
Abstract
Brucellosis is one of the most common bacterial zoonoses worldwide affecting not only livestock and wildlife but also pets. Canine brucellosis is characterized by reproductive failure in dogs. Human Brucella canis infections are rarely reported but probably underestimated due to insufficient diagnostic surveillance. To improve diagnostics, we investigated dogs in a breeding kennel that showed clinical manifestations of brucellosis and revealed positive blood cultures. As an alternative to the time-consuming and hazardous classical identification procedures, a newly developed species-specific intact-cell matrix-assisted laser desorption/ionization–time of flight mass spectrometry analysis was applied, which allowed for rapid identification of B. canis and differentiation from closely related B. suis biovar 1. High-throughput sequencing and comparative genomics using single nucleotide polymorphism analysis clustered our isolates together with canine and human strains from various Central and South American countries in a distinct sub-lineage. Hence, molecular epidemiology clearly defined the outbreak cluster and demonstrated the endemic situation in South America. Our study illustrates that MALDI-TOF MS analysis using a validated in-house reference database facilitates rapid B. canis identification at species level. Additional whole genome sequencing provides more detailed outbreak information and leads to a deeper understanding of the epidemiology of canine brucellosis.
Similar content being viewed by others


Introduction
Brucellosis is a widespread zoonosis in livestock, mainly in cattle, sheep as well as goats, and pigs caused by Brucella abortus, Brucella melitensis, and Brucella suis, respectively. Humans are usually infected through direct animal contact or by the consumption of unpasteurized dairy products and undercooked meat. Both, the lack of surveillance and control of the classical Brucella species in some regions1 and the lack of awareness of novel and atypical species2,3 contribute to an underestimation of the actual prevalence of brucellosis worldwide, especially in developing countries.
Canine brucellosis was first described in the late 1960s4 and Brucella canis has been identified as the most common cause of brucellosis in dogs with breeding kennels showing high prevalence rates all over the world5,6,7. The infection is transmitted among dogs by intercourse, ingestion of abortion material or reproductive secretions. Moreover, urine of affected dogs is infectious due to contamination with semen8,9. The control of canine brucellosis is difficult because dogs maintain close contacts within the population and common management practices in breeding kennels favor the spreading of disease10.
Besides B. canis, Brucella suis has been reported as another cause of canine brucellosis11,12,13. Especially B. suis biovar (bv) 1, commonly isolated from feral pigs (Sus scrofa)14,15, was found in infected dogs12,16. In contrast to B. canis, which mainly infects domestic dogs, B. suis infections occur in hounds that participate in wild boar hunting or are fed with raw boar meat16,17,18. B. suis bv 4, the causative agent of reindeer brucellosis in Alaska, Canada and Russia was isolated from wolves, the native relative of dogs, and other carnivores like foxes and wolverines19,20.
Brucella canis is presumed to be less pathogenic to humans than B. suis. However, B. canis may be transmitted from dogs to humans, and not only mild clinical symptoms, such as fever, headache, anorexia and fatigue, have been reported in patients, but also severe and/or chronic manifestations, such as peritonitis21, endocarditis22, osteomyelitis23 and arthralgia24. Likewise, the virulence of B. suis varies between its five biovars, with biovar 1 being highly pathogenic to humans and responsible for severe and chronic courses of the disease25,26,27.
Considering the emerging zoonotic potential of B. canis as well as B. suis bv 1 and 4, diagnostic tools are necessary to monitor and control canine brucellosis. Unfortunately, infected dogs cannot be easily distinguished from non-infected animals by clinical examination due to the high number of asymptomatic dogs8,28. Therefore, fast and reliable laboratory tests are indispensable for the identification of dogs infected with B. canis or B. suis to contain outbreaks within the dog population and prevent transmission to humans. Since B. canis lacks the immunodominant O-polysaccharide of the smooth Brucella species29, the standard serological tests based on a B. abortus-antigen, which are routinely used for the diagnosis of human brucellosis cases, cannot detect anti-B. canis antibodies30. Hence, the diagnosis of a B. canis infection in humans remains a challenge.
The objective of our study was to compare the proficiency of traditional and new omics-based methods in identifying the infectious agent in Brazilian dogs that showed brucellosis-like disease manifestations.
Results
Serological examination of dogs with reproductive failure
In 2013, the pregnancy of a female dog in a breeding kennel in São Paulo, Brazil, comprising 17 adult pugs, 2 males and 15 females, resulted in abortion. Another 5 pregnancies ended in abortion in the following four months (Fig. 1A). To clarify whether the observed abortions were a consequence of B. canis infections, all animals were clinically examined. While three dogs showed no clinical manifestations related to canine brucellosis, 14 dogs suffered from at least one brucellosis-like symptom (Fig. 1B). Vaginal discharge and general lymphadenopathy were the most common clinical manifestations, followed by abortion/stillbirth and infertility (defined as inability to get pregnant and produce viable offspring) (Fig. 1B).

Suspected outbreak of canine brucellosis in a breeding kennel in São Paulo, Brazil. (A) Schedule of clinical investigations to clarify the suspected canine brucellosis outbreak. (B) Clinical manifestations observed in 14 out of 17 dogs in the kennel.
To test whether the animals had developed an immune response against Brucella antigens, we collected serum samples from each dog at three consecutive time points (Fig. 1A and Supplementary Table S1) and subjected them to classical serological tests, namely the Immunochromatographic Test (ICT), the Rapid Slide Agglutination Test (RSAT) and a B. canis IgG ELISA. In all 17 dogs, we detected antibodies towards rough Brucella antigens at each sampling date with at least one of the three conducted serological tests (Fig. 2). In several cases, serological results were equivocal, because the tests revealed contradictory results. For example, the serum from the bacteremic dog D09 did not consistently react against the rough antigen of Brucella in the ICT at every sampling date, but in RSAT and ELISA. At the same time, when 2-mercaptoethanol (2ME-RSAT) was applied, which inactivates IgM in the RSAT, we could not detect any antibody reaction in the serum of this dog (Fig. 2).

Blood culture and serological test results of dogs from a kennel with a suspected outbreak of canine brucellosis. To determine whether the 17 dogs from the kennel under study were infected with Brucella, whole blood samples were incubated in selective medium (BC). Additionally, dogs were serologically tested using an immunochromatographic test (ICT), the rapid slide agglutination test (RSAT), the 2-mercapthoetanol rapid slide agglutination test (2ME-RSAT) and an enzyme-linked immunosorbent assay (ELISA). Positive test results are shown in dark and negative ones in light color. A few tests gave inconclusive results (i) or were not conducted (nc); ID: identification code of the dogs; G: gender (M: male, F: female); S1, S2, S3: sampling dates in March, August and November 2014, respectively; *no serum sample available.
Phenotypic characterization of Brucella isolates
To assess the infection status of the kennel and to check whether the disease symptoms were indeed a result of an infection with B. canis, blood samples were taken from each individual animal at three different time points and aliquots were plated on Brucella-selective agar after enrichment broth culture. Gram-negative, short rod-shaped, non-motile bacteria were isolated from 15 out of 17 dogs at least once. A total of 29 isolates from 51 blood samples were obtained (Fig. 2 and Supplementary Table S1) and identified by genus-specific PCR as Brucella. Subsequently, genomic DNA was extracted from the 22 blood samples with negative culture results. In three of these blood samples (13.6%) the IS711 marker gene of Brucella could be amplified by PCR (Supplementary Fig. S1), which suggests Brucella infection in these dogs although the isolation of the bacterium had failed.
