome system containing PsbS and LHCII33,34,35,36. LHCII is a trimeric complex, and as the main antenna of PSII is responsible for the majority of NPQ37. The advantage of a proteoliposome system is that it avoids the complexity of the thylakoid membrane whilst allowing interprotein interactions within the plane of the membrane. Surprisingly, however, the results of these studies are at variance with what is known to occur in vivo. Most observe quenching at neutral pH suggesting the quenching is an artefact33,34,35, or no quenching at all 36. In the current study, we improve upon existing proteoliposomes systems by more closely mimicking the native thylakoid environment. We aim to reproduce NPQ as it occurs in vivo i.e. fluorescence quenching that is both pH- and PsbS-dependent. This will allow determination of the minimal components necessary for NPQ and shed light on the action mechanism of PsbS.
Results and discussion
Purification of PsbS from the thylakoid membrane
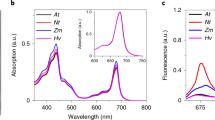
Arabidopsis thaliana PsbS knockout plants were transformed with PsbS modified to contain a strep-tag at the C terminus. These plants have a ~ 50% increase in the amount of qE compared to WT plants and a corresponding ~ 65% increase in the stoichiometry of PsbS to the PSII core, confirming that the NPQ level increases with the increase of the amount of PsbS in the membrane and indicating that the strep-tag has minimal effect on PsbS function (Supplementary Fig. S1). PSII-enriched membranes (BBY) isolated from the mutant plants were solubilized with a mild detergent, and PsbS was isolated in a single step via affinity chromatography. The purity of PsbS in the eluate was determined to be approximately 90% based on SDS-PAGE analysis, and the impurities appear to be a non-specific array of PSII components (Fig. 1a). This is consistent with the highly hydrophobic properties of PsbS, and its tendency to form non-specific interactions with complexes upon membrane solubilisation38. Given that PsbS is not a pigment-binding protein, the pigments in the eluate are likely bound to the protein impurities. This is supported by the resemblance of the absorption spectra to LHC monomers (Lhcb1-6; found in band 2 of a sucrose gradient7) and the low absorption intensity relative to protein concentration (Fig. 1b). The absence of any strong fluorescence emission in the 720–740 nm range at 77 K, excludes the presence of PSI or LHCI (Fig. 1c).

Protein composition and spectral characteristics of the PsbS eluate following strep-tactin affinity chromatography. (a) SDS-PAGE of WT BBY membranes and PsbS eluate. Lanes are cropped from different parts of the same gel. The uncropped gel is presented in Supplementary Figure S4. (b) absorption spectra of PsbS eluate and LHC monomers normalized to the Qy maximum. The inset shows the absorption spectra normalized to protein concentration. (c) 77 k fluorescence emission spectra of PsbS eluate and LHC monomers normalized to the maximum. Results are representative of three independent experiments with similar results.
LHCII was obtained by solubilisation of the thylakoid membrane with α-DM. The preparation has been previously characterized39. The complex contains 3.8 carotenoids (0.9 neoxanthin, 0.3 violaxanthin and 2.6 luteins) per 14 Chls (8 Chl a and 6 Chl b). No zeaxanthin was detected in the preparation (Supplementary Fig. S2) in agreement with the fact that the complex was purified from dark-adapted, non-stressed plants.
LHCII folds correctly in proteoliposomes at neutral and low pH
Liposomes were prepared from native thylakoid lipids, MGDG, DGDG, SQDG and PG in a molar ratio similar to what is found in vivo40. The liposomes were destabilized in 0.03% α-DM, and LHCII was added to the liposome preparation in a lipid:protein molar ratio of 133:1. The proteins were incorporated into the lipid bilayer by removing detergent with polystyrene beads. Any unincorporated protein was removed from the final preparation via centrifugation. A decrease in the absorption of the final preparation indicates that LHCII reconstitution efficiency is approximately 70% at pH 7.5 and 40% at pH 5.5 (Supplementary Fig. S2). A trypsin digest revealed that there is no preferential orientation of LHCII in the liposome, so the interaction between complexes with opposite orientation cannot be excluded (Supplementary Fig. S2).
