
Abstract
Post-stroke cognitive impairment (PSCI) is a major source of disability, affecting up to two thirds of stroke survivors with no available therapeutic options. The condition remains understudied in preclinical models due to its delayed presentation. Although hypertension is a leading risk factor for dementia, how ischemic stroke contributes to this neurodegenerative condition is unknown. In this study, we used a model of hypertension to study the development of PSCI and its mechanisms. Spontaneously hypertensive rats (SHR) were compared to normotensive rats and were subjected to 1-h middle cerebral artery occlusion or sham surgery. Novel object recognition, passive avoidance test and Morris water maze were used to assess cognition. In addition, brain magnetic resonance images were obtained 12-weeks post-stroke and tissue was collected for immunohistochemistry and protein quantification. Stroked animals developed impairment in long-term memory at 4-weeks post-stroke despite recovery from motor deficits, with hypertensive animals showing some symptoms of anhedonia. Stroked SHRs displayed grey matter atrophy and had a two-fold increase in apoptosis in the ischemic borderzone and increased markers of inflammatory cell death and DNA damage at 12 weeks post-stroke. This indicates that preexisting hypertension exacerbates the development of secondary neurodegeneration after stroke beyond its acute effects on neurovascular injury.
Similar content being viewed by others


Introduction
Stroke has recently become the fifth leading cause of death in the United States1. While stroke mortality has been steadily declining over the last decade due to the continuous improvement in health care standards2, the number of stroke survivors with residual disability is steadily increasing3. Ischemic stroke is a condition characterized by an initial ischemic event that deprives brain tissue from blood supply and oxygenation, that is sometimes followed by reperfusion, leading to irreversible brain damage and subsequent motor and cognitive impairment4.
Post-stroke cognitive impairment (PSCI) is a condition that affects up to two-thirds of patients following ischemic stroke, with up to one third eventually developing dementia5,6. Although PSCI is highly prevalent among stroke survivors, there is evidence suggesting that the criteria for diagnosis may underestimate the frequency of dementia and cognitive decline among stroke survivors7,8. While it was traditionally thought that the cognitive impairment results from the recurrence of ischemic insults, newer evidence suggests that a very substantial portion of this impairment results from neuronal pathogenesis9. Published data from a large, NIH-funded, epidemiologic trial showed that patients, in addition to acute changes, suffer from a slowly progressive cognitive decline after a single-stroke lesion10,11. This continuous deterioration occurs even in the absence of any evidence of new ischemic injuries12,13.
Diagnosis and characterization of clinically apparent PSCI has proven to be a challenging task, owing to the heterogeneity of the condition itself. The incidence of PSCI and its severity depends largely upon the morphology of the vascular injury (focal or multifocal; large or small vessel), volume of brain tissue affected by ischemia and, most importantly, the location and number of lesions14. Histopathological studies have shown a clear link between the vascular aspects of brain injuries affecting cognition and the neurodegenerative aspects that resemble Alzheimer’s disease pathology15.
Hypertension is the most commonly occurring modifiable risk-factor for stroke worldwide and is being increasingly recognized as a risk factor for the development of PSCI15,16. Chronic hypertension, particularly midlife high blood pressure (BP), has been associated with an increased risk for cognitive decline, vascular dementia and Alzheimer’s disease17. One of the mechanisms by which hypertension is believed to contribute to the development of cognitive impairment is exposing the cerebral microvasculature to pulsatile pressure causing tearing of the brain vascular endothelium and smooth muscle cells leading to lipohyalinosis and fibrinoid necrosis18. Clinical studies on hypertensive patients have shown that acute disruption of blood perfusion can lead to the formation of lacunar infarcts, while chronic ischemia can lead to the development of white matter lesions which are associated with the development of cognitive impairment19,20.
In this study, we aimed to determine the role of hypertension in the development of cognitive impairment following experimental stroke in rats by comparing normotensive (Wistar) rats to spontaneously hypertensive rats (SHR). We hypothesized that hypertension plays a key role in exacerbating PSCI by inducing neurodegenerative processes, causing SHRs to be more susceptible to PSCI compared to normotensive animals.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request. All animal experiments were approved by the Institutional Animal Care and Use Committee of the Charlie Norwood VA Medical Center, Augusta, Georgia. All methods were performed in accordance with the relevant guidelines and regulations. Overall experimental design can be found in (Fig. 1A).

Hypertension increases stroke mortality and reduces spontaneous recovery. (A) Experimental design showing different time points for behavioral testing. (B) The mean arterial blood pressure of a cohort of SHR animals of the same age and weight of our stroke animals was followed for 2 weeks to establish their blood pressure (n = 6). (C) 63% of the Wistar animals achieved complete spontaneous post-stroke recovery after 24 h of MCAO, compared to 0% for SHR animals. (P < 0.0001, Fischer’s exact test) (D) & (E) Body weight was recorded for 12 weeks following MCAO comparing sham animals from both strains to stroke animals (n = 6–12 per group).
Data analysis
Statistical analyses were performed using SAS version 9.4 (SAS Institute, Inc, Cary, NC) by a biostatistician (Johnson MH). All data are presented as means ± SEM. Unless otherwise mentioned, all results presented were analyzed by 2-way ANOVA (2 Strain (Wistar vs. SHR) X 2 Surgery (sham vs. stroke)), with p-values for the tests presented as tables on the graphs. Letters in the graphs represent the results of Tukey post-hoc multiple comparisons tests for significant ANOVA results where pairs of means with different letters are significantly different. Otherwise, comparisons of two groups were analyzed by student t-test for parametric values, and Wilcoxon’s signed rank test for non-parametric values. Results are considered significant at a Type I error rate of 5%.
Experimental animals
Both male Wistar and SHR animals were purchased from Charles River (Wilmington, MA) and were housed (1 rat per cage) in pathogen free, temperature-controlled facility (24 ± 1 C; 12–12-h light–dark cycle) with access to standard chow and water ad libitum.