The isolated bacteria were further characterized with phenotyping methods commonly used for the identification of the genus Brucella and subtyping of its species and biovars (Supplementary Table S2). The isolates did not require CO2 for growth and could be cultured in ambient air (21% O2) on blood agar as well as on Brucella Agar with or without serum. The bacteria showed oxidase, catalase and urease activity but no H2S production similar to the reference strain B. canis RM 6/66. Moreover, the isolates grew on thionine and basic fuchsin-supplemented Brucella Agar plates comparable to B. canis RM 6/66. Additional phage typing experiments revealed the same lysis patterns for the isolated bacteria (constantly lysed by phage R/C, but not by any other tested phage) and B. canis RM 6/66 (Fig. 3 and Supplementary Table S3). The isolates did not agglutinate with anti-A and anti-M but with anti-R sera, specific for Brucella species expressing rough lipopolysaccharide (LPS). Likewise, LPS phenotyping with crystal violet staining revealed that all isolates expressed a rough LPS, which is characteristic of B. canis and B. ovis.

Phage typing of Brucella isolates. Presented are the phage lysis patterns of the B. canis isolates after incubation with the bacteriophages Tb, Wb, BK2, F1, F25, Iz, Fi, and R/C. Black squares indicate lysis of the bacteria, and white squares indicate no lysis; v: variable. The phage lysis patterns of the reference strains B. canis RM6/66, B. melitensis 16 M, B. abortus 544, and B. suis 1330 are shown as determined in our study.
Taken together, the classical microbiological methods clearly diagnosed an infection of the dogs with Brucella and suggested B. canis as the disease-causing agent.
MALDI-TOF MS-based identification of the canine Brucella isolates
To establish an alternative to the traditional time-consuming Brucella diagnostic methods that are based on phenotypic testing, we took advantage of MALDI-TOF MS. Identifying closely related Brucella species beyond the genus level with MALDI-TOF MS has been challenging31 since only few Brucella entries are available in the Security-Relevant reference library used by the Bruker Biotyper software (Bruker Daltonics). Consequently, the here described Brucella isolates (Supplementary Table S4) were initially misidentified as B. melitensis with scores between 2.0 and 2.3, although their mass spectral patterns were noticeably different from that of B. melitensis 16 M (Supplementary Fig. S2) but highly similar to B. canis RM 6/66 (Fig. 4A). It has been shown that in particular the discrimination of B. canis from B. suis bv 3 and bv 4 by MALDI-TOF MS is very difficult32,33,34 even with customized spectral libraries. We therefore improved MALDI-TOF MS-based identification of Brucella spp. by optimizing a previously established in-house spectral library32 with modified scoring weights for discriminant m/z peaks in the Biotyper software. Our B. canis isolates shared a highly similar mass spectral profile with B. suis bv 4 strains (Fig. 4B) as described32, but could be clearly distinguished from B. suis bv 1 (Fig. 4B), which is one of the frequently found Brucella species in dogs apart from B. canis.

Identification of Brucella canis from diseased dogs by MALDI-TOF MS. (A) Comparison of pre-processed and normalized spectra of the reference strain B. canis RM 6/66 and a representative isolate from the kennel under investigation in the m/z range 3–12 kDa; (B) Gel view depiction of averaged group spectra. Arrows indicate m/z positions with high divergence in either intensity or mass-to-charge ratio between B. canis, B. suis bv 4 and B. suis bv 1; (C) A biomarker for discrimination of B. canis and B. suis bv 4 versus B. suis bv 1 found by Karger et al.32 and its double (D) and triple charged (E) ions (both from this study); (F) The mass peak at m/z 7073 is a unique biomarker for the group B. canis and B. suis bv 4; (G) B. canis and B. suis bv 4 may be discriminated from B. suis bv 1 by their higher intensity mass peaks at m/z 7661 (single charged) and (H) m/z 3830 (double charged). (I + J) Peaks at m/z 5900 and m/z 3926 discriminate B. canis from B. suis bv 4.
By visual comparison of the MALDI-TOF MS spectra we were able to identify additional discriminant biomarker signals for B. canis and B. suis besides the previously described biomarkers at m/z 5833 and m/z 9076 specific to canis-vs-suis bv 132, corresponding to m/z 5834 (Supplementary Fig. S3A) and m/z 9075 (Fig. 4C), respectively. Both peaks represent single charged molecules and we could also detect their corresponding double or triple charged ions at m/z 2915, m/z 4539 and m/z 3024 (Supplementary Fig. S3B and Fig. 4D,E), respectively. In particular, the peak signals at m/z 9109 (single charged) and m/z 4555 (double charged) occurred prominently in B. suis bv 1 but not in B. canis or B. suis bv 4.
Brucella canis and B. suis bv 4 share a unique biomarker at m/z 7073 allowing for distinction from B. suis bv 1 (Fig. 4F) and other B. suis biovars (Supplementary Fig. S4). Further comparison of spectra revealed many differences in peak intensities rather than a strict peak absence or presence. Hence, the peaks at m/z 7661 (single charged) and m/z 3830 (double charged) can be used for the discrimination of B. canis and B. suis bv 4 against B. suis bv 1 (Fig. 4G,H) as well as other B. suis biovars (Supplementary Fig. S4). While an accurate and unambiguous distinction between B. canis and B. suis bv 4 by MALDI-TOF MS has been difficult in previous studies32, the isolates used in this study exhibited two signals at m/z 5900 and m/z 3926 that were mainly found in B. canis but not in B. suis bv 4 strains (Fig. 4I,J).
In summary, our MALDI-TOF MS-based characterization of the Brucella isolates from diseased dogs enabled fast and reliable identification beyond the Brucella genus level and had sufficient discriminatory power to distinguish B. canis from the closely related B. suis bv 1 as well as B. suis bv 4 isolates.
Genomic and phylogenetic characterization of the Brucella canis isolates
Besides the serological, biochemical and MALDI-TOF MS-based identification tools described above, molecular methods like ribotyping, pulsed-field gel electrophoresis (PFGE), multilocus sequence typing (MLST) or multiple locus variable number of tandem repeats (MLVA) analysis are used for subtyping of Brucella spp.35,36,37 and for the differentiation of B. canis subclades38. As a consequence of the high genetic homology among Brucella species39, we performed next-generation sequencing (NGS) analyses for a more sophisticated phylogenetic characterization of the Brucella isolates in our study. Analyzing the assembled DNA sequencing short reads clearly identified all isolated bacteria as B. canis with an average draft genome size of 3,295,525 bp and an average G + C content of 57.27% (Fig. 5 and Supplementary Table S5). Excluding lower quality sequences, the applied short read sequencing technique allowed de novo genome assemblies with 24 to 57 contigs. Repetitive elements like IS711, ISBm2 and ISBm3, IS1953 and IS2020 insertion sequences or rRNA operons hampered the completion of contiguous, circular chromosome sequences, a typical drawback of Illumina and other next-generation sequencing techniques based on short read lengths40.