The resulting proteoliposomes have absorption spectra very similar to LHCII in detergent, demonstrating there is no pigment loss during the procedure (Fig. 2a). The 600–700 nm region of the CD spectra indicate that pigment organization is similar to that of LHCII in detergent (Fig. 2c). Differences in the CD spectra at shorter wavelengths are expected as this region is highly sensitive to protein environment41. Overlap of the low-temperature fluorescence emission spectra upon excitation of Chl a (440 nm), Chl b (475 nm) and carotenoids (500 nm) demonstrate efficient excitation energy transfer between pigments and proper folding of complexes (Fig. 2b). Overall, the results confirm correct folding and incorporation of LHCII into the lipid bilayer at both neutral and low pH.

Spectral characteristics of LHCII in detergent (at pH 7.5) compared to LHCII reconstituted into liposomes at pH 7.5 or pH 5.5. (a) Absorption spectra normalized to the Qy maximum (b) circular dichroism spectra normalized to the negative ~ 680 nm peak (c) 77 K emission spectra with excitation wavelengths of 440 nm, 475 nm and 500 nm, normalized to the maximum (d) fluorescence decay traces upon excitation at 468 nm and detection at 680 nm. Proteoliposome preparations had lipid:protein molar ratios of 133:1. Results are representative of three independent experiments with similar results.
LHCII is pre-clustered and pre-quenched in the proteoliposome
Bulk LHCII can exist in a continuum between light-harvesting and quenched states. In detergent micelles, LHCII is the fully light-harvesting state, whereas, in the thylakoid membrane, it is poised between light-harvesting and quenched states42,43. This partial quenching is a result of protein crowding and LHCII oligomerization, rather than direct lipid-protein interactions18,44. In proteoliposomes, the position of LHCII on the continuum can be finely tuned by controlling the lipid:LHCII ratio, where low lipid:LHCII ratios increase the propensity of LHCII to aggregate, resulting in a shift towards the quenched state18. The extent of oligomerization can be monitored by the intensity of a low-temperature fluorescence emission peak at ~ 700 nm and the degree of quenching can be monitored directly via the rate of LHCII fluorescence decay18.
In the present study, LHCII in the proteoliposome displays a substantial increase in the low-temperature fluorescence emission at ~ 700 nm (Fig. 2c) and a faster rate of fluorescence decay (Fig. 2d) compared to LHCII in detergent. Average fluorescence lifetimes were obtained by fitting fluorescence decay curves to a sum of exponential decay components (Supplementary Table S1). LHCII in detergent has a lifetime of 3.3 ns, whereas LHCII in the proteoliposome prepared at pH 7.5 has an average lifetime of 2.4 ns, similar to the ~ 2 ns lifetime observed in the thylakoid membrane42. This is further shortened to 1.6 ns when proteoliposomes were prepared at pH 5.5, supporting the notion that LHCII has some intrinsic pH sensing capacity when in the aggregated state45. While this effect is almost negligible in terms of contribution to NPQ in WT plants, it is thought to be the cause of a slowly forming NPQ (on the order of 1 h) in the PsbS knockout mutant46. Also note that low pH in the absence of aggregation is insufficient to induce quenching in isolated LHCII47.
PsbS induces quenching in LHCII at low pH
To investigate the PsbS-LHCII interaction, PsbS was added in a 1:1 molar ratio with LHCII trimers. This stoichiometry is higher than what naturally occurs in the thylakoid membrane but is intended to increase the probability of PsbS-LHCII interaction when liposome occupancy is known to be heterogeneous48. Immunoblotting confirmed successful reconstitution of PsbS into the liposome, with the lipid bilayer appearing to stabilize the dimeric state (Fig. 3a). The lipid:LHCII ratio of this sample is 133:1, and the total lipid:protein ratio is 100:1. To distinguish the effect of PsbS and increased protein concentration, we have also used an LHCII-only control with a lipid:protein ratio of 100:1. In this instance, the increase in protein concentration did not have a large effect on results (Fig. 3b–d).