Blood pressure monitoring
SHR animals are known to develop hypertension in a progressive fashion. To prove that our SHR animals have developed an elevated mean arterial blood pressure, we used an age matched cohort of 6 male SHR animals implanted with telemetry devices, measuring their mean arterial blood pressure, using continuous BP telemetry with transmitters (Data Sciences International, St Paul, MN, USA). Description of this method is adapted from our previously published work Ahmed et al.21. The transmitters were implanted in rats, under 3–5% isoflurane inhalation anesthesia, as reported previously according to the manufacturer’s specifications22. In brief, a midline incision was performed to expose the abdominal aorta, which was shortly occluded to allow insertion of the transmitter catheter, which was secured in place using tissue glue. The incision was closed using non-absorbable suture (3–0). Rats were returned to their individual cages and allowed to recover from surgery for 10 days. By placing cages on top of the telemetry receivers, arterial pressure waveforms were continuously recorded throughout a 14-day period.
Transient middle cerebral artery occlusion (tMCAO) surgery
Male Wistar and SHR rats (age: 10–14 weeks) (body weight: 320–400 g) were subjected to 60-min of tMCAO using 4–0 silicon coated nylon suture (Doccol 403756)) as previously described22. The original source of method description is our previously published work, Eldahshan et al.23 . Briefly, the animals were anesthetized using 2–5% isoflurane, a ventral mid-line neck incision was made, the right common carotid artery (CCA) was exposed, and the external carotid artery (ECA) was ligated and cut. The suture was advanced from a small nick at the ECA into the internal carotid artery (ICA) until a mild resistance was encountered, indicating the branching of the anterior and middle cerebral artery. The suture was tied in place for the duration of the occlusion and the animals were allowed to recover from anesthesia. Several minutes before the end of the occlusion time, the animals were re-anesthetized, the suture was removed for reperfusion and the small nick at the ECA was permanently ligated. In sham surgeries, the CCA was isolated and manipulated without cutting or insertion of the suture and the skin was closed.
We defined a failed MCAO surgery as: “Insertion of the suture that did not succeed in producing an ischemic damage as evident from the lack of motor deficits after reperfusion, possibly due to an incomplete occlusion of the origin of the MCA”.
Randomization and blinding
Animals were randomized to either the sham or stroke group using a stratified block randomization method, in which animals were arranged into strata based on body weight, with each stratum being divided equally between groups, to achieve an equal distribution of body weight (sham vs. stroke) within strain, before surgery. All behavioral, histochemical and molecular assessments and analyses were performed by blinded investigators. Animals were assigned numbers 1–50 after randomization, with their strain and surgery unknown to all investigators except the surgeon. Animals were assigned to groups A-D based on strain and surgery, and group assignments were revealed to the investigators after the conclusion of the tests and analyses.
Inclusion and exclusion
To ensure the success of the surgery, animals were tested for unilateral paresis immediately following reperfusion to exclude any animals that did not show a significant deficit. No animals were excluded for failed MCAO. Animals that showed spontaneous recovery at 24 h post-surgery were excluded. Our inclusion criteria were: (1) At least 5% loss in body weight at 24 h after stroke and (2) a score of 6 out of 8 or less on a 4-task neurological score based on the Bederson test. Animals that failed to satisfy one of these criteria at 24 h post-stroke were deemed spontaneously recovered.
Assessment of functional outcome
Body weight
As we described in our previously published work Ahmed et al.21, weight monitoring is an extremely important tool that serves as an independent and unambiguous measure of an animal’s overall health and welfare, specifically after stroke. For our studies, animals were weighed before surgery and then daily after stroke for the first 14 days, then once a week until the day of sacrifice. All animals selected for the study were in the range of 300–400 g body weight at baseline.
Neurobehavioral testing
All neurobehavioral tests were conducted, recorded, and analyzed in a blinded manner.
Sensorimotor testing
To assess sensorimotor function, animals underwent an 8-point modified neurological assessment modified from the Bederson protocol23 at days 1, 3, 5, 7 and 14 post surgery. Furthermore, motor recovery was assessed via measuring the locomotor activity during the open field and Morris water maze (MWM) tests.
Modified Bederson score
Animals were assessed neurologically on an 8-point scale measuring 4 basic functions (spontaneous rotation, resistance to lateral push and fore and back paw flexion) with a score of 2 points awarded in each category for an animal exhibiting a natural response, a score of 1 point for mildly impaired animals and a score of 0 for strongly impaired animals. Higher scores indicate better performance with a maximum possible score of 8/8 and a minimum possible score of 0/8.
Cognitive testing
Cognitive tests were performed according to the design in (Fig. 1). Special consideration was taken for cognitive tests to allow sufficient period of time to prevent different tests from affecting one another. Open Field and Novel Object Recognition (NOR) were allowed to be performed on the same days as these tests were of minimal invasiveness, measuring the spontaneous activity of the animals. However, invasive tests such as passive avoidance (PAT) and exhausting tests such as the MWM were done individually. The NOR was performed to evaluate non-spatial working memory24,25,26,27, while the passive avoidance test (PAT) assessed associative learning and reference memory28,29.
The novel object recognition (NOR) test
The original source of method description is our previously published work, Ahmed et al.21. The NOR test was performed to evaluate non-spatial working memory related to frontal-subcortical circuits. This test was based on the spontaneous tendency of animals to interact with a novel object more than a familiar one and consisted of 2 trials separated by a retention period. On the designated test day, animals were first subjected to an acquisition/sample trial, where the animal is presented with 2 identical (sample) objects and allowed to explore for 15 min. Following sample object exposure, the animal was returned to its home cage for a 1-h retention period. The 2nd preference trial/test session (5 min), which follows the retention period, was conducted in the same manner as the 1st trial, except that a new/novel object replaces one of the familiar/sample objects. The arena and objects were cleaned after each session with 70% ethanol. The time spent in exploring each object during the preference trial/test session was recorded and the discrimination index, which is the difference in exploration time for the objects divided by total time of exploration, was calculated. The discrimination index (DI) and the recognition index (RI), which is the time spent exploring the novel object relative to the total time of exploration, were taken as indicators of working memory.
▪ Time spent interacting with the familiar object (TF).
▪ Time spent interacting with the novel object (TN).