Consensus genome sequence of the Brucella canis outbreak strain from a kennel in São Paulo, Brazil. Shown are the two chromosomes of the B. canis BfR-SPBR-consensus genome derived from whole genome sequencing results of four B. canis isolates. The genomes were sequenced using Illumina NGS technology. The open reading frames (CDS) determined by Prokka v1.1289 are presented in the two outer circles with grey (+ strand) and black (-strand) boxes. Positions with nucleotide sequence variations in the B. canis BfR-SPBR-consensus genome compared to the reference strain B. canis ATCC 23365 are depicted in the inner circle and marked with the colors red (stop-loss variants), orange (missense variants), blue (upstream or downstream variants) and green (synonymous variants). Open reading frames affected by missense variants or stop-loss variants are shown in red together with the corresponding protein IDs given as NCBI numbers (WP_xxxxxxxxx.1).
Detailed genome sequence comparisons revealed a high clonality, with only seven SNPs among the B. canis isolates (Supplementary Table S6). Apart from isolate I16, which harbored mutations at two different positions, five B. canis isolates (I5, I8, I19, I22, I23) differed only by one nucleotide within their genome sequences, which strongly indicates a clonal origin of these bacteria. In four cases, nucleotide variations were identified in Brucella isolates from dogs at later sampling dates (Supplementary Tables S1 and S6). Two SNPs, A2198519T in I8 and C642523T in I23, may be the result of post-isolation, lab-acquired mutations or an artefact of the low sequencing coverage (5 ×) for these nucleotide positions. To create an improved consensus genome, sequence reads of four high-quality draft genomes without SNP deviations among the Brucella isolates were concatenated by SPAdes de novo assembly. The resulting B. canis BfR-SPBR-consensus genome consists of 23 contigs (≥ 500 bp) and has a size of 3,293,143 bp, which is about 19.6 kb shorter than the reference genome sequence of B. canis ATCC 23,365 with 3,312,769 bp.
Sequence variants between the B. canis consensus genome described here and the B. canis ATCC 23365 genome were determined by applying the SNP calling strategies of BioNumerics and Mauve/ParSNP. The BioNumerics read mapping approach revealed 179 positions with high sequence read coverage and five positions with low sequence read coverage SNPs, whereas the analysis with Mauve identified 183 SNPs in the assembly-based approach. Analyses with these two different bioinformatics tools identified 180 SNP positions in common that distinguish the Brucella isolates found in dogs of the kennel from the reference strain Brucella ATCC 23365 (Fig. 5). Besides various SNPs in non-coding regions of B. canis BfR-SPBR-consensus, 57 and 47 SNPs were identified in coding regions of chromosome 1 and chromosome 2, respectively.
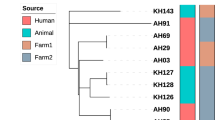
To illustrate the relationship of B. canis BfR-SPBR-consensus with previously sequenced strains, we performed a core genome multi-alignment with ParSNP using all publicly available B. canis draft or complete genome sequences and included the B. suis bv 4 reference strain 40 as the outgroup (Fig. 6). This analysis clearly placed the B. canis BfR-SPBR-consensus in a clade together with other Brazilian B. canis isolates but also with strains from neighboring countries. The most closely related strains to B. canis BfR-SPBR-consensus were B. canis 10469 and B. canis 07-2859-6070, also isolated from kennels in São Paulo in 2005 and 199841, respectively, whereas the genome sequences of B. canis strains from Chile and Colombia revealed more genetic variations. Interestingly, the human B. canis CNGB 1324 isolate from Argentina clustered within the group of Brazilian B. canis isolates from dogs (Fig. 6).

Phylogenetic comparison of the Brucella canis BfR-SPBR-consensus strain newly identified in São Paulo with isolates from worldwide outbreaks. The genetic relationship between the outbreak strain B. canis BfR-SPBR-consensus and previously sequenced B. canis strains was determined by SNP analysis. The genome sequences were analyzed with ParSNP, FastTree2 and iTol. The B. suis outgroup strain and the B. canis outbreak strain under study are marked in bold. Shown is a neighbor-joining phylogenetic tree with the branch length displaying the relative genetic distance. All bootstrap support values were either below 0.5 or above 0.8.
Detailed comparison of the SNP distribution in B. canis BfR-SPBR-consensus and other South American B. canis strains found several conserved missense and synonymous mutations in coding regions, which were absent in B. canis ATCC 23365 (Fig. 7). SNPs leading to stop-loss mutations in genes were conserved in most of the South American B. canis isolates as well (Table 1). In contrast, SNPs resulting in stop-gain mutations were less frequent and occurred especially in B. canis strains from Colombia and Chile (Table 1). The described stop-loss and stop-gain mutations affected genes with physiological functions like nutrient utilization but also potential classical virulence traits such as an autotransporter protein of the type V secretion family, including adhesins, invasins, toxins and proteases, found in various bacterial pathogens42.

Distribution of single-nucleotide polymorphisms (SNPs) in South American Brucella canis isolates. The positions of SNPs in the genomes of South American B. canis strains are shown in comparison to the reference strain B. canis ATCC 23365. The two chromosomes of the B. canis isolates are color-coded and the background colors show the geographical origin for each strain. The strains are presented in the following order from inside to outside: 10469 (1), B. canis BfR-SPBR-consensus (2), 07-2859-6070 (3), CNBG 1324 (4), 07-2859-6071 (5), CNGB 513 (6), SCL (7), Oliveri (8), CNGB 1172 (9) and ATCC 23365 (10). SNPs are shown as colored strokes in red (stop-loss variants), in light pink (stop-gain variants), orange (missense variants), blue (upstream or downstream variants) and green (synonymous variants).
Summarizing, our genetic analysis clearly reveals the clonality of the Brucella isolates from the diseased kennel in São Paulo and suggests a single-strain outbreak. Moreover, we identified SNPs in the South American B. canis isolates that may indicate diverse phenotypic properties of representatives within this subclade in spite of their close genetic relationship.
Discussion
Infections of dogs with B. canis and B. suis may be asymptomatic or lead to similar clinical manifestations in the infected animals like reproductive failures due to infertility, abortion, stillbirth, orchitis, epididymitis and prostatitis10,43,44. In recent years, not only dog-to-dog and pig-to-dog transmissions of B. canis and B. suis, respectively, have been reported, but both species have also been transmitted from dogs to humans. Hence, canine brucellosis mainly caused by B. canis but also by B. suis is an emerging zoonosis worldwide18,26,27,45,46,47,48. Human infection may lead to various unspecific symptoms such as prolonged fever, shivering, chills, night sweats, weight loss, overall weakness or malaise, headache, enlarged lymph nodes, back pain, and arthralgia10. Therefore, fast and reliable laboratory methods for the identification of canine Brucella species and for epidemiological investigations on single strains are essential to prevent the spread of infection among dogs and transmission to humans.
For this purpose, we examined a potential brucellosis outbreak in a kennel of 17 dogs by comparing the traditional Brucella diagnostic tools with the latest proteomics- and genomics-based methods. Fourteen out of 17 animals exhibited clinical manifestations associated with canine brucellosis. One female and two male dogs were asymptomatic, which raised the question of whether these animals were subclinically infected (healthy carriers) or not infected. Subsequent blood cultures allowed us to isolate Brucella from two out of the three dogs confirming previous reports that infected dogs do not necessarily develop disease-specific symptoms28,43. Therefore, the isolation of bacteria is considered the gold standard method and the most reliable way to confirm canine brucellosis. Moreover, bacterial isolates are required for phenotypic characterization49,50 in order to differentiate between the Brucella species potentially infecting dogs8. By performing the microbiological and biochemical tests commonly used for the classification of Brucella spp., we were able to identify all bacteria isolated from the kenneled dogs as B. canis. The phenotypic traits of our isolates were consistent with those of the reference strain B. canis RM6/66.
