
Abstract
The South Korean government effectively contained the coronavirus disease-2019 (COVID-19) outbreak primarily associated with a religious group. We conducted SARS-CoV-2 whole genome sequencing of 66 cases to investigate connections among the initial South Korean cases and the religious group outbreak. We assessed the accuracy of genomic investigation by comparing the whole genome sequences with comprehensive contact tracing records. Five transmission clusters were estimated among the 15 initial cases. The six close-contact cases and two potential exposure pairs identified by contact tracing showed two or fewer nucleotide base differences. Additionally, we identified two transmission clusters that were phylogenetically distinct from the initial clusters, sharing common G11083T, G26144T, and C14805T markers. The strain closest to the two additional clusters was identified from a pair of identical sequences isolated from individuals who traveled from Wuhan to Italy. Our findings provide insights into the origins of community spread of COVID-19.
Similar content being viewed by others


Introduction
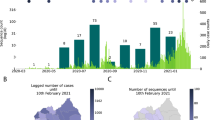
After identifying a massive outbreak of coronavirus disease-2019 (COVID-19) primarily within a religious group on February 18, 2020, the South Korean government immediately began contact tracing and mandated self-isolation for all members of the church (~ 240,000 individuals). As a result, the government was able to halt the spread of the virus at approximately 10,000 cases for more than 1 month (Fig. 1). Notably, the government of South Korea did not issue a country-wide stay at home order and only concentrated on individuals specifically identified through contact tracing.

South Korea’s COVID-19 outbreak and intervention timeline.
In retrospect, the prompt responses of the South Korean government were effective in containing the COVID-19 spread. However, this method required extensive in-person questionnaires that not only were susceptible to human error but were also often met with resistance owing to privacy concerns1. Herein, we propose performing genomic investigation as a supplementary tool to epidemiological contact tracing, which could help determine the current and future spread of the disease. With genomic investigation, we could detect the source of infection directly from the patient’s viral genotype and reduce uncertainties in transmission route identification, thereby guiding more effective intervention implementations2,3,4,5. In this study, we evaluated the accuracy of genomic investigation by directly comparing South Korea’s comprehensive contact tracing data with the genomic associations among severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) whole genome sequences.
Results
South Korea COVID-19 outbreak and response
When the Wuhan government first reported 27 cases of pneumonia on December 31, 2019, the South Korean government immediately issued the lowest threat alert (blue) and started to screen air travelers from Wuhan using infrared thermometers and a pan-corona virus PCR kit. Upon discovery of the first confirmed case on January 20, the government promptly raised the threat level to yellow and subsequently to orange on January 27 following the identification of a fourth case (Fig. 1). By February 17, 30 cases were identified, and 10 had already recovered. Additionally, 11 cases were confirmed to be caused by close contact, and 19 were confirmed to be imported.
The first instance of a massive outbreak in the church was identified on February 18, 2020. This case was a member of the church without any international travel records. Ten close contacts of this individual, mainly including church members, were confirmed on the following day, and positive cases from the church subsequently increased rapidly (Fig. 1). On February 20, another outbreak was identified at a hospital complex, and subsequently, the first COVID-19-related death was reported. By February 22, the total number of cases rose to 346, among which 169 (48.8%) and 108 (31.2%) cases were related to the church outbreak and the regional hospital outbreak, respectively.
The government started screening all church members in Daegu and all individuals at the hospital complex. When the cases rose to 600 on February 23, the government raised the alert level to red, officially declaring a national emergency (Fig. 1). On February 25, the Korean Government started massive contact tracing and real-time RT-PCR testing on the church members in South Korea, completing around 240,000 tests in just 16 days by March 12 (Fig. 1). The resulting exponential increase in cases, however, led to a shortage of negative pressure isolation rooms and overall burden on the South Korean healthcare system. Following this peak, there were several minor outbreaks related to the known cases, but all were successfully contained owing to rapid screening efforts and preemptive response measures for vulnerable groups. Additionally, instances continued to be imported, particularly from Europe and North America, and travel-related cases became greater than 10% of the total daily confirmed cases on March 31. In response, the government enforced a mandatory 14-day self-quarantine for all travelers on April 1 (Fig. 1). On May 8, one local case was identified, whereas 11 were deemed imported.
Demographics of patients with COVID-19
As of May 8, approximately 60% of total confirmed cases were female; however, the mortality rate was higher in males than in females (3.0% versus 1.9%, respectively; Table 1). Mortality rates also increase with age (0% and 1% for individuals below 30 years old and below 60 years old, respectively; 25% for individuals 80 years old and older). Additionally, 80.5% of total cases were part of local cluster outbreaks, and 10.8% were imported. Notably, 5212 confirmed cases related to the church outbreak were localized around Daegu (dark blue in Fig. 2), accounting for 48.1% of total confirmed cases in South Korea.