The required exploratory criteria was that animals should spend between 20–80% of the time exploring the objects out of the 5 min. Objects used were chosen as previously described30. Briefly, objects used were unified between animals and chosen according to recommendations of Heyser and Chemero in that they were symmetrical and transparent, and made of odorless, durable, and easy to clean plastic and glass.
The passive avoidance test (PAT)
The original source of method description is our previously published work, Ahmed et al.22. The passive avoidance test was used to assess aversive associative learning and related reference memory. For this test, one of the compartments of a Y-maze was equipped with a metal floor connected to an electric circuit box, adjusted to deliver brief, moderate intensity electric shocks (3 s duration, 0.75 mA). For the acquisition trial, the shock compartment/arm was blocked, and the animal placed in one of the “safe” arms and allowed 10 min to explore the 2 open arms. Upon completion of 10 min, the door blocking the shock arm was opened allowing the animal to enter. Once the animal had fully entered the shock arm, its initial latency was recorded, and it received a brief electric shock before being returned to its cage. After a 72-h retention period, the test trial was conducted. This was performed in a manner similar to that of the acquisition trial except that the foot shock was omitted, and all 3 arms were accessible to the animal from the start. The difference, between training and test sessions, the latency to enter the shock arm was used as a measure of retention. This latency was recorded for up to 300 s, as the index of long-term aversive associative memory consolidation.
The Morris water maze (MWM)
The MWM test was used to assess spatial learning and long-term memory. The original source of method description is our previously published work, Ahmed et al.24. All water maze tests were conducted in a large circular pool of water, 120 cm in diameter, 55 cm height, filled to a depth of 35 ± 1 cm with water at 25 ± 2 °C. This was separated into quadrants designated northeast (NE), northwest (NW), southeast (SE) and south-west (SW), based on the 4 equally spaced cardinal points N (North), S (South), E (East), and W (West) around the edge of the pool. One of these quadrants contained a transparent escape platform (10.5 cm diameter), submerged 1.5 cm below the water surface and obscured from view. Visual extra-maze cues were mounted to aid spatial navigation.
MWM training/learning sessions
The initial training consisted of a single daily session of 8 trials (60 s each) for the first day, followed by a daily session of 4 trials per day for 3 consecutive days, for a total of 20 training trials. Each trial consisted of releasing the rat into the water from 1 of the 4 starting locations and allowing it to find the platform. If they did not reach the platform within 60 s, they were gently guided to it and kept there for 10 s, then removed. Trials were spaced at least 1 min apart. All trials were recorded, and video tracked by the computerized tracking system Etho-Vision XT 7 (Noldus, Leesburg, VA, USA). This automated system monitored animals’ swim patterns and calculated mean escape latency (s), total distance travelled to target (cm), and velocity to target (cm/s). Data from all training sessions were pooled for each individual animal, evaluated and compared between groups at the different time points.
MWM spatial reference memory test
Spatial reference memory was assessed with a probe test 24 h after the last daily session. For this test, all procedures were kept the same as during training, except that the platform was removed, and rats were allowed to swim for 60 s in an attempt to find it. Performance was evaluated by measuring time spent in the target quadrant/zones, proximity to the target location, and initial latency to the target zone. The target zone was centered on the platform location and was 3 times bigger.
Magnetic resonance imaging (MRI)
Method description is adapted, with changes, from our previously published work, Ahmed et al.25. To determine the changes in the brain, ventricular volume, and the presence of white matter hyperintensities, animals underwent T2 -weighted and fluid attenuated inversion recovery (FLAIR), 8 weeks after MCAO. This was performed using a horizontal 7.0 T BioSpec MRI spectrometer (Bruker Instruments, Billerica, MA) equipped with an 8.9-cm micro imaging gradient insert (100 G/cm. All T2-weighted MRI and FLAIR images were obtained at Augusta University by the Core Imaging Facility for Small Animals (CIFSA). All MRI images were registered DICOM sequences, analyzed using FIJI. Total brain volume as well as hemispheric and ventricular volumes (regions of interest), were determined on binarized sequences obtained by thresholding. For each region of interest, the volumes were calculated by adding the areas measured on each slice (11 slices total) and multiplying it by the slice thickness (1 mm in all cases).
Animal sacrifice and tissue collection
The original source of method description is our previously published work, Ahmed et al.21. At week 12, animals were anesthetized with IP ketamine/xylazine and transcardially perfused with 300 ml of PBS. Animals were decapitated, and their brains collected. Brains were sliced into 2 mm coronal sections, using a glass slicer matrix (Braintree Scientific, Braintree, MA, USA) and sections were labeled from A to F, anterior to posterior. Sections A and B, from brain matrix, were snap frozen and kept for molecular testing. The remaining brain tissue was immersed in 10% formalin (Fischer Scientific, Waltham, MA, USA) for 48 h and then transferred to a 30% sucrose solution until taken for frozen sectioning.
Immunohistochemistry (IHC)
Frozen brain sections (5 μm thick) were processed and stained following a standard technique. Method description is adapted from our previously published work, Jackson et al.26. Briefly, sections were washed in PBS plus 0.03%/Triton X-100 with gentle agitation, blocked for 2 h in 10% normal goat serum/1% BSA, and incubated overnight at 4 °C with anti-IBA-1 (Ionized calcium-binding adaptor molecule 1, 1:500, Wako, Japan) and anti-GFAP (Glial fibrillary acidic protein, 1:500, Sigma-Aldrich, Burlington, MA). After washing, slides were incubated with their appropriate fluorophore-conjugated (Texas Red, Alexa 488) secondary antibodies (1:1000; Abcam) for 1 h at room temperature and washed and cover-slipped with Fluoroshield mounting medium with DAPI (Millipore-Sigma, Burlington, MA). Imaging was performed using the Keyence Microscope (Itasca, IL). 20× magnification images were obtained from the ischemic border zone (penumbra) of the stroked animals and compared to the cortex of the sham animals. Images were analyzed by using the ImageJ software (NIH).