The effect of PsbS on LHCII spectral characteristics in the liposome at pH 7.5 and pH 5.5. (a) Immunoblot of PsbS in detergent and in LHCII + PsbS proteoliposomes where M and D denote the monomeric and dimeric forms of PsbS, respectively. The full-length blot is presented in Supplementary Figure S4. (b) Absorption spectra normalized to the Qy maximum (c) 77 K fluorescence spectra upon excitation at 440 nm, normalized to the maximum. (d) Fluorescence decay traces upon excitation at 468 nm and detection at 680 nm. LHCII-only proteoliposome preparations have lipid:protein ratios of 100:1 and 133:1. LHCII + PsbS proteoliposome preparations have total lipid:protein ratios of 100:1, lipid:LHCII ratios of 133:1 and PsbS:LHCII ratios of 1:3. Results are representative of three independent experiments with similar results, which can be viewed in Supplementary Figure S3.
The addition of PsbS had no significant impact on LHCII spectral characteristics at pH 7.5; however, at pH 5.5, a number of differences are observed. There is a broadening of the Qy absorption peak and a relative increase in absorption at 650 nm (Fig. 3b), a significant increase in low-temperature fluorescence emission at 700 nm (Fig. 3c) and a shortening of LHCII fluorescence lifetime to 1.1 ns (Fig. 3d). Using proteoliposomes containing PsbS at neutral and low pH as representative of the unquenched (Fm) and quenched (Fm') state respectively, we can approximate the level of NPQ obtained in this system is around 1, a value very similar to what is reported for isolated thylakoids and chloroplasts28,39,49.
The requirement of both PsbS and low pH to induce quenching shows that our system mimics the properties of NPQ in vivo. This is in contrast to previous PsbS-LHCII proteoliposome systems, where quenching was observed at neutral pH33,34,35. This discrepancy is likely due to the effect of lipid:protein ratio on LHCII lifetime. As mentioned, increasing LHCII concentration in the liposome increases the tendency to cluster and shortens fluorescence lifetime18. It is conceivable that increasing the protein concentration via the addition of PsbS could have the same effect, highlighting the importance of adequate protein concentration controls. The lack of additional quenching at low pH in these studies can presumably be attributed to the use of recombinant PsbS, which may have a different tertiary structure to that of PsbS folded in vivo.
The data also demonstrates that a minimal system composed of thylakoid membrane lipids, PsbS, LHCII and low pH is sufficient for non-photochemical quenching. This excludes the possibility that other components of the thylakoid membrane are essential for quenching and discounts the hypotheses that quenching centres are created by the transient association of pigments with PsbS 12,30,31. The low pH, PsbS-induced increase in fluorescence emission at 700 nm suggests that PsbS triggers clustering of LHCII. This is fully consistent with the observations of enhanced PsbS-LHCII interactions upon NPQ activation in intact chloroplasts50,51 and further supports the role for aggregation-related quenching in vivo14,17,29.
It is important to note that red fluorescence emission is not a direct spectroscopic marker of the quencher, but of aggregation. Oligomerized LHCII can exist in a variety of conformational states characterized by different emission spectra24. The red-emitting state is relatively long-lived and results from a Chl–Chl charge transfer state that acts as a trap for excitation energy24,52. Clustering of LHCII leads to an increase of the population of these forms, an effect that is particularly visible at low temperature. The quenched conformation has been shown to emit at 680 nm23,24 where the quencher is a carotenoid dark state, which is populated by excitation energy transfer from neighbouring Chls23,32. Whether PsbS increases the probability of this quenched conformation or influences the physical quenching mechanism requires further spectroscopic analysis. This will be aided by the simplicity of the proteoliposome system, which lacks the overlapping spectral and kinetic signals from Photosystems and other LHCs present in the thylakoid membrane.
The flexibility of the system will permit the investigation of mutants of both PsbS and LHCII to identify the pigments/protein domains involved in the quenching process. Furthermore, the addition of zeaxanthin, and/or LHCII binding zeaxanthin in the V1 site will provide valuable insight into zeaxanthin dependent quenching.
Methods
Plant growth
Arabidopsis thaliana WT (Col-0) and PsbS KO (npq4) lines were obtained from the Nottingham Arabidopsis Stock Centre (NASC). All plants were grown under 120 μmol photons m−2 s−1, 16 h/8 h day/night cycle for 4–5 weeks. The research involving plants complies with relevant institutional and national guidelines and legislation.