In about 40% of the analyzed blood samples, especially from later sampling dates, the isolation of bacteria was unsuccessful although Brucella could be recovered from preceding blood samples of respective animals. Obviously, diagnosis by culture may produce false-negative results, particularly in the later stage of infection when bacteremia ceases or becomes intermittent51,52. Although we were unable to isolate Brucella from the blood of several infected dogs at later sampling dates, we could still detect Brucella DNA by genus-specific PCR analysis, which further confirms that these dogs had been infected with Brucella. Our results verify previous observations that blood culture may be insufficient to diagnose canine brucellosis, especially since attempts to recover B. suis bv 1 from blood and urine had failed while bacteria could be isolated from the semen of dogs with clinical manifestations of brucellosis53.
Because of the drawbacks of blood culture for diagnostic purposes, serodiagnosis is commonly used to detect Brucella infections in animals. Serological approaches take advantage of seroconversion in infected hosts and detect host antibodies that react with Brucella antigens. These anti-Brucella antibodies can still be detected several years after the acute stage of infection28,43,54. Our serological screening of the 17 dogs using ICT, ELISA and RSAT suggested that all animals were infected with Brucella, even the two dogs with negative blood culture results at any sampling date. However, the results of the three serological tests were inconclusive in various cases, since some serum samples with positive ELISA results showed negative results with ICT or RSAT. Moreover, we observed negative serological test results in samples from which Brucella had been isolated. Hence, our analysis confirmed previous studies on the serological diagnosis of canine brucellosis demonstrating that although the detection of antibodies against rough Brucella species is widely used, misdiagnosis due to sensitivity and specificity failures may occur when these tests are applied43,54,55,56,57,58. False-negative results can be a consequence of testing during the initial phase of infection prior to seroconversion, or of low levels of circulating antibodies in chronically infected dogs, after bacteremia has ceased51,59. Serological titers may also wax and wane during bacteremia leading to false-negative results28. False-positive results, for instance, may occur as a consequence of cross-reactivity of anti-Brucella antibodies directed against other pathogens. For B. canis harboring a rough LPS, cross-reactivity with Streptococcus, Staphylococcus, Bordetella, and Pseudomonas has been described, whereas the food- and waterborne pathogens Escherichia coli O157:H7, Yersinia enterocolitica O:9, Salmonella Typhimurium (group N; O:30), and Vibrio cholerae O1 induce immune responses that generate antibodies reacting against the smooth LPS of B. suis43,60,61,62.
The species identification of our Brucella strains as B. canis with traditional microbiological, serological, biochemical and phage typing methods required several working days. Therefore, one focus of our study was to compare MALDI-TOF MS, as a more reliable and faster identification and differentiation tool for Brucella spp., with the classical phenotyping tests. Previous studies have shown that the application of MALDI-TOF MS with the currently available public libraries is a highly sensitive and specific technique for genus identification33,63,64,65. However, due to the close genetic relationship among Brucella spp., MALDI-TOF MS analyses has not yet allowed for a robust and unambiguous discrimination of Brucella species or their biovars. With our present work we significantly improved the current MALDI-TOF MS-based diagnostics of canine Brucella species by determining new biomarkers that enabled us not only to distinguish between B. canis and B. suis bv 1 but also between B. canis and the phylogenetically much closer B. suis bv 4. Brucella canis has previously been characterized by MALDI-TOF MS from blood cultures of infected dogs63, but these isolates could only be correctly identified at species level by adding customized main spectra of B. canis to the Bruker SR library containing B. melitensis as the sole representative of the genus Brucella. The obtained Bruker Biotyper identification scores consequently supported identification of B. canis with high probability, since the lack of complementary main spectra from other Brucella spp. constrains identification results to the species available in the used library. Purvis et al.63 did not evaluate the discriminatory power of MALDI-TOF MS analysis for B. canis identification against an expanded database containing reference spectra from closely related species, which, in our opinion, is a prerequisite for the reliable identification of B. canis and discrimination from B. suis. Our here presented weighted pattern matching approach overcomes this shortcoming, and its ability to identify and distinguish B. suis bv 1 from B. suis bv 4 and B. canis provides a valuable benefit for the diagnosis of canine brucellosis. The discriminatory power of MALDI-TOF MS can also be of public health importance because the transmission of B. suis has been reported not only from feral pigs and hares to dogs but also from feral pigs to domestic pigs and to humans18,27,66,67. The implementation of our optimized MALDI-TOF MS identification approach for B. canis and B. suis will speed up the specific diagnosis of canine brucellosis as described for the new diagnostic routines with other pathogens68.
When we complemented our MALDI-TOF MS analysis with whole genome sequencing data of Brucella isolates from the different dogs of the kennel, we were able to determine that the diverse clinical presentations of the animals were indeed the result of a B. canis single-strain outbreak. A detailed SNP comparison of the consensus sequence derived from our B. canis isolates with published B. canis genome sequences clustered the outbreak clone with various strains from South America comprising a clade that was clearly distinguishable from B. canis isolates from other parts of the world41. The genetic proximity of the Brazilian and Argentinian B. canis strains indicates that the infection circulates in kennels by cross-border trade of dogs, which may finally lead to a high risk for public health due to the close contact of humans with dogs and the lack of surveillance for canine brucellosis. The close genetic relationship of certain B. canis strains isolated from dogs and humans hints to a yet underestimated zoonotic transmission of this pathogen. Such transmission was recently documented with the infection of a 3-year old child by the same B. canis strain found in the blood of the child’s puppy46.
The close phylogenetic relationship of the South American B. canis strains based on the SNPtree analysis suggested that they represent a homogenous group with similar properties. However, our detailed and genome-wide analysis of single nucleotide mutations revealed that various SNPs affect coding regions either resulting in gene inactivation or activation of pseudogenes. Consequently, this genetic microdiversity might lead to more pronounced functional differences than their genetic homology may imply. Future studies have to address the extent to which such phenotypic differences exist and how they might affect the virulence of specific B. canis strains.
Our study supports previous notions7 that B. canis is prevalent in Brazilian breeding kennels and can be readily spread. These cases highlight the need to assess potential public health risks associated with the trade of kennel dogs or the handling of hunting dogs. In this context, new surveillance and control measures are indispensable to protect animal and human health. The rapid identification of the pathogen involved in kennel outbreaks is an important step to prevent the spreading of canine brucellosis. When the infection is confirmed in a dog population, the detailed characterization of the Brucella strains is required to trace back the infection. Since the members of the genus Brucella show high DNA sequence identity and clonal evolution, high-resolution typing methods are necessary to perform epidemiological analyses. Our study illustrates that proteomics and genomics approaches are a necessary complement to the traditional tests used in Brucella diagnostics because these novel techniques may speed up the reliable diagnosis of canine brucellosis.