Results
Hypertension increases stroke mortality and worsens stroke outcome
As SHRs are known to develop hypertension over the course of the first few months of their life, it was important to make sure that our animals had already developed hypertension at the time of stroke (10–12 weeks of age). For this purpose, we assessed the mean arterial blood pressure of a cohort of 6 male SHR animals over the course of 2 weeks, via telemetry. All of the animals developed elevated mean arterial blood pressure with an average ranging between 147–150 mm Hg compared to a normal expected value of 96.5 ± 10.7 for Wistar rats under normothermia27 (Fig. 1B).
Next, we evaluated the impact of ischemic stroke. Based on preset criteria (see “Methods”), 12 out of 19 animals from the Wistar group spontaneously recovered at 24 h while there was no spontaneous recovery in the SHR group (P < 0.0001, Fisher’s exact test) (Fig. 1C).
Animals were weighed regularly over the course of the study to assess their recovery after the surgery. Animals from both strains started regaining weight 3 days after stroke, with Wistar animals continuing to gain weight until the end of the study, while SHR animals plateaued at an average of less than 400 g (Fig. 1D,E). Three animals from the Stroke SHR group were sacrificed prior to the completion of the study due to excessive weight loss (> 30%), in accordance with the study protocol. Survival curves are displayed in (Supp. Fig. 1) (P = 0.03, Log-rank test for trend).
To assess motor recovery, animals were assessed neurologically, in a blinded manner, on a modified Bederson score, as described in the methods. Based on this assessment, both strains of animals recovered to near their baseline during the course of the first 14 days of follow-up (Fig. 2A). All animals were able to ambulate freely. Animals were assessed at the end of week 1 post-stroke using the open field test, without significant differences from sham (Supp. Fig. 2).

Hypertension causes slower sensorimotor recovery. (A) Both strains had significant neurological impairment (Modified Bederson’s Score) starting 24 h after stroke and up to day 14. (n = 6–12 per group) (One sample t-test compared to the maximum score of 8). (B) There was no significant difference between the 4 groups in object discrimination in the novel object recognition test at 2- and 12-weeks post MCAO. (n = 6–12 per group). (C) SHRs showed a trend for reduced total object exploration time at 2-weeks post stroke (p = 0.058, 2-way ANOVA). The difference disappeared at 12-weeks post-stroke. (n = 6–12 per group).
Hypertension does not affect working memory in the novel object recognition test
Short-term memory was assessed using the novel object recognition test (NOR). Animals that spent less than 20% of the total time exploring the objects, had their trials excluded from the results (0 trials). Neither Wistar nor SHR animals showed a significant impairment in recognizing the novel object at weeks 2 or 12 post-stroke (Fig. 2B). Total exploration time was reduced in SHR animals (Fig. 2C) but it did not achieve significance.
Hypertension induced depressive symptoms without affecting memory and learning in Morris water maze
To assess the spatial learning and memory, animals were tested using the Morris Water Maze at week 4. All groups showed a significant learning behavior (P < 0.0001) (Fig. 3A) and similar average swim speeds over the course of the experiment (Fig. 3B). There was no significant difference between the four groups during the probe-trial in the time spent in the platform zone or the time spent in the target quadrant, indicating an intact memory function. (Fig. 3C,D) Despite this, SHR animals exhibited a significant reduction in the total distance traveled during the probe test, regardless of the surgery, indicating a significantly impaired exploratory behavior, a sign of “behavioral despair” and depression33 (Fig. 3E).

Stroke causes long-term fear associated memory dysfunction and hypertension induces behavioral despair. (A) Time to reach the platform during the acquisition of the Morris Water Maze was recorded. All groups showed a consistent learning curve (P for time < 0.001, 2-way ANOVA). (B) Average swim speed over the course of the experiment was measured for all groups. (P > 0.05, 2-way ANOVA) (n = 6–12 per group). (C) Time spent in the platform zone and (D) time spent in the target quadrant were measured in the probe trial to examine the memory function (P > 0.05, 2-Way ANOVA). (E) Total distance travelled during the probe test was a measurement for behavioral despair and depression. (P for strain < 0.001, 2-way ANOVA). (a,b: Tukey post-hoc multiple comparisons, pairs of means with different letters are significantly different) (F) Passive Avoidance showed a decrease in the average latency to enter the shock arm for stroked animals. (P < 0.01, 2-way ANOVA) (n = 6–12 per group).
Stroke induces long-term memory impairment in both hypertensive and normotensive animals
Long-term memory was assessed using the passive avoidance test starting at week 4 post-stroke. Both strains of stroked animals showed a significant reduction in their latency to enter the shock arm, 3 days after receiving the shock, indicating an impairment in their avoidance-driven long-term memory (P = 0.0054). Animals that entered the shock arm immediately after being introduced into the arena (< 30 s latency) had their trials excluded from the results of this study (1 animal) (Fig. 3F). Since this test has a pronounced learning effect, it was not repeated.
Stroke accelerates grey matter atrophy in hypertensive animals
The animals were assessed via T2-weighted MRI on the brain at 12 weeks post-stroke to assess brain morphological changes (Fig. 4A). Due to small numbers in the Wistar group, the larger ischemic infarct size in the SHR animals did not achieve statistical significance (Fig. 4B). The total volume of lateral ventricles, an indicator of cerebral atrophy, was significantly larger in SHR animals compared to Wistars, and this was more prominent in the stroked animals (Fig. 4C). Interestingly, this difference was not apparent when comparing the size of the ipsilesional ventricles alone (Fig. 4D), where both strains showed a marked enlargement over their sham counterparts. This indicates that the loss of tissue in SHR animals is not confined to the stroked hemisphere, but also affects the contralesional hemisphere in a significant way. As the total ventricular volume quantified includes the two lateral ventricles, as well as the third central ventricle; and by subtracting the volume of the ipsilateral lateral ventricle from the total ventricular space, we can see that the significant difference found in Fig. 4C was due to the difference in the size of the contralateral ventricle and the central third ventricle, indicating a generalized atrophy effect for stroke in hypertensive animals. In both strains, as expected, the ipsilesional hemisphere was significantly atrophied compared to the contralesional one (Fig. 4E).