PsbS-strep construction
The PsbS promoter region was amplified by PCR from genomic DNA isolated from A. thaliana with the forward primer 5′ TTCAAGCTTAGGGGTTTAATGTATGTACA 3′ and reverse primer 5′ TTCCTCGAGTCTTTCTGAGGATGAGAGAA 3′. The product was cloned into pORE-O353, using HindIII and XhoI restriction sites. The PsbS coding region was amplified by PCR from genomic DNA isolated from A. thaliana with the forward primer 5′ TAGAATTCAAGAATGGCTCAAACCATGCTGCTTACTTCAG 3′ and reverse primer 5′ TAGCGGCCGCTTATTTTTCAAACTGCGGATGGCTCCACGCGCTGCTTTCTTCACCATCATCGG 3′. The PCR product was digested with EcoRI and NotI before re-amplification with the forward primer 5′ AGAAGGCCTTGGATCCAGAATGGCTCAAACCATGCTGCT 3′ and reverse primer 5′ CTAGCGGCCGCTTATTTTTCAAACTGCGGATGGCTC 3′. The reverse primer was designed to add a two amino acid spacer SA and a tail of WSHPQFEK, which constitutes the strep-tag. The product was cloned into pORE-O3 containing the PsbS promoter, using BamHI and NotI restriction sites. Agrobacterium tumefaciens strain GV3101 was transformed with sequence confirmed constructs. PsbS KO (npq4) plants were transformed by floral dip54,55. Transformants were selected with the herbicide glufosinate. Expression of the PsbS transgene was assessed by immunoblotting, and a homozygous line was identified in subsequent generations. Thylakoids were prepared according to39. PsbS activity was confirmed by measuring NPQ of leaves in a modular Dual PAM-100 apparatus (Walz) as in37. NPQ is calculated as (Fm − Fm) · Fm−1.
Protein isolation
Grana-enriched membranes (BBY) were prepared from WT and PsbS-strep plants as in7. WT BBY were used to prepare LHCII trimers according to ref7 and HPLC was performed as in39 to ensure the retention of ≥ 3.8 xanthophylls per 14 Chls (Supplementary Figure S2). PsbS-strep BBY were unstacked in 5 mM EDTA; 10 mM Hepes (pH 8) and pelleted by centrifugation at 15,000 rpm for 10 min. The unstacked membranes were solubilized with a final concentration of 0.6% α-DM; 10 mM Hepes (pH 8) at a chlorophyll concentration of 0.5 mg ml−1. Unsolubilized material was removed via 2 × centrifugation at 15,000 rpm for 30 min. The supernatant was loaded onto a Strep-Tactin gravity flow column (IBA), with all pre-made buffers being supplemented with 0.03% α-DM. The eluate was mixed with a sucrose buffer (2 M sucrose, 0.03% α-DM, 10 mM Hepes pH 7.5) to have a final sucrose concentration of 0.3 M. All procedures were performed at 4 °C in near darkness. Samples were aliquoted, snap-frozen in liquid N2 and stored at − 80 °C.
Protein quantification, SDS-PAGE, immunoblotting, trypsin digest
PsbS protein concentration was determined via BCA assay (Pierce, Thermo Scientific). LHCII protein concentration was determined based on a Chl/protein (w/w) of 0.5. Chl concentration was determined using the method of56 but modified such that the absorption spectra are first fitted with the spectra of individual pigments57. SDS-PAGE was performed using a tris-tricine system with a 4% stacking and 12% running gel58. Immunoblotting was performed using antibodies from Agrisera. Images of gels/blots were taken with an ImageQuant LAS 4000 (GE). Relative protein levels were determined using Image Studio Lite (LI-COR). Enzymatic cleavage experiments were performed with an LHCII to Trypsin (from bovine pancreas; Merck) ratio of 50:1. The digest was carried out at 37 °C and stopped at 1 h with the addition of SDS-PAGE loading buffer and heating to 100 °C for 5 min.