Regional distribution of COVID-19 cases by May 8, 2020 in South Korea. The majority of cases (6859 of 10,822 cases) occurred in Daegu (dark blue), and the total number of imported cases (marked by the airplane) was 470.
Genomic investigation cohort
We performed SARS-CoV-2 whole genome sequencing on 66 cases to investigate associations between the church outbreak and the initial cases within South Korea (Table 2). Short-read high-throughput sequencing was performed for whole genome coverage. The total number of filtered reads, number of reads mapped to the reference genome (Wuhan-Hu-1, NCBI Reference Sequence: NC_045512, GenBank accession number: MN908947.3), base coverage, and average sequencing depth are presented in Supplementary Table S1. As detailed in Table 2, we obtained viral sequences from (i) 15 initial cases (out of 30) prior to the church outbreak, (ii) 37 cases (out of 311) within the first 5 days of the church outbreak, and (iii) 14 other cases presumed to be related to the church outbreak by contact tracing records.
Initial cases
Our genomic investigation estimated five transmission clusters among the 15 cases prior to the church outbreak (Fig. 3). The first cluster (C1) contained four cases, i.e., cases 5174, 7156, 1534, and 7834, where all but case 5174 showed sequences identical to that of one of the earliest cases (Wuhan-Hu-1 strain: NCBI Reference Sequence: NC_045512, GenBank accession number: MN908947.3), and case 5174 harbored one nonsynonymous mutation (G26144T)6. This aligned with the reported contact tracing results (Fig. 3a), as follows: (i) case 7156 had interacted with a Wuhan resident and was a family member of case 1534; (ii) case 7834 had met a confirmed case from Wuhan, and (iii) case 5174 was from Wuhan. Taken together, all four individuals in C1 were either closely linked to Wuhan or had close contact of Wuhan cases, revealing a strong correlation between our genomic investigation and the epidemiological tracing.

Contact tracing record arc diagram (a) and maximum likelihood tree (b) of initial 15 cases prior to a church outbreak in South Korea. (a) Confirmed close contacts via epidemiological tracing are linked with a solid curve, with the transmission directionality (from case 8123 to case 7098, for example) indicated by width (wide: source; narrow: recipient). Potentially exposed pairs of individuals (within the same relative area) are marked with faded curves (for example, between Case 4536 and Case 9273). Imported (local) cases are presented by solid (faded) numbers. (b) Five transmission clusters inferred via maximum likelihood tree analysis on the SARS-CoV-2 whole genome sequences from the 15 cases: from top to bottom, cluster 1 (blue), cluster 2 (light blue), cluster 3 (green), cluster 4 (brown), cluster 5 (light brown). A star denotes sequences obtained by virus inoculation and cell culture. The length of the black bar indicates a Hamming distance (HD) of 1 or a one nucleotide base difference. The branch support values of the tree using the SH-like likelihood ratio test were estimated as around 1.
The second cluster (C2) consisted of cases 9273, 3542, 5431, 4536, 2314, a and 4023 (Fig. 3b). Case 9273 had been working in Wuhan when case 3542 had visited, and the sequence of the virus from case 9273 showed two mutations compared with case 3542 and case 5431, but shared two common mutations from the C1 founder sequence. Case 3542 had met case 5431, and the genomic sequences of the virus for these cases were identical. Case 4023 was a close-contact of Case 5431, with the sequences showing a difference of only one mutation. Case 4536 had worked in Wuhan prior to diagnosis in South Korea and interacted with case 2314; the genomic sequence of the virus from case 4536 showed one mutation compared with that of case 3542, whereas cases 4536 and 2314 had two nucleotide substitutions. Therefore, all confirmed close-contact cases had either identical sequences or sequence pairs with one nucleotide difference, whereas potential exposure pairs showed only two mutations.
The three other sequence clusters were also consistent with contact tracing data (Fig. 3). The third cluster (C3) has only one member, Case 3401, a Wuhan resident who did not have close contact with any of the other cases in South Korea. The fourth (C4) and fifth (C5) clusters each consisted of two members, who shared six and eight common mutation markers from the first cluster, respectively. Thus, our maximum likelihood tree constructed from mutation signatures was consistent with epidemiological contact tracing records, demonstrating the effectiveness of genomic sequencing in discerning close-contact transmission clusters.