Methods
Serodiagnosis of canine brucellosis
Blood sampling from dogs was performed according to a protocol approved by the Ethics Committee of the Faculty of Veterinary Medicine and Animal Science at the University of São Paulo, Brazil, under protocol CEUA 3113091015/2015, and in accordance with the relevant guidelines and regulations of the National Council for Control of Animal Experimentation (CONCEA). All blood samples were collected after mutual consensus between the owner of the kennel and the veterinarians responsible. Blood samples (~ 5 ml) were aseptically taken by cephalic or jugular venipuncture from dogs in a kennel located in São Paulo, Brazil. Half of the collected blood volume was mixed with sodium citrate as an anticoagulant. The remaining blood clotted at room temperature, was centrifuged and serum was stored at − 20 °C until use in serological tests. To test for anti-B. canis antibodies, we performed immunochromatographic tests (ICT; Rapid Canine Brucella Ab Test Kit, Bionote, Hwaseong, South Korea), ELISA (Novateinbio Kit, Cambridge, MA, USA) and rapid slide agglutination tests (RSAT; Canine Brucellosis Antibody Test Kit, D-TEC CB, Kansas City, MO, USA) with or without 2-mercaptoethanol (2ME) according to manufacturers’ instructions. The 2ME-RSAT was only conducted on serum samples tested positive by RSAT.
Isolation of B. canis from blood culture and phenotypic characterization
Full blood samples containing sodium citrate were used for culture as previously described69. Briefly, enrichment culture was performed in tryptose phosphate broth (Difco) including 5% fetal calf serum (FCS; Cultilab) at 37 °C for 30 days, followed by subcultures every four days on solid tryptose agar, also supplemented with 5% FCS. The isolated bacteria were primarily screened to confirm Brucella spp. colonies using the genus-specific PCRs targeting IS71170 and bcsp3171. All Brucella isolates collected on the first and second sampling date (Supplementary Table S1) were further characterized using morphological, biochemical and metabolic tests, such as Gram, Stamp and crystal violet staining, CO2 requirement, H2S production, oxidase and catalase activity, urea hydrolysis, agglutination with monospecific sera (anti-A, anti-M, and anti-R), dye sensitivity (basic fuchsin and thionine), bacterial motility (triphenyl tetrazolium chloride solid agar), and phage lysis (F1, F25, Tb, BK2, Iz, Wb, R/C) in order to identify and sub-differentiate Brucella spp.30.
Polymerase chain reaction (PCR) to detect Brucella DNA in canine blood samples
The whole blood samples were also used for direct detection of Brucella DNA by IS711 PCR when bacterial isolation was not successful. Briefly, the extraction of the bacterial DNA was based on mechanical and enzymatic pre-lysis of leukocytes, using zirconia/silica beads and lysozyme, respectively, followed by overnight enzymatic lysis using proteinase K and sodium dodecyl sulphate (SDS). The DNA was finally purified by phenol/chloroform and alcohol precipitation as described previously71,72. DNA gel electrophoresis images of the PCR products were edited using Adobe Photoshop CS5 (Adobe Systems, San Jose, CA, USA).
Matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS) to sub-differentiate Brucella species
Brucella isolates were primarily inactivated in 75% ethanol for at least 90 min and stored at − 20 °C until use. Samples were prepared for mass spectrometry by ethanol-formic acid extraction according to manufacturer’s instructions before being spotted on a 96-spot steel plate target and covered with alpha-cyano-4-hydroxy-cinnamic acid (HCCA) matrix solution (Bruker Daltonics). Mass spectra were acquired using a Bruker Microflex LT MALDI-TOF MS system (Bruker Daltonics). Spectra were initially analyzed using the Bruker Biotyper 3.1 software with MSP library version MBT 7311 (7311 entries) and the Security-Relevant (SR) Database (104 entries). None of the commercial libraries contained reference spectra of B. canis. Therefore, we complemented the database with in-house mass spectra of various B. canis and B. suis strains (obtained from Karger et al. (2013)32 and this study).
For each sample, a total of 24 spectra were measured, analyzed and visualized with the commercial Biotyper software as well as the open source statistical computing environment R v3.6.373 using the packages MALDIquant v1.19.374 and Tidyverse v1.3.075.
Bruker Biotyper identification scores for each sample were calculated from a main spectrum (MSP) composed of the 20 best quality technical replicate spectra following manufacturer’s instructions. Identification scores were interpreted accordingly, with scores ≥ 2.3 indicating highly probable species identification, scores between 2 and 2.3 indicating secure genus identification and probable species identification, scores between 1.7 and < 2 indicating probable genus identification and scores < 1.7 indicating no reliable identification.
Spectra processing with MALDIquant was performed similar to MSP creation by calculating an averaged spectrum of all technical replicate spectra of a sample. Averaged group spectra represent the mean of all available Brucella field isolates and reference strains for the respective species or biovar.
Next generation sequencing (NGS) of the Brucella canis isolates
Sample preparation
Brucella canis was grown on Brucella Agar and the DNA of the bacteria was extracted using the PureLink Genomic DNA extraction kit (Invitrogen) according to the manufacturer’s protocol. The libraries for whole-genome sequencing were prepared with a Nextera XT sample preparation kit. An Illumina MiSeq platform with a 2 × 300 cycle, paired-end configuration was used for sequencing.
Bioinformatics analysis
Sequence read quality was analyzed with FastQC v0.11.5 (Babraham Bioinformatics). The sequence reads and publicly available read files from the Sequencing Read Archive (https://www.ncbi.nlm.nih.gov/sra/) were assembled with SPAdes v3.10.0 (BayesHammer read error correction) and the options –careful (post-processing mismatch corrector with BWA) and –cov-cutoff auto76. Assembly quality was analyzed using Quast v4.577 by comparison to the type strain B. canis ATCC 23,365 genome (GenBank file GCF_000018525.1, accession no. NC_010103.1 for chromosome 1 and accession no. NC_010104.1 for chromosome 2 with a genome size of 3,312,769 bp in total). Sequence reads were mapped with BowTie2 v2.3.078 using default parameters against the complete genome of B. canis ATCC 23365 or the consensus genome of the B. canis outbreak strain under study, assembled from concatenated read files of high quality samples (I9, I13, I14 and I15), hereafter named “Brucella canis BfR-SPBR-consensus”.
Mapping statistics were analyzed with QualiMap v2.2.179. To determine the average nucleotide identity (ANI), Gegenees v2.2.180 was applied with a fragment size of 200. Insertion sequences (IS) were identified by online BLASTn analysis against the IS database of the ISfinder website81. Inspection of the De Bruijn assembly graph was done with Bandage v1.082. For molecular genotyping, in silico multi-locus variable number of tandem repeat analysis (MLVA-16) was calculated with a Python script83 and in silico multi-locus sequence typing (MLST-21) was calculated with the mlst program by Torsten Seemann (https://github.com/tseemann/mlst).
Genetic relatedness between the Brazilian isolates and publicly available sequences was determined with ParSNP v1.084. To this end, all available sequences including B. canis genomes in the PATRIC (www.patricbrc.org) or NCBI (https://www.ncbi.nlm.nih.gov/genome/microbes/) database as well as draft genomes assembled from read files retrieved from the Sequencing Read Archive (https://www.ncbi.nlm.nih.gov/sra/) and the B. canis BfR-SPBR-consensus genome were collected. Maximum-Likelihood trees were calculated by FastTree285. The origin of strains was extracted from genome metadata and integrated into the phylogeny with the EMBL interactive tree of life, iTOL v486, for visualization.