Proteoliposome preparation
Monogalactosyldiacylglycerol (MGDG), digalactosyldiacylglycerol (DGDG), sulphoquinovosyldiacylglycerol (SQDG) and l-α-phosphatidylglycerol (Soy PG) were purchased from Avanti Polar Lipids and suspended in 1:1 methanol:chloroform. A lipid mixture was prepared from a 40:30:15:15 molar ratio of MGDG, DGDG, SQDG and PG in a glass vial. Organic solvent was evaporated under N2 followed by 1 h under a low vacuum in a SpeedVac vacuum concentrator (Thermo Scientific). The lipid film was hydrated with either a 20 mM MOPS (pH 7.0) or 20 mM MES (pH 5.5) buffer containing 10 mM NaCl to give a final lipid concentration of 2.5 μg μL−1. The solution was vortexed for 1 h at RT. The hydrated lipids were then freeze-thawed 10× by alternating between liquid nitrogen and a 37 °C water bath before extrusion 10× through a 0.2-μm polycarbonate filter (Mini-Extruder, Avanti Polar Lipids). The liposomes were destabilized in 0.03% α-DM; 20 mM MOPS/MES (pH 7.5/5.5); 10 mM NaCl at a lipid concentration of 0.25 μg μL−1. The lipid-detergent mixture was equilibrated for 2 h at RT before being separated into three batches. Protein was added to the three batches according to the following lipid:protein molar ratios: (1) LHCII; 100:1 (2) LHCII; 133:1 (3) LHCII; 133:1 and PsbS; 400:1. Unless specified otherwise, the ratios refer to the LHCII monomer. Following addition of protein, the mixture was equilibrated for 30 min at RT. Absorbent beads (Bio-Beads SM2, Bio-Rad) were activated in methanol and rinsed thoroughly in hydration buffer before 20 mg mL−1 were added for 2 h and an additional 40 mg mL−1 for 30 min. During this time, samples were continuously mixed on an Eppendorf rotator at RT. Protein aggregates not incorporated into the liposome were sedimented by centrifugation at 15,000×g for 15 min. The proteoliposomes were subsequently stored at 4 °C and analyzed within 72 h.
Steady-state spectroscopy
Absorptions spectra were recorded at RT on a 4000 UV–Vis-spectrophotometer (Varian). Fluorescence spectra were recorded at 77 K on a Fluorolog 3.22 spectrofluorimeter (Jobin Yvon-Spex). All fluorescence spectra were measured at an OD of < 0.05 cm−1 at the Qy maximum. The circular-dichroism (CD) spectra were recorded on a Chirascan-Plus spectropolarimeter (Applied Photophysics). When necessary, the samples were diluted with a 20 mM MOPS (pH 7.5) or 20 mM MES (pH 5.5) buffer containing 10 mM NaCl. Plots were generated in OriginPro 2020 (OriginLab).
Time-correlated single-photon counting (TCSPC)
Fluorescence decay kinetics were recorded on a FluoTime 200 fluorometer (PicoQuant). Excitation was provided by a 5 μW 468 nm laser diode with a repetition rate of 10 MHz. Fluorescence emission was detected at 680 nm. Samples were diluted to an OD of < 0.05 cm−1 at the Qy maximum and stirred in a cuvette with a path length of 1 cm. All measurements were performed at 10 °C. Fluorescence decay curves were analyzed using FluoFit software (PicoQuant), following deconvolution of the instrument response function (IRF; 88 ps full width half maximum) measured from the ~ 6 ps decay of pinacyanol iodide‐dye dissolved in methanol59. The data were fitted to multi-exponential decay functions with amplitudes Ai and associated fluorescence decay times τi. The average fluorescence lifetimes were calculated according to τavg = ΣAi × τi/ΣAi. Plots were generated in OriginPro 2020 (OriginLab).
Data availability
The datasets and materials generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Croce, R. & van Amerongen, H. Light harvesting in oxygenic photosynthesis: Structural biology meets spectroscopy. Science 369(6506), eaay2058 (2020).
Krieger-Liszkay, A. Singlet oxygen production in photosynthesis. J. Exp. Bot. 56, 337–346 (2005).
Briantais, J.-M., Vernotte, C., Picaud, M. & Krause, G. H. A quantitative study of the slow decline of chlorophyll a fluorescence in isolated chloroplasts. Biochim. Biophys. Acta Bioenerg. 548, 128–138 (1979).
Li, X.-P. et al. A pigment-binding protein essential for regulation of photosynthetic light harvesting. Nature 403, 391–395 (2000).