Church outbreak analysis
Figure 4 shows the maximum likelihood tree of 51 cases after the onset of the church outbreak alongside the 15 initial cases that were sequenced. Most cases after the onset of the outbreak were phylogenetically distinct from the prior cases and formed two closely related, yet independent clusters: C6 (Fig. 4a, dark blue brackets), which mainly consisted of church members in Daegu; and C7 (Fig. 4a, light blue brackets), which consisted of all and only cases from the hospital.

Maximum likelihood tree of all 66 cases. (a) Maximum likelihood tree of all 66 South Korean cases that were sequenced. The church cluster (cluster 6) is marked by blue brackets, and the hospital cluster (cluster 7) is marked by light blue. The branch support values of the tree using the SH-like likelihood ratio test were estimated as around 1. The most recent common ancestor sequence of clusters 6 and 7 is presented by blue circle and the strain that shared two common mutation markers with this ancestor is presented by red circle. (b) Star-like phylogeny of the 37 church cluster cases (cluster 6, dark blue in (a)) (c) Star-like phylogeny of the 11 hospital cluster cases (cluster 7, light blue in (a)).
Additionally, both clusters displayed star-like phylogeny (Fig. 4b,c), a signature of random neutral evolution7. Both clusters had multiple identical sequences at the tree center (13 out of 37 for cluster 6 and six out of 11 for cluster 7), an evolutionary pattern frequently observed in acute-stage human immunodeficiency virus sequences within an infected individual8,9. As listed in Table 2, this group of identical sequences is a putative founder of each transmission cluster and only one mutation away (G5572T (NSP3-M951I) nonsynonymous mutation for cluster 6 and C26681T synonymous mutation for cluster 7) from their most recent common ancestor sequence (Fig. 4a, blue circle). The independent and random evolution patterns observed with the star-like phylogeny suggested allopatric speciation or diversification under geographic isolation. This was plausible given that most cases from the hospital occurred within the long-term care unit, which was isolated from the rest of the complex. Thus, the initial transmission of the two clusters may have occurred simultaneously at the hospital, based on the following observations: (i) the founder strains of the two clusters were one base substitution only from their most probable common ancestor; (ii) the common ancestor was not identified, despite the abundance of the two founders; and (iii) the hospital had an isolated setting.
Additionally, the church cluster (C6) and hospital cluster (C7) were unique, sharing common markers from the Wuhan-Hu-1 strain (two nonsynonymous mutations, G11083T (NSP6-L37F) and G26144T (ORF3a-G251V), and one synonymous mutation, C14805T), as presented in Table 2. As of May 8, 2020, no sequences within the two clusters were identical to any of the those registered in GISAID. Moreover, no GISAID sequences were identical to that of the most probable ancestor. However, we were able to identify a pair of identical strains that shared two common mutation markers (Fig. 4a, red circle) with this ancestor. These strains (GISAID access numbers: EPI_ISL_410546 and EPI_ISL_412974) were isolated from individuals who traveled from Wuhan to Italy, suggesting that Wuhan may have been the origin of the church and hospital clusters (see Supplementary Fig. S1).
The church cluster (C6) was a likely transmission origin of the other outbreaks that occurred outside Daegu, displaying genomic linkage with cases in Gwagju cluster (Case 2397, 8 total), Busan cluster (Case 2058, 4 total), and Gyeongsangbuk-do cluster (Case 7239, 3 total). Similarly, our genomic investigation suggested that multiple sporadic cases, such as Case 2697 in Gangwon-do and Case 4327 in Gyeonggi-do, could also be related to the church cluster (cluster 6).
Discussion
Genomic investigations can compensate for the limitations of epidemiological contact tracing. First, this method can identify hidden relationships among new cases and previous clusters. Epidemiological contact tracing was unable to find a plausible transmission source for case 6303, an individual who resides in Seoul. However, the genomic analyses in our study suggested that this case shared a possible ancestor with cluster 2. Second, genomic investigations can provide clarity in the event that there are multiple possible transmission scenarios. Case 2058 and 3495 had travelled to China and thus they might be the potential transmission source of the church cluster (C6). However, we estimated that these cases were not a transmission origin of the church cluster, given that the strain of these two cases had one respective additional mutation than the founder strain of cluster 6. Finally, this method could mitigate human bias and possibly override human error. For example, cases 9234 and 6712 were presumed to be within the church cluster as they were both church members who had traveled to Daegu. However, we observed that case 9234 belonged to cluster 5, whereas case 6712 belonged to cluster 2, suggesting that these individuals were infected from other sources prior to their visit to Daegu; additional tracing efforts may have revealed new information regarding the spread.