Single nucleotide polymorphism (SNP) analysis
Whole genome single nucleotide polymorphism (wgSNP) analysis was conducted using two different strategies, a read-based approach with BioNumerics v7.6 (Applied Maths, Sint-Martens-Latem, Belgium) and an assembly-based approach with Mauve v2015-02-1387.
For the BioNumerics procedure, reads were trimmed in a consecutive manner: first with the configuration “structural trimming (remove stretches: homopolymer ≤ 20; polyA > 90%; polyGC < 10% & > 90%), tail quality trimming (tail q ≥ 18; windowed average q ≥ 15; rolling average q ≥ 15), length trimming (exclude < 100 bp & > 250 bp)”, and second with the configuration “overall quality trimming (exclude minimum q window = 5; average q < 20; replace with N for q < 15)”, and were mapped to either the complete genome of B. canis ATCC 23365 or the B. canis BfR-SPBR-consensus genome.
The resulting SNP matrix was filtered for positions with a minimum coverage of five reads in total, with at least one SNP on the forward and the reverse strand. Due to the filter limitations of BioNumerics, SNP positions were extracted and manually curated in Microsoft Excel 2013 to differentiate between false base calls in the de novo assembly and SNPs at positions present in all 24 sequences or at positions with ambiguous base calls or insufficient coverage (N). Based on the final SNP matrix the pairwise distance between the isolates was calculated and clustering of isolates was performed using the Neighbor Joining algorithm in BioNumerics.
A comparison of B. canis BfR-SPBR-consensus against reference sequences including variant calling was performed in Mauve v2015-02-13. An identical SNP list was extracted using ParSNP v1.0 and hence could be used for SNP annotation by SnpEff88 with the RefSeq annotation of reference GCF_000018525.1.
Nucleotide sequence accession numbers
The lllumina MiSeq raw sequence reads used to generate the draft B. canis BfR-SPBR-consensus genome sequence have been deposited in the database of the European Nucleotide Archive (ENA) under the accession numbers ERX4130724, ERX4130725, ERX4130726 and ERX4130727 within the BioProject ERP121782.
References
Gwida, M. et al. Brucellosis—regionally emerging zoonotic disease?. Croat. Med. J. 51, 289–295. https://doi.org/10.3325/cmj.2010.51.289 (2010).
Muhldorfer, K. et al. The role of “atypical” Brucella in amphibians: are we facing novel emerging pathogens?. J. Appl. Microbiol. 122, 40–53. https://doi.org/10.1111/jam.13326 (2017).
El-Sayed, A. & Awad, W. Brucellosis: evolution and expected comeback. Int. J. Vet. Sci. Med. 6, S31-s35. https://doi.org/10.1016/j.ijvsm.2018.01.008 (2018).
Carmichael, L. E. & Kenney, R. M. Canine abortion caused by Brucella canis. J. Am. Vet. Med. Assoc. 152, 605–616 (1968).
Brennan, S. J., Ngeleka, M., Philibert, H. M., Forbes, L. B. & Allen, A. L. Canine brucellosis in a Saskatchewan kennel. Can. Vet. J. 49, 703–708 (2008).
Buhmann, G. et al. Canine brucellosis: insights into the epidemiologic situation in Europe. Front. Vet. Sci. 6, 151. https://doi.org/10.3389/fvets.2019.00151 (2019).
Keid, L. B. et al. Brucella canis infection in dogs from commercial breeding kennels in Brazil. Transbound. Emerg. Dis. 64, 691–697. https://doi.org/10.1111/tbed.12632 (2017).
Wanke, M. M. Canine brucellosis. Anim. Reprod. Sci. 82–83, 195–207. https://doi.org/10.1016/j.anireprosci.2004.05.005 (2004).
Carmichael, L. E. & Joubert, J. C. Transmission of Brucella canis by contact exposure. Cornell Vet. 78, 63–73 (1988).
Kauffman, L. K. & Petersen, C. A. Canine brucellosis: old foe and reemerging scourge. Vet. Clin. N. Am. Small Anim. Pract. 49, 763–779. https://doi.org/10.1016/j.cvsm.2019.02.013 (2019).
Woldemeskel, M. Zoonosis due to Brucella suis with special reference to infection in dogs (carnivores): a brief review. Open J. Vet. Med. 3, 213–221. https://doi.org/10.4236/ojvm.2013.33034 (2013).
Ramamoorthy, S. et al. Brucella suis infection in dogs, Georgia USA. Emerg. Infect. Dis. 17, 2386–2387. https://doi.org/10.3201/eid1712.111127 (2011).
van Dijk, M. A. M. et al. Brucella suis infection in dog fed raw meat, the Netherlands. Emerg. Infect. Dis. 24, 1127–1129. https://doi.org/10.3201/eid2406.171887 (2018).
Lama, J. K. & Bachoon, D. S. Detection of Brucella suis, Campylobacter jejuni, and Escherichia coli strains in feral pig (Sus scrofa) communities of Georgia. Vector Borne Zoonotic Dis. 18, 350–355. https://doi.org/10.1089/vbz.2017.2187 (2018).
Pedersen, K. et al. Identification of Brucella suis from feral swine in selected states in the USA. J. Wildl. Dis. 50, 171–179. https://doi.org/10.7589/2013-09-235 (2014).
James, D. R. et al. Clinical management of Brucella suis infection in dogs and implications for public health. Aust. Vet. J. 95, 19–25. https://doi.org/10.1111/avj.12550 (2017).
Franco-Paredes, C., Chastain, D., Taylor, P., Stocking, S. & Sellers, B. Boar hunting and brucellosis caused by Brucella suis. Travel. Med. Infect. Dis. 16, 18–22. https://doi.org/10.1016/j.tmaid.2017.03.006 (2017).
Mor, S. M. et al. Emergence of Brucella suis in dogs in New South Wales, Australia: clinical findings and implications for zoonotic transmission. BMC Vet. Res. 12, 199. https://doi.org/10.1186/s12917-016-0835-0 (2016).
Forbes, L. B. Isolates of Brucella suis biovar 4 from animals and humans in Canada, 1982–1990. Can. Vet. J. 32, 686–688 (1991).
Kosoy, M. & Goodrich, I. Comparative ecology of Bartonella and Brucella infections in wild carnivores. Front. Vet. Sci. 5, 322–322. https://doi.org/10.3389/fvets.2018.00322 (2019).
Javeri, H., Jamieson, S., Sehgal, R. & Cadena, J. Brucella canis peritonitis. Infection 42, 195–197. https://doi.org/10.1007/s15010-013-0505-0 (2014).
Ying, W., Nguyen, M. Q. & Jahre, J. A. Brucella canis endocarditis: case report. Clin. Infect. Dis. 29, 1593–1594. https://doi.org/10.1086/313545 (1999).
Piampiano, P., McLeary, M., Young, L. W. & Janner, D. Brucellosis: unusual presentations in two adolescent boys. Pediatr. Radiol. 30, 355–357. https://doi.org/10.1007/s002470050760 (2000).
Wallach, J. C., Giambartolomei, G. H., Baldi, P. C. & Fossati, C. A. Human infection with M-strain of Brucella canis. Emerg. Infect. Dis. 10, 146–148. https://doi.org/10.3201/eid1001.020622 (2004).