Li, X.-P., Muller-Moule, P., Gilmore, A. M. & Niyogi, K. K. PsbS-dependent enhancement of feedback de-excitation protects Photosystem II from photoinhibition. Proc. Natl. Acad. Sci. USA 99, 15222–15227 (2002).
Kereïche, S., Kiss, A. Z., Kouřil, R., Boekema, E. J. & Horton, P. The PsbS protein controls the macro-organisation of photosystem II complexes in the grana membranes of higher plant chloroplasts. FEBS Lett. 584, 759–764 (2010).
Caffarri, S., Kouril, R., Kereïche, S., Boekema, E. J. & Croce, R. Functional architecture of higher plant photosystem II supercomplexes. EMBO J. 28, 3052–3063 (2009).
Crepin, A., Santabarbara, S. & Caffarri, S. Biochemical and spectroscopic characterization of highly stable photosystem II supercomplexes from arabidopsis. J. Biol. Chem. 291, 19157–19171 (2016).
McKenzie, S. D., Ibrahim, I. M., Aryal, U. K. & Puthiyaveetil, S. Stoichiometry of protein complexes in plant photosynthetic membranes. Biochim. Biophys. Acta Bioenerg. 1861, 148141 (2020).
Fan, M. et al. Crystal structures of the PsbS protein essential for photoprotection in plants. Nat. Struct. Mol. Biol. 22, 729–735 (2015).
Ruban, A. V. Light harvesting control in plants. FEBS Lett. 592, 3030–3039 (2018).
Barros, T., Royant, A., Standfuss, J., Dreuw, A. & Kühlbrandt, W. Crystal structure of plant light-harvesting complex shows the active, energy-transmitting state. EMBO J. 28, 298–306 (2009).
Lokstein, H., Härtel, H., Hoffmann, P. & Renger, G. Comparison of chlorophyll fluorescence quenching in leaves of wild-type with a chlorophyll-b-less mutant of barley (Hordeum vulgare L.). J. Photochem. Photobiol. B Biol. 19, 217–225 (1993).
Horton, P. et al. Control of the light-harvesting function of chloroplast membranes by aggregation of the LHCII chlorophyll-protein complex. FEBS Lett. 292, 1–4 (1991).
Mullineaux, C. W., Pascal, A. A., Horton, P. & Holzwarth, A. R. Excitation-energy quenching in aggregates of the LHC II chlorophyll-protein complex: A time-resolved fluorescence study. BBA Bioenerg. 1141, 23–28 (1993).
Betterle, N. et al. Light-induced dissociation of an antenna hetero-oligomer is needed for non-photochemical quenching induction. J. Biol. Chem. 284, 15255–15266 (2009).
Goral, T. K. et al. Light-harvesting antenna composition controls the macrostructure and dynamics of thylakoid membranes in Arabidopsis. Plant J. 69, 289–301 (2012).
Natali, A. et al. Light-harvesting Complexes (LHCs) cluster spontaneously in membrane environment leading to shortening of their excited state lifetimes. J. Biol. Chem. 291, 16730–16739 (2016).
Pascal, A. A. et al. Molecular basis of photoprotection and control of photosynthetic light-harvesting. Nature 436, 134–137 (2005).
Valkunas, L., Chmeliov, J., Krüger, T. P. J., Ilioaia, C. & Van Grondelle, R. How photosynthetic proteins switch. J. Phys. Chem. Lett. 3, 2779–2784 (2012).
Liguori, N., Periole, X., Marrink, S. J. & Croce, R. From light-harvesting to photoprotection: Structural basis of the dynamic switch of the major antenna complex of plants (LHCII). Sci. Rep. 5, 1–10 (2015).
Schlau-Cohen, G. S. et al. Single-molecule identification of quenched and unquenched states of LHCII. J. Phys. Chem. Lett. 6, 860–867 (2015).
Mascoli, V. et al. Capturing the quenching mechanism of light-harvesting complexes of plants by zooming in on the ensemble. Chem 5, 2900–2912 (2019).
Chmeliov, J. et al. The nature of self-regulation in photosynthetic light-harvesting antenna. Nat. Plants 2, 20 (2016).