Stroke induces enlarged ventricles and grey matter atrophy in SHRs, as measured by MRI at 12-weeks post-stroke. (A) Representative images for diffusion weighted MRI, week 12 post-stroke. White indicates areas with high water content. (B) Ischemic tissue as % of the volume of the contralesional hemisphere (P > 0.05, t-test) (n = 6–12 per group). (b) Total volume of lateral ventricles as % of total brain volume, with SHR showing significant bilateral enlargement of lateral ventricles (P for strain < 0.01, 2-way ANOVA). (D) Volume of the ipsilesional ventricle as % of the contralesional ventricle, with significant enlargement for stroked animals in both strains. (P for surgery < 0.001, 2-way ANOVA). (E) Total volume of the ipsilesional hemisphere as % of total brain volume, decreased for stroked animals in both strains. (P for surgery < 0.001, 2-way ANOVA). (a,b,c: Tukey post-hoc multiple comparisons, pairs of means with different letters are significantly different).
We performed an MRI scan on all the excluded animals and found that only 1 out of the 12 excluded animals showed signs of a very minor ischemic damage in the brain, which added to our confidence in the validity of our exclusion criteria. (data not shown).
Hypertension contributed to heightened apoptosis in the ischemic borderzone at 12 weeks following stroke
Quantification of TUNEL staining in the ischemic borderzone (Fig. 5A), showed increased apoptosis in SHR animals compared to Wistars (Fig. 5D), and both stroke groups exhibited a marked increase in apoptosis compared to their sham counterparts. Stroke exacerbated the effect of hypertension significantly when comparing the stroked SHRs to the stroked Wistars (Fig. 5B).

Stroke induces neuronal apoptosis in the ischemic border zone region of brain tissue, which is exacerbated by hypertension. (A) Representative image of the positions from which the TUNEL and IBA1 images were taken. The polygon outline represents the ischemic tissue, the squares represent fields for quantitation. (DAPI) (B) SHRs displayed a significant increase in apoptosis (P < 0.05, 2-way ANOVA) compared to normotensive animals. Stroked animals displayed a significant increase in apoptosis (P < 0.01, 2-way ANOVA) compared to Sham animals. (n = 4–8 per group). (C) Stroked animals displayed a significant increase in IBA1 staining compared to shams (P < 0.001, 2-way ANOVA) (n = 4–8 per group). (a,b: Tukey post-hoc multiple comparisons, pairs of means with different letters are significantly different.) (D) Representative images from TUNEL staining. (E) Representative images from IBA1 staining (false color).
IBA1 is a marker of microglia/macrophage activation. To assess whether the increase in apoptosis was associated with increased inflammation, IBA1 positive cells from the ischemic borderzone (Fig. 5A) were quantified (Fig. 5E). Even 12 weeks after stroke, there was ongoing inflammation, IBA1 positive cells, in both hypertensive and normotensive animals to a similar extent (Fig. 5C). No significant difference was found in the levels of the pro-inflammatory cytokines IL1B and TNFa between groups using ELISA on the whole-brain homogenate of the ipsilesional brain hemisphere. (Supp. Fig. 3).
Hypertension increases the late expression of markers of DNA damage and cell death after stroke, which are not increased in normotensive animals
Pharmacological poly(ADP-ribose)polymerase-1 (PARP1) is a marker of DNA damage that is associated with increased inflammation and the accumulation of reactive oxygen species28,29,30. We found that stroke caused a significant increase in the expression of PARP1 in the brain homogenate of SHRs, an effect that was not present in normotensive animals (Fig. 6A). There was no difference in the expression level of cleaved (inactive) PARP1 (data not shown).

SHRs display increases in markers of neuronal damage following stroke that are not increased in normotensive animals. (A) Representative image for Western blot data. (B) Stroked SHR show increase in HMGB1 (p (interaction) < 0.05, 2-way ANOVA). (n = 4–8, per group). (C) and (D) Stroked SHRs show increase in activated MMP9 (p (interaction) < 0.01 for upper band and < 0.001 for lower band, 2-way ANOVA). Stroked SHRs show an increase of activated MMP9 (p < 0.05 for upper band and < 0.01 for lower band, Bonferroni post-hoc test) (n = 4–8, per group). (E) SHR show increase in full-length PARP1 after stroke (p (interaction) < 0.01, 2-way ANOVA). Stroked SHR show an increase of PARP1 compared to stroked Wistars (p < 0.05, Bonferroni post-hoc test) (n = 4–8, per group). (a,b: Tukey post-hoc multiple comparisons, pairs of means with different letters are significantly different).
In SHRs, increased High mobility group box 1 protein (HMGB1) expression has been shown to be associated with neuronal damage, increased cell death and increased inflammation31. We found that stroke significantly increased the expression of HMGB1 in the brain homogenates of SHRs compared to shams, but not in normotensive animals (Fig. 6B).
Matrix metalloproteinase 9 (MMP-9) is a member of the zinc-metalloproteinases family involved in the degradation of the extracellular matrix. In this study, hypertension was associated with increased expression of the cleaved active forms of MMP9 at 12 weeks after stroke, but this was not the case in normotensive animals. Furthermore, stroked SHRs displayed a significant increase in the expression of activated MMP9 compared to their sham counterparts, showing that this was not due to hypertension alone. (Fig. 6C,D).