Kutlu, M. et al. The first report of Brucella suis biovar 1 isolation in human in Turkey. J. Infect. Public Health 9, 675–678. https://doi.org/10.1016/j.jiph.2016.01.011 (2016).
Zange, S. et al. A headache with surprising outcome: first case of brucellosis caused by Brucella suis biovar 1 in Germany. Infection 47, 863–868. https://doi.org/10.1007/s15010-019-01312-7 (2019).
Olsen, S. C. & Tatum, F. M. Swine brucellosis: current perspectives. Vet. Med. (Auckl.) 8, 1–12. https://doi.org/10.2147/vmrr.S91360 (2017).
Hollett, R. B. Canine brucellosis: outbreaks and compliance. Theriogenology 66, 575–587. https://doi.org/10.1016/j.theriogenology.2006.04.011 (2006).
Moreno, E., Jones, L. M. & Berman, D. T. Immunochemical characterization of rough Brucella lipopolysaccharides. Infect. Immun. 43, 779–782 (1984).
Alton, G. G., Jones, L. M. & Pietz, D. E. Laboratory techniques in Brucellosis. Monographs Series 1–163 (World Health Organization, Geneva, 1975).
Rudrik, J. T. et al. Safety and accuracy of matrix-assisted laser desorption ionization-time of flight mass spectrometry for identification of highly pathogenic organisms. J. Clin. Microbiol. 55, 3513–3529. https://doi.org/10.1128/jcm.01023-17 (2017).
Karger, A. et al. Interlaboratory comparison of intact-cell matrix-assisted laser desorption ionization-time of flight mass spectrometry results for identification and differentiation of Brucella spp. J. Clin. Microbiol. 51, 3123–3126. https://doi.org/10.1128/jcm.01720-13 (2013).
Mesureur, J. et al. A MALDI-TOF MS database with broad genus coverage for species-level identification of Brucella. PLoS Negl. Trop. Dis. 12, e0006874. https://doi.org/10.1371/journal.pntd.0006874 (2018).
Sali, M. et al. Rapid and safe one-step extraction method for the identification of Brucella strains at genus and species level by MALDI-TOF mass spectrometry. PLoS ONE 13, e0197864. https://doi.org/10.1371/journal.pone.0197864 (2018).
Whatmore, A. M. et al. Extended multilocus sequence analysis to describe the global population structure of the genus Brucella: phylogeography and relationship to biovars. Front. Microbiol. 7, 2049. https://doi.org/10.3389/fmicb.2016.02049 (2016).
Scholz, H. C. & Vergnaud, G. Molecular characterisation of Brucella species. Rev. Sci. Tech. 32, 149–162. https://doi.org/10.20506/rst.32.1.2189 (2013).
Al Dahouk, S. et al. Identification of Brucella species and biotypes using polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP). Crit. Rev. Microbiol. 31, 191–196. https://doi.org/10.1080/10408410500304041 (2005).
Yang, Y. et al. Genotyping Brucella canis isolates using a highly discriminatory multilocus variable-number tandem-repeat analysis (MLVA) assay. Sci. Rep. 7, 1067. https://doi.org/10.1038/s41598-017-01114-7 (2017).
Whatmore, A. M. Current understanding of the genetic diversity of Brucella, an expanding genus of zoonotic pathogens. Infect. Genet. Evol. 9, 1168–1184. https://doi.org/10.1016/j.meegid.2009.07.001 (2009).
Treangen, T. J. & Salzberg, S. L. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46. https://doi.org/10.1038/nrg3117 (2011).
Ferreira Vicente, A. et al. New insights into phylogeography of worldwide Brucella canis isolates by comparative genomics-based approaches: focus on Brazil. BMC Genom. 19, 636. https://doi.org/10.1186/s12864-018-5001-6 (2018).
Henderson, I. R., Navarro-Garcia, F. & Nataro, J. P. The great escape: structure and function of the autotransporter proteins. Trends Microbiol. 6, 370–378. https://doi.org/10.1016/s0966-842x(98)01318-3 (1998).
Cosford, K. L. Brucella canis: an update on research and clinical management. Can. Vet. J. 59, 74–81 (2018).
Makloski, C. L. Canine brucellosis management. Vet. Clin. N. Am. Small Anim. Pract. 41, 1209–1219. https://doi.org/10.1016/j.cvsm.2011.08.001 (2011).
Hensel, M. E., Negron, M. & Arenas-Gamboa, A. M. Brucellosis in dogs and public health risk. Emerg. Infect. Dis. 24, 1401–1406. https://doi.org/10.3201/eid2408.171171 (2018).
Dentinger, C. M. et al. Human Brucella canis infection and subsequent laboratory exposures associated with a puppy, New York City, 2012. Zoonoses Public Health 62, 407–414. https://doi.org/10.1111/zph.12163 (2015).
Lucero, N. E. et al. Human Brucella canis outbreak linked to infection in dogs. Epidemiol. Infect. 138, 280–285. https://doi.org/10.1017/s0950268809990525 (2010).
Swenson, R. M., Carmichael, L. E. & Cundy, K. R. Human infection with Brucella canis. Ann. Intern. Med. 76, 435–438. https://doi.org/10.7326/0003-4819-76-3-435 (1972).
Al Dahouk, S., Tomaso, H., Nockler, K., Neubauer, H. & Frangoulidis, D. Laboratory-based diagnosis of brucellosis—a review of the literature. Part II: serological tests for brucellosis. Clin. Lab. 49, 577–589 (2003).
Al Dahouk, S., Tomaso, H., Nockler, K., Neubauer, H. & Frangoulidis, D. Laboratory-based diagnosis of brucellosis—a review of the literature. Part I: techniques for direct detection and identification of Brucella spp. Clin. Lab. 49, 487–505 (2003).
Zoha, S. J. & Carmichael, L. E. Serological responses of dogs to cell wall and internal antigens of Brucella canis (B. canis). Vet. Microbiol. 7, 35–50. https://doi.org/10.1016/0378-1135(82)90004-9 (1982).
Flores-Castro, R. & Carmichael, L. E. Canine brucellosis. Current status of methods for diagnosis. Cornell Vet. 68(Suppl 7), 76–88 (1978).
Barr, S. C., Eilts, B. E., Roy, A. F. & Miller, R. Brucella suis biotype 1 infection in a dog. J. Am. Vet. Med. Assoc. 189, 686–687 (1986).
Keid, L. B., Diniz, J. A., Oliveira, T. M., Ferreira, H. L. & Soares, R. M. Evaluation of an immunochromatographic test to the diagnosis of canine brucellosis caused by Brucella canis. Reprod. Domest. Anim. 50, 939–944. https://doi.org/10.1111/rda.12612 (2015).
Keid, L. B. et al. Comparison of agar gel immunodiffusion test, rapid slide agglutination test, microbiological culture and PCR for the diagnosis of canine brucellosis. Res. Vet. Sci. 86, 22–26. https://doi.org/10.1016/j.rvsc.2008.05.012 (2009).
Carmichael, L. E., Zoha, S. J. & Flores-Castro, R. Problems in the serodiagnosis of canine brucellosis: dog responses to cell wall and internal antigens of Brucella canis. Dev. Biol. Stand. 56, 371–383 (1984).