By overcoming the limitations of epidemiological contact tracing, our genomic investigation could lead to new outlooks regarding the spread of COVID-19. One key observation of our study was the incongruous spread throughout Seoul, specifically regarding the newly identified members of cluster 2. Epidemiological tracing records initially identified 15 close-contact cases associated with cluster 2 in Seoul and three of these close-contact cases were sequenced in this study (cases 4023, 5431, and 3542). However, case 6303, as described above, had sequence that was identical to that of cases 5431 and 3542, strongly suggesting that case 6303 and its 12 other related cases reported by contact tracing may belong to cluster 2. Moreover, cases 9273, 4536, 2314, and 6712 were also categorized into cluster 2 by genomic sequencing, as discussed above. Thus the size of this cluster becomes double, compared to the size estimated by contact tracing. These newly discovered connections suggested that there could be another unaddressed transmission source that could have caused the various outbreaks in Seoul.
In summary, our genomic investigation of the COVID-19 outbreak in South Korea was highly consistent with our comprehensive and rigorous contact tracing records. Furthermore, genomic investigation revealed missing linkages among individuals who had not been initially traced. By providing more insight into the source and direction of transmission, genomic investigation can help public health officials implement more targeted and specific policies that prevent the spread of the disease at minimal cost to society.
Methods
Ethical considerations
This study has been approved by the Institutional Review Board at the Korea Centers for Disease Control and Prevention (2020-03-01-P-A) and is considered to be a public health’s act to the outbreak. Thus, the board has waived the requirement for written consent as outlined in the Title Laboratory Respondence to COVID-19. All the methods presented in this study were conducted in accordance with the relevant guidelines and regulations.
Contact tracing and isolation
Following the strict regulations set forth by the Infection Disease Control and Prevention Act, the South Korean government created a comprehensive travel record of those with confirmed cases by collecting data via in-person questionnaire surveys and cross-checking with phone GPS traces, medical records, closed-circuit television, and credit-card transactions10. All asymptomatic close contacts (within 2 m) and potentially exposed individuals (within the same relative area) were mandated to self-quarantine for 14 days and were monitored twice a day by public health workers to check for the presence of fever or respiratory symptoms. A smartphone-based self-assessment application was implemented to further monitor the development of COVID-19 symptoms.
Specimen collection and processing
Remnant specimens from COVID-19 test-positive cases in the form of nasopharyngeal and oropharyngeal (NP/OP) swabs and sputum were used for SARS-CoV-2 RNA isolation5. RNA was extracted from 140 µL of these specimens using a Qiagen Viral RNA Mini kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol11.
Viral culture of SARS-CoV-2 was conducted in a Biosafety Level-3 facility according to the laboratory guidelines of Korea Centers for Disease Control and Prevention7. NP/OP swab specimens were inoculated into Vero E6 cells, and the cells were then cultured at 37 °C in an atmosphere containing 5% CO2 with 1× Dulbecco’s modified Eagle’s medium supplemented with 2% fetal bovine serum and penicillin–streptomycin. Virus replication and isolation were confirmed through cytopathic effects and gene detection by real-time reverse transcription polymerase chain reaction (RT-PCR).
SARS-CoV-2 whole genome sequencing
In total, 10–100 ng of the extracted viral RNA with a maximum volume of 8.5 µL was subjected to target enrichment using a Truseq RNA library prep for enrichment (Illumina. San Diego, CA, USA) and Truseq RNA Enrichment (Illumina). The enriched products were size-selected and evaluated for quality and concentration using a D1000 screen tape (Agilent Technologies) on a TapeStation 4200 (Agilent Technologies). The selected libraries were quantified using a Library Quantification Kit (KAPA Biosystems) on a Quantstudio 6 Flex Real-Time PCR System (ThermoFisher Scientific). The final loading concentration was 4–14 pM, with PhiX. High-throughput sequencing was performed on a MiSeq and NextSeq 500 sequencer with 2 × 150 base pairs (Illumina).
Dual-index filtering and adapter trimming were conducted on the sequences using our in-house scripts. For quality control, low-quality bases were removed by FaQCs software with the Q30 threshold. The NCBI Bacteria RefSeq and Human GRCh38 databases were used to remove host reads. Filtered reads were then aligned to the reference genome sequence of Wuhan-Hu-1 (NCBI Reference Sequence: NC_045512, GenBank accession number: MN908947.3) using Burrows-Wheeler Aligner6. SAMtools and VCFtools were used to generate the SARS-CoV-2 whole genome consensus sequence of each specimen.