Discussion
Post-stroke cognitive and psychological problems are one of the most prevalent yet understudied aspects of stroke-related disability in human patients. In order to be able to develop treatments for PSCI, it is important to develop reliable animal models suitable for translational studies. Although numerous studies investigated the effects of stroke and MCAO on the development of post-stroke cognitive impairment32,33, most studies used relatively healthy animals and concluded by 30 days post-stroke. In humans, most patients suffer from comorbid diseases like hypertension and diabetes and although maximum motor recovery is usually achieved within 6 months to one year after stroke, stroke survivors display increased incidence of cognitive impairment and dementia for decades after the initial event34. We now know that this is likely due to long term progressive neurodegeneration, which is exacerbated by comorbidities12. The deleterious effect of hypertension on the development of cognitive impairment, even in the absence of stroke is well established18. However, it is usually studied in the context of assessing the effect of blood-pressure lowering medications on preventing these deleterious effects. Our study is one of the very few studies to compare normotensive and SHRs, with and without stroke, in order to determine the extent of contribution of hypertension and stroke to the development of cognitive impairment. Hypertension is known to worsen stroke outcome and increase mortality acutely following MCAO35,36. In this study, we started by measuring the mean arterial blood pressure of a separate cohort of animals, of the same weight and age to our main SHR cohort, using telemetry, to prove that the animals had already developed hypertension before the initiation of the MCAO. This was done because the stress generated by telemetry was expected to impact the behavioral data. Although our lab has extensive experience working on both normotensive and hypertensive animals, comparing Wistar and SHR animals directly in a head-to-head fashion presented a unique challenge. Although a 90-min MCAO was found to reliably produce cognitive deficits in Wistar rats, it resulted in a high rate of mortality in SHRs, and while 60-min MCAO produced very little mortality in SHRs, Wistars had a high rate of spontaneous, 24-h recovery. We elected to use the 60-min MCAO to reduce animal mortality, excluding spontaneously recovering Wistar animals from further analysis. Wistar rats were selected for this comparison instead of Wistar Kyoto, as Wistar Kyoto animals are known to have intrinsic problems in neurobehavior, regardless of hypertension37.
The choice of our timepoints for cognitive assessment was made taking into consideration the suitability of those behavioral tests for repetition, as well as the effect of the different tests on each other. For example, with more repetition we found that animals lost interest in exploring the objects in the NOR, which prompted us to retire the test from week 3 to week 12. Even with this long break, overall object exploration was decreased for all animals at week 12. Similarly, we found that the PAT and the MWM produced a great physical and psychological stress for the animals, thus potentially affecting other behavioral tests that could have been done in the same time period. Therefore, no other tests were administered concurrently with the PAT and MWM.
Our main hypothesis was that hypertension would exacerbate the deleterious effects of MCAO on memory and learning, based on our previously published findings in SHRs21,25. Both SHRs and Wistars in this study developed impairments in learning and memory at 4 weeks after stroke, as demonstrated in the passive avoidance task, but the binary outcome (yes/no) was not conducive to detecting differences between groups with impairment. We were unable to demonstrate deficits due to stroke in NOR task in either strain, however, when tested at 3 and 12 weeks after stroke. Our previous investigation revealed profound deficits in NOR at 30 days in a similarly young SHR cohort21. Whether our failure to replicate that finding was due to the differences in timing of the testing (3 and 12 weeks vs. 30 days) or the experimental set-up remains unclear. It is also possible that depression-like behavior animals develop has a big impact on NOR and water maze tests which limits our ability to detect cognitive deficits. Additional tests designed to evaluate the depression-like behavior need to be incorporated into future studies that monitor long term outcomes. While our previous study did not include a Wistar comparison group21, in the current study the PAT findings remained robust, assuring us that both strains developed PSCI. It is worth noting that our animals were housed individually following MCAO due to concerns that housing the animals in pairs would be dangerous due to the wounds present, and concerns regarding post-stroke feeding competition and recovery. We consider this to be one of the limitations of our study as social isolation is known to be an influencing factor on animal behavior in cognitive tests38. However, the differences in behavior we found between the experimental groups were all among animals of the same housing conditions, leading to our conclusion that the behavioral differences observed were a result of strain differences rather than the housing conditions.
This study is one of the first studies to establish the ongoing deleterious long-term effects of hypertension on recovery of the stroked brain. Although others have reported changes over time after stroke, they failed to include sham groups, so the effects of hypertension alone could not be teased out39,40. It is known that SHR animals develop enlarged brain ventricles over time41, however in our study, the ventricular volume was not significantly different between the Wistar and SHR sham groups, which points to the fact that our SHR animals have not yet spontaneously developed larger ventricles at the time of the study. Similarly, the increase in the ventricular size in the ipsilateral hemisphere was similar in both strains. We found that SHRs had a significant increase in the total ventricular size of both hemispheres, which did not disappear after standardizing on the ventricle size of the strain using the sham animals. We found that SHR animals had profound brain atrophy, in both hemispheres, at 12-weeks post-stroke, when compared to normotensive animals and even to sham-operated SHRs. This indicates that hypertension prolongs the ongoing neurogenic damage of stroke, and this continues to the late time-points. Our findings point to SHR animals being uniquely more vulnerable to the stroke induced ventricular enlargement, specifically in the contralateral hemisphere, which can possibly be attributed to the increased apoptosis post stroke. Our original hypothesis was that this damage was immunologic in nature due to increased inflammation in SHRs, however, our results obtained 12 weeks post-stroke indicate similar levels of post-stroke inflammation. Flow cytometry may have been able to better describe the nature of the activated microglia and macrophages in our samples, but we did not collect the tissue in a way that would allow that analysis. In our previous investigations, we showed that PSCI was associated with ongoing microglial activation and cell death at 30 days in SHRs, and it could be therapeutically targeted21. Here we see that a long-term increase in apoptotic cell death is more pronounced in SHRs. It was ideal to perform detailed histological analyses on the hippocampus to determine the extent of hippocampal damage. However, in most of our animals, the ischemic lesion was big enough to reach sub-cortical areas including destroying all or parts of the ipsilateral hippocampus, which made quantifiable analyses of hippocampal injury impossible for these animals.
The increase in PARP1, MMP9 and HMGB1 associated with stroke in SHRs points to the unique relationship between hypertension and the enhancement of post-stroke cell death (Fig. 7). In addition, PARP1 plays a role in the development of a caspase-independent form of cell death, termed as parthanatos, in response to ischemia/reperfusion damage42,43. Chronic inhibition of full-length (active) PARP1 in SHRs was found to reduce hypertension-related tissue damage in the brain and vascular tissue without affecting the blood pressure44. Both PARP1 knock-out and PARP1 inhibition are known to provide anti-inflammatory and neuroprotective effects in models of traumatic brain injury45 and cerebral ischemia46.

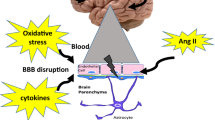
Stroke induces long term neurodegeneration in hypertensive animals. Ischemic stroke causes a marked inflammation in stroked animals. In hypertensive animals, which already suffer from an increase in ROS production, the ischemic insult and the oxidative stress result in chronic increase in DNA damage and neuronal cell death, marked with an increase in PARP1 and HMGB1. PARP1 increase induces the transcription of MMP9, which triggers apoptosis. The result of these processes is chronic neuronal loss in the form of grey matter atrophy.