Mol, J. P. S. et al. Diagnosis of canine brucellosis: comparison of various serologic tests and PCR. J. Vet. Diagn. Invest. 32, 77–86. https://doi.org/10.1177/1040638719891083 (2020).
Carmichael, L. E. & Shin, S. J. Canine brucellosis: a diagnostician’s dilemma. Semin. Vet. Med. Surg. Small Anim. 11, 161–165. https://doi.org/10.1016/s1096-2867(96)80028-4 (1996).
Wanke, M. M. et al. Preliminary study of an immunochromatography test for serological diagnosis of canine brucellosis. Reprod. Domest. Anim. 47, 370–372. https://doi.org/10.1111/rda.12108 (2012).
Jungersen, G., SØRensen, V., Giese, S. B. & Stack, J. A. Riber U (2006) Differentiation between serological responses to Brucella suis and Yersinia enterocolitica serotype O[ratio ]9 after natural or experimental infection in pigs. Epidemiol. Infect. 134, 347–357. https://doi.org/10.1017/S095026880500511X (2006).
Corbel, M. J., Stuart, F. A. & Brewer, R. A. Observations on serological cross-reactions between smooth Brucella species and organisms of other genera. Dev. Biol. Stand. 56, 341–348 (1984).
Goicochea, C. E., Gotuzzo, E. & Carrillo, C. Cholera-Brucella cross-reaction: a new potential diagnostic problem for travelers to latin America. J. Travel Med. 3, 37–39. https://doi.org/10.1111/j.1708-8305.1996.tb00694.x (1996).
Purvis, T. J. et al. Detection of Brucella canis infection in dogs by blood culture and bacterial identification using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J. Vet. Diagn. Invest. 29, 586–588. https://doi.org/10.1177/1040638717704652 (2017).
Lasch, P. et al. Identification of highly pathogenic microorganisms by matrix-assisted laser desorption ionization-time of flight mass spectrometry: results of an interlaboratory ring trial. J. Clin. Microbiol. 53, 2632–2640. https://doi.org/10.1128/jcm.00813-15 (2015).
Ferreira, L. et al. Identification of Brucella by MALDI-TOF mass spectrometry. Fast and reliable identification from agar plates and blood cultures. PLoS ONE 5, e14235. https://doi.org/10.1371/journal.pone.0014235 (2010).
Wu, N. et al. Risk factors for contacts between wild boar and outdoor pigs in Switzerland and investigations on potential Brucella suis spill-over. BMC Vet. Res. 8, 116. https://doi.org/10.1186/1746-6148-8-116 (2012).
Kreizinger, Z. et al. Genetic relatedness of Brucella suis biovar 2 isolates from hares, wild boars and domestic pigs. Vet. Microbiol. 172, 492–498. https://doi.org/10.1016/j.vetmic.2014.05.031 (2014).
Patel, R. MALDI-TOF MS for the diagnosis of infectious diseases. Clin. Chem. 61, 100–111. https://doi.org/10.1373/clinchem.2014.221770 (2015).
Keid, L. B. et al. Comparison of a PCR assay in whole blood and serum specimens for canine brucellosis diagnosis. Vet. Rec. 167, 96–99. https://doi.org/10.1136/vr.c3811 (2010).
Redkar, R., Rose, S., Bricker, B. & DelVecchio, V. Real-time detection of Brucella abortus, Brucella melitensis and Brucella suis. Mol. Cell. Probes 15, 43–52. https://doi.org/10.1006/mcpr.2000.0338 (2001).
Baily, G. G., Krahn, J. B., Drasar, B. S. & Stoker, N. G. Detection of Brucella melitensis and Brucella abortus by DNA amplification. J. Trop. Med. Hyg. 95, 271–275 (1992).
Batinga, M. C. A. et al. Comparative application of IS711-based polymerase chain reaction (PCR) and loop-mediated isothermal amplification (LAMP) for canine brucellosis diagnosis. Mol. Cell. Probes 39, 1–6. https://doi.org/10.1016/j.mcp.2018.02.003 (2018).
Team, R. C. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2018).
Gibb, S. & Strimmer, K. MALDIquant: a versatile R package for the analysis of mass spectrometry data. Bioinformatics 28, 2270–2271. https://doi.org/10.1093/bioinformatics/bts447 (2012).
Wickham, et al. Welcome to the Tidyverse. J. Open Source Softw. 4, 1686. https://doi.org/10.21105/joss.01686 (2019).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. https://doi.org/10.1093/bioinformatics/btt086 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. https://doi.org/10.1038/nmeth.1923 (2012).
Okonechnikov, K., Conesa, A. & Garcia-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294. https://doi.org/10.1093/bioinformatics/btv566 (2016).
Agren, J., Sundstrom, A., Hafstrom, T. & Segerman, B. Gegenees: fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS ONE 7, e39107. https://doi.org/10.1371/journal.pone.0039107 (2012).
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J. & Chandler, M. ISfinder: the reference centre for bacterial insertion sequences. Nucl. Acids Res. 34, D32-36. https://doi.org/10.1093/nar/gkj014 (2006).
Wick, R. R., Schultz, M. B., Zobel, J. & Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. https://doi.org/10.1093/bioinformatics/btv383 (2015).
Vergnaud, G. et al. Genotypic expansion within the population structure of classical Brucella species revealed by MLVA16 typing of 1404 Brucella isolates from different animal and geographic origins, 1974–2006. Front. Microbiol. 9, 1545. https://doi.org/10.3389/fmicb.2018.01545 (2018).
Treangen, T. J., Ondov, B. D., Koren, S. & Phillippy, A. M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15, 524. https://doi.org/10.1186/s13059-014-0524-x (2014).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490. https://doi.org/10.1371/journal.pone.0009490 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucl. Acids Res. 47, W256-w259. https://doi.org/10.1093/nar/gkz239 (2019).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5, e11147. https://doi.org/10.1371/journal.pone.0011147 (2010).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. https://doi.org/10.4161/fly.19695 (2012).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. https://doi.org/10.1093/bioinformatics/btu153 (2014).
Acknowledgements
We thank Jane Jatsch and Anne Stephan for technical assistance and Alyssa Ingmundson for critical reading of the manuscript. Our work was funded by a grant of the German Federal Ministry of Education and Research (BMBF) within the joint project Ess-B.A.R (FKZ 13N13982). Further support was provided by the São Paulo Research Foundation (FAPESP) with the grants #2016/01276-8 and #2016/13804-9 and from the Brazilian National Council for Scientific and Technological Development (CNPq).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
D.D.S., H.B., L.K., R.D. and S.A.D. conceived and designed the experiments. D.D.S., J.G., R.S., J.D.L. and L.K. performed the laboratory experiments. H.B., D.H., D.D.S., J.G., L.K. and S.A.D. analyzed and interpreted the data. H.B. and J.G. performed genomic analysis. H.B. and R.D. analyzed the proteomic data. S.A.D. and D.H. outlined the manuscript. D.H., H.B., D.D.S. and S.A.D. wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
da Silva, D.A.V., Brendebach, H., Grützke, J. et al. MALDI-TOF MS and genomic analysis can make the difference in the clarification of canine brucellosis outbreaks. Sci Rep 10, 19246 (2020). https://doi.org/10.1038/s41598-020-75960-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-75960-3
This article is cited by
-
Laboratory methods to decipher epigenetic signatures: a comparative review
Cellular & Molecular Biology Letters (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