Hybridization probes were designed to cover the whole genome of SARS-CoV-2 using the Wuhan-Hu-1 strain6. The biotinylated probes were 120 bp in length with 3× tiling (Celemics). In total, 745 conserved probes were generated for target enrichment during sequencing library preparation.
Maximum likelihood tree analysis
Maximum likelihood trees were generated using the PHYML program, which simultaneously adjusted the tree structure and branch length12. The general time-reversible model was used with the ‘ML’ option, and invariable sites were estimated with 12 substitution rate categories. The branch support values were estimated using the SH-like likelihood ratio test in PHYML3.313. The tree was generated by the BIONJ option and presented using FigTree.
Analysis of global SARS-CoV-2 strains
In total, 16,508 full-length SARS-CoV-2 sequences that were registered by May 7, 2020 were downloaded from GISAID (Supplementary Table S2)14,15. The sequences were globally aligned using MUSCLE16, and sequences at both the 5′- and 3′-ends were trimmed, yielding a 597–29,340 segment of the Wuhan-Hu-1 reference strain. Sequences shorter than this region and those with one or more ambiguous bases were filtered out, yielding 7645 fully aligned SARS-CoV-2 sequences.
Data availability
The SARS-CoV-2 whole genome sequences described are available in GISAID (Accession IDs EPI_ISL_407193, 412869, 412870–412873, 425117, 425118, 426163, 426164, 426166, 426168, 426169, 426171, 426173, 426180–426183, 426187, 427809–427813, 471425, 471426, 471438–471455, 481370–481379, 497951–497961, 497968–497971, 497974).
Code availability
Pre-existing software was used for our analysis.
References
Zastrow, M. South Korea is reporting intimate details of COVID-19 cases: Has it helped? Nature. https://doi.org/10.1038/d41586-020-00740- (2020).
Gudbjartsson, D. F. et al. Spread of SARS-CoV-2 in the Icelandic population. N. Engl. J. Med. 382, 2302–2315 (2020).
Pung, R. et al. Investigation of three clusters of COVID-19 in Singapore: Implications for surveillance and response measures. Lancet 395, 1039–1046 (2020).
Peters, P. J. et al. HIV infection linked to injection use of oxumorphone in Indiana, 2014–2015. N. Engl. J. Med. 375, 229–239 (2016).
Gardy, J. L. et al. Whole genome sequencing and social-network analysis of a tuberculosis outbreak. N. Engl. J. Med. 364, 730–739 (2011).
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Wakeley, J. Coalescent Theory, an Introduction (Roberts & Co., 2008).
Keele, B. F. et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. U.S.A. 105, 7552–7557 (2008).
Lee, H. Y. et al. Modeling sequence evolution in acute HIV-1 infection. J. Theor. Biol. 261, 341–360 (2009).
COVID-19 National Emergency Response Center, Epidemiology & Case Management Team & Korea Centers for Disease Control & Prevention. Contact transmission of COVID-19 in South Korea. Osong Public Health Res. Perspect. 11, 60–63 (2020).
Kim, J. M. et al. Identification of coronavirus isolated from a patient in Korea with COVID-19. Osong Public Health Res. Perspect. 11, 3–7 (2020).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Anisimova, M. & Gascuel, O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst. Biol. 55, 539–552 (2006).
Elbe, S. & Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 1, 33–46 (2017).
Shu, Y. & McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Euro. Surveill. 22, 30494 (2017).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Acknowledgements
This study was funded by the Korea Centers for Disease Control and Prevention (Grant No.: 4800-4837-301). We thank all those who assisted in the collection and transport of patient specimens, the many staff at the Korea Centers for Disease Control and Prevention and local government for their contribution to controlling COVID-19. We also thank the authors and laboratories of the sequences archived at GISAID’s EpiFlu Database listed in our acknowledgment table (Supplementary Table S2).
Author information
Authors and Affiliations
Contributions
J.K., S.P., H.L. and M.H. conceived this study. J.K., D.L., J.K., D.S., S. J., Y.C., C.Y. and M. H. performed SARS-CoV-2 whole genome sequencing and bioinformatics analysis. S.P. and H.L. conducted maximum-likelihood tree analysis and global SARS-CoV-2 whole genome sequence analysis. Y.P., J.G. and M.K. performed contact tracing. All authors wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, JM., Park, S.Y., Lee, D. et al. Genomic investigation of the coronavirus disease-2019 outbreak in the Republic of Korea. Sci Rep 11, 6009 (2021). https://doi.org/10.1038/s41598-021-85623-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-85623-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