In addition to its well-known deleterious effects on acute ischemic stroke, including disruption of the blood brain barrier (BBB), increased risk of hemorrhagic complications, and worsened stroke outcome47,48, MMP9 expression has been found to be increased in SHR brains after MCAO, and associated with increased deficits in memory and learning49. In addition, it plays an important role in the cleavage of PARP150. Although the increase in MMP-9 after acute ischemia has been well documented, the increased activity has been reported as transient, returning to baseline in the week following stroke51,52. However, MMP9 expression is increased in several chronic inflammatory CNS pathologies such as multiple sclerosis and Devic's neuromyelitis optica42. It has been reported that PARP1 inhibitors also inhibit MMPs, indicating that the beneficial effects of PARP1 inhibitors after stroke may be in part due to the inhibition of MMPs53. MMP9 is secreted as an inactive pro form, which is cleaved and activated, appearing as two bands of cleaved MMP954. MMP9 was found to play a critical role in the development of age-dependent post-operative cognitive decline, and MMP9 knockout mice were found to be protected from this phenomenon55. Lastly, MMP9 plays an important role in the secretion of pro-inflammatory cytokines and can act as one itself, particularly in response to tissue injury56.
HMGB1 is a protein involved in DNA organization and transcription regulation in the nucleus57. In the brain, HMGB1 acts on microglia to mediate chronic neuroinflammation that drives progressive neurodegeneration58. HMGB1 was also found to be elevated in models of traumatic brain injury, neuroinflammation, epilepsy, and cognitive dysfunction59,60. Finally, HMGB1 was found to be released from necrotic cells in the ischemic core, activating an early inflammatory response and its concentrations were found to correlate with disease severity and outcome after brain injury61.
Our study demonstrated that comorbid hypertension not only worsens the initial injury due to stroke, it exacerbates ongoing tissue damage that occurs months after motor recovery. Although cognitive impairment was evident in both Wistars and SHRs at 30 days after stroke, molecular evidence of active neurodegeneration was more than twofold higher in the SHRs at 12 weeks. In fact, the statistically significant “interaction” we report suggests that the presence of hypertension actually reverses the normal response of these mediators to ischemia and reperfusion. While we found significant histological differences between the normotensive and hypertensive animals, these histological findings did not directly correlate to a difference in cognitive impairment, probably due to the relative insensitivity of the testing methods. It is still likely that hypertension contributes to progressive post-stroke cognitive impairment and differences may have been demonstrated with more sensitive tests or longer-term follow-up.
Perspectives
Our study aimed to establish the contribution of hypertension to the clinically relevant phenomenon of delayed PSCI. We found that delayed cognitive impairment develops in both normotensive and SHRs. However, SHRs showed an increase in delayed DNA damage and cell death, resulting in overall tissue atrophy which was greater than that seen in hypertension alone. Neurodegeneration is a LATE target for intervention after stroke in hypertensive individuals.
References
Benjamin, E. J. et al. Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation 137(12), e67–e492 (2018).
Towfighi, A. & Saver, J. L. Stroke declines from third to fourth leading cause of death in the United States: historical perspective and challenges ahead. Stroke 42(8), 2351–2355 (2011).
Sun, J. H., Tan, L. & Yu, J. T. Post-stroke cognitive impairment: epidemiology, mechanisms and management. Ann. Transl Med. 2(8), 80 (2014).
Woitzik, J., Back, T. & Thome, C. Flow-dependent versus spreading-like impairment of brain tissue integrity during focal cerebral ischemia and its consequences for neuroprotective strategies. Front. Biosci. 13, 1500–1506 (2008).
Leys, D. et al. Poststroke dementia. Lancet Neurol 4(11), 752–759 (2005).
Obaid, M., et al. Long-Term Outcomes in Stroke Patients with Cognitive Impairment: A Population-Based Study. Geriatrics (Basel), 2020. 5(2).
Pendlebury, S. T. et al. Underestimation of cognitive impairment by Mini-Mental State Examination versus the Montreal Cognitive Assessment in patients with transient ischemic attack and stroke: a population-based study. Stroke 41(6), 1290–1293 (2010).
Bour, A. et al. How predictive is the MMSE for cognitive performance after stroke?. J. Neurol. 257(4), 630–637 (2010).
Arba, F. et al. Enlarged perivascular spaces and cognitive impairment after stroke and transient ischemic attack. Int. J. Stroke 13(1), 47–56 (2018).
Delavaran, H. et al. Cognitive function in stroke survivors: A 10-year follow-up study. Acta Neurol. Scand 136(3), 187–194 (2017).
Rouch, L. et al. Antihypertensive drugs, prevention of cognitive decline and dementia: a systematic review of observational studies, randomized controlled trials and meta-analyses, with discussion of potential mechanisms. CNS Drugs 29(2), 113–130 (2015).
Wiesmann, M., Kiliaan, A. J. & Claassen, J. A. Vascular aspects of cognitive impairment and dementia. J. Cereb. Blood Flow Metab. 33(11), 1696–1706 (2013).
Filipska, K., et al. Are depression and dementia a common problem for stroke older adults? A review of chosen epidemiological studies. Psychiatr Q, 2020.
Jellinger, K. A. The pathology of “vascular dementia”: a critical update. J. Alzheimers Dis. 14(1), 107–123 (2008).
Sahathevan, R., Brodtmann, A. & Donnan, G. A. Dementia, stroke, and vascular risk factors; a review. Int. J. Stroke 7(1), 61–73 (2012).
Tu, J. V. Reducing the global burden of stroke: INTERSTROKE. Lancet 376(9735), 74–75 (2010).
Qiu, C., Winblad, B. & Fratiglioni, L. The age-dependent relation of blood pressure to cognitive function and dementia. Lancet Neurol. 4(8), 487–499 (2005).