Mascoli, V., Gelzinis, A., Chmeliov, J., Valkunas, L. & Croce, R. Light-harvesting complexes access analogue emissive states in different environments. Chem. Sci. 11, 5697–5709 (2020).
Miloslavina, Y. et al. Far-red fluorescence: A direct spectroscopic marker for LHCII oligomer formation in non-photochemical quenching. FEBS Lett. 582, 3625–3631 (2008).
Farooq, S. et al. Dynamic feedback of the Photosystem II reaction centre on photoprotection in plants. Nat. Plants 4, 225–231 (2018).
Johnson, M. P. et al. Photoprotective energy dissipation involves the reorganization of Photosystem II light-harvesting complexes in the grana membranes of spinach chloroplasts. Plant Cell 23, 1468–1479 (2011).
Ware, M. A., Giovagnetti, V., Belgio, E. & Ruban, A. V. PsbS protein modulates non-photochemical chlorophyll fluorescence quenching in membranes depleted of photosystems. J. Photochem. Photobiol. B Biol. 152, 301–307 (2015).
Niyogi, K. K., Li, X.-P., Rosenberg, V. & Jung, H.-S. Is PsbS the site of non-photochemical quenching in photosynthesis?. J. Exp. Bot. 56, 375–382 (2005).
Croce, R. PsbS is the plants’ pick for sun protection. Nat. Struct. Mol. Biol. 22, 650–652 (2015).
Ruban, A. V. et al. Identification of a mechanism of photoprotective energy dissipation in higher plants. Nature 450, 575–578 (2007).
Wilk, L., Grunwald, M., Liao, P. N., Walla, P. J. & Kühlbrandt, W. Direct interaction of the major light-harvesting complex II and PsbS in nonphotochemical quenching. Proc. Natl. Acad. Sci. USA 110, 5452–5456 (2013).
Liu, C. et al. Simultaneous refolding of denatured PsbS and reconstitution with LHCII into liposomes of thylakoid lipids. Photosynth. Res. 127, 109–116 (2016).
Pawlak, K. et al. On the PsbS-induced quenching in the plant major light-harvesting complex LHCII studied in proteoliposomes. Photosynth. Res. 144, 195–208 (2020).
Crisafi, E., Krishnan, M. & Pandit, A. Time-resolved fluorescence analysis of LHCII in the presence of PsbS at neutral and low pH. bioRxiv https://doi.org/10.1101/456046 (2018).
Nicol, L., Nawrocki, W. J. & Croce, R. Disentangling the sites of non-photochemical quenching in vascular plants. Nat. Plants 5, 1177–1183 (2019).
Teardo, E. et al. Evidences for interaction of PsbS with photosynthetic complexes in maize thylakoids. Biochim. Biophys. Acta Bioenerg. 1767, 703–711 (2007).
Xu, P., Tian, L., Kloz, M. & Croce, R. Molecular insights into zeaxanthin-dependent quenching in higher plants. Sci. Rep. 5, 13679 (2015).
Sakurai, I. et al. Lipids in oxygen-evolving photosystem II complexes of cyanobacteria and higher plants. J. Biochem. 140, 201–209 (2006).
Georgakopoulou, S. et al. Understanding the changes in the circular dichroism of light harvesting complex II upon varying its pigment composition and organization. Biochemistry 46, 4745–4754 (2007).
Belgio, E., Johnson, M. P., Jurić, S. & Ruban, A. V. Higher plant photosystem II light-harvesting antenna, not the reaction center, determines the excited-state lifetime—both the maximum and the nonphotochemically quenched. Biophys. J. 102, 2761–2771 (2012).
Tian, L., Dinc, E. & Croce, R. LHCII populations in different quenching states are present in the thylakoid membranes in a ratio that depends on the light conditions. J. Phys. Chem. Lett. 6, 2339–2344 (2015).
Crisafi, E. & Pandit, A. Disentangling protein and lipid interactions that control a molecular switch in photosynthetic light harvesting. Biochim. Biophys. Acta Biomembr. 1859, 40–47 (2017).
Petrou, K., Belgio, E. & Ruban, A. V. PH sensitivity of chlorophyll fluorescence quenching is determined by the detergent/protein ratio and the state of LHCII aggregation. Biochim. Biophys. Acta Bioenerg. 1837, 1533–1539 (2014).
Johnson, M. P. & Ruban, A. V. Arabidopsis plants lacking PsbS protein possess photoprotective energy dissipation. Plant J. 61, 283–289 (2010).
Liguori, N., Roy, L. M., Opacic, M., Durand, G. & Croce, R. Regulation of light harvesting in the green alga chlamydomonas reinhardtii: The c-terminus of lhcsr is the knob of a dimmer switch. J. Am. Chem. Soc. 135, 18339–18342 (2013).
Akhtar, P., Görföl, F., Garab, G. & Lambrev, P. H. Dependence of chlorophyll fluorescence quenching on the lipid-to-protein ratio in reconstituted light-harvesting complex II membranes containing lipid labels. Chem. Phys. 522, 242–248 (2019).
Crouchman, S., Ruban, A. & Horton, P. PsbS enhances nonphotochemical fluorescence quenching in the absence of zeaxanthin. FEBS Lett. 580, 2053–2058 (2006).
Correa-Galvis, V., Poschmann, G., Melzer, M., Stühler, K. & Jahns, P. PsbS interactions involved in the activation of energy dissipation in Arabidopsis. Nat. Plants 2, 15225 (2016).
Sacharz, J., Giovagnetti, V., Ungerer, P., Mastroianni, G. & Ruban, A. V. The xanthophyll cycle affects reversible interactions between PsbS and light-harvesting complex II to control non-photochemical quenching. Nat. Plants 3, 16225 (2017).
Gelzinis, A., Chmeliov, J., Ruban, A. V. & Valkunas, L. Can red-emitting state be responsible for fluorescence quenching in LHCII aggregates?. Photosynth. Res. 135, 275–284 (2018).
Coutu, C. et al. PORE: A modular binary vector series suited for both monocot and dicot plant transformation. Transgenic Res. 16, 771–781 (2007).
Clough, S. J. & Bent, A. F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998).
Zhang, X., Henriques, R., Lin, S. S., Niu, Q. W. & Chua, N. H. Agrobacterium-mediated transformation of Arabidopsis thaliana using the floral dip method. Nat. Protoc. 1, 641–646 (2006).
Porra, R. J., Thompson, W. A. & Kriedemann, P. E. Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents: Verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. BBA Bioenerg. 975, 384–394 (1989).
Croce, R., Canino, G., Ros, F. & Bassi, R. Chromophore organization in the higher-plant photosystem II antenna protein CP26. Biochemistry 41, 7334–7343 (2002).
Schägger, H. Tricine–SDS-PAGE. Nat. Protoc. 1, 16–22 (2006).
Van Oort, B. et al. Picosecond fluorescence of intact and dissolved PSI-LHCI crystals. Biophys. J. 95, 5851–5861 (2008).
Acknowledgements
The authors thank Laura M. Roy for constructing the PsbS-Strep plants and Vincent M. Friebe for helpful technical advice. The project was supported by the Dutch organization for Scientific research via a VICI Grant to R.C. L.N. received financial support from the New Zealand Government through the Royal Society of New Zealand Te Apārangi Rutherford Foundation.
Author information
Authors and Affiliations
Contributions
Conceptualization, R.C.; methodology, L.N.; investigation, L.N.; writing—original draft, L.N.; writing—review and editing, R.C.; supervision, R.C.; funding acquisition, R.C.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nicol, L., Croce, R. The PsbS protein and low pH are necessary and sufficient to induce quenching in the light-harvesting complex of plants LHCII. Sci Rep 11, 7415 (2021). https://doi.org/10.1038/s41598-021-86975-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-86975-9
This article is cited by
-
The nature of carotenoid S* state and its role in the nonphotochemical quenching of plants
Nature Communications (2024)
-
Cryo-EM structures of LHCII in photo-active and photo-protecting states reveal allosteric regulation of light harvesting and excess energy dissipation
Nature Plants (2023)
-
The origin of pigment-binding differences in CP29 and LHCII: the role of protein structure and dynamics
Photochemical & Photobiological Sciences (2023)
-
Light quality as a driver of photosynthetic apparatus development
Biophysical Reviews (2022)
-
A different perspective for nonphotochemical quenching in plant antenna complexes
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