O’Rourke, M. F. & Safar, M. E. Relationship between aortic stiffening and microvascular disease in brain and kidney: cause and logic of therapy. Hypertension 46(1), 200–204 (2005).
Au, R. et al. Association of white matter hyperintensity volume with decreased cognitive functioning: the Framingham Heart Study. Arch. Neurol. 63(2), 246–250 (2006).
de Groot, J. C. et al. Cerebral white matter lesions and subjective cognitive dysfunction: the Rotterdam Scan Study. Neurology 56(11), 1539–1545 (2001).
Ahmed, H. A. et al. RAS modulation prevents progressive cognitive impairment after experimental stroke: a randomized, blinded preclinical trial. J. Neuroinflamm. 15(1), 229–229 (2018).
Kozak, W. et al. Vascular protection with candesartan after experimental acute stroke in hypertensive rats: a dose-response study. J. Pharmacol. Exp. Ther. 326(3), 773–782 (2008).
Eldahshan, W. et al. Angiotensin II type 2 receptor stimulation with compound 21 improves neurological function after stroke in female rats: a pilot study. Am. J. Physiol. Heart Circ. Physiol. 316(5), H1192-h1201 (2019).
Ahmed, H. A. et al. Angiotensin receptor (AT2R) agonist C21 prevents cognitive decline after permanent stroke in aged animals-A randomized double- blind pre-clinical study. Behav. Brain Res. 359, 560–569 (2019).
Ahmed, H. A. et al. Role of angiotensin system modulation on progression of cognitive impairment and brain MRI changes in aged hypertensive animals - A randomized double- blind pre-clinical study. Behav. Brain Res. 346, 29–40 (2018).
Jackson, L., et al. Delayed administration of Angiotensin II Type 2 receptor (AT2R) agonist compound 21 prevents the development of post-stroke cognitive impairment in diabetes through the modulation of microglia polarization. Transl Stroke Res. 2019.
Wang, Y. et al. Comparison of invasive blood pressure measurements from the caudal ventral artery and the femoral artery in male adult SD and Wistar rats. PLoS ONE 8(4), e60625 (2013).
Ba, X. & Garg, N. J. Signaling mechanism of poly(ADP-ribose) polymerase-1 (PARP-1) in inflammatory diseases. Am. J. Pathol. 178(3), 946–955 (2011).
Bai, P. & Virag, L. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett. 586(21), 3771–3777 (2012).
Ha, H. C. Defective transcription factor activation for proinflammatory gene expression in poly(ADP-ribose) polymerase 1-deficient glia. Proc. Natl. Acad. Sci. USA 101(14), 5087–5092 (2004).
Masson, G. S. et al. Aerobic training normalizes autonomic dysfunction, HMGB1 content, microglia activation and inflammation in hypothalamic paraventricular nucleus of SHR. Am. J. Physiol. Heart Circ. Physiol. 309(7), H1115–H1122 (2015).
Barbay, M. et al. Vascular cognitive impairment: advances and trends. Rev. Neurol. (Paris) 173(7–8), 473–480 (2017).
Kalaria, R. N., Akinyemi, R. & Ihara, M. Stroke injury, cognitive impairment and vascular dementia. Biochim. Biophys. Acta 1862(5), 915–925 (2016).
Levine, D. A. et al. Trajectory of cognitive decline after incident stroke. JAMA 314(1), 41–51 (2015).
Ishitsuka, K. et al. High blood pressure after acute ischemic stroke is associated with poor clinical outcomes: Fukuoka Stroke Registry. Hypertension 63(1), 54–60 (2014).
Ergul, A. et al. Impact of comorbidities on acute injury and recovery in preclinical stroke research: focus on hypertension and diabetes. Transl Stroke Res. 7(4), 248–260 (2016).
Nam, H., et al. Learned helplessness and social avoidance in the Wistar-Kyoto rat. 2014. 8(109).
Zorzo, C. et al. Adult social isolation leads to anxiety and spatial memory impairment: Brain activity pattern of COx and c-Fos. Behav. Brain Res. 365, 170–177 (2019).
Kono, S. et al. Neurovascular protection by telmisartan via reducing neuroinflammation in stroke-resistant spontaneously hypertensive rat brain after ischemic stroke. J. Stroke Cerebrovasc. Dis. 24(3), 537–547 (2015).
Sato, K. et al. Telmisartan reduces progressive oxidative stress and phosphorylated alpha-synuclein accumulation in stroke-resistant spontaneously hypertensive rats after transient middle cerebral artery occlusion. J. Stroke Cerebrovasc. Dis. 23(6), 1554–1563 (2014).
Ritter, S. et al. Cerebroventricular dilation in spontaneously hypertensive rats (SHRs) is not attenuated by reduction of blood pressure. Brain Res. 450(1–2), 354–359 (1988).
Halmosi, R. et al. Effect of poly(ADP-ribose) polymerase inhibitors on the ischemia-reperfusion-induced oxidative cell damage and mitochondrial metabolism in Langendorff heart perfusion system. Mol. Pharmacol. 59(6), 1497–1505 (2001).
Li, W. H. et al. Baicalein attenuates caspase-independent cells death via inhibiting PARP-1 activation and AIF nuclear translocation in cerebral ischemia/reperfusion rats. Apoptosis 25(5–6), 354–369 (2020).
Eros, K. et al. Chronic PARP-1 inhibition reduces carotid vessel remodeling and oxidative damage of the dorsal hippocampus in spontaneously hypertensive rats. PLoS ONE 12(3), e0174401 (2017).
Besson, V. C. et al. Deleterious poly(ADP-ribose)polymerase-1 pathway activation in traumatic brain injury in rat. Brain Res. 989(1), 58–66 (2003).
Noh, M. Y. et al. Regulatory T cells increase after treatment with poly (ADP-ribose) polymerase-1 inhibitor in ischemic stroke patients. Int. Immunopharmacol. 60, 104–110 (2018).
Turner, R. J. & Sharp, F. R. Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front. Cell Neurosci. 10, 56 (2016).
Mechtouff, L. et al. Matrix metalloproteinase-9 and monocyte chemoattractant protein-1 are associated with collateral status in acute ischemic stroke with large vessel occlusion. Stroke 51(7), 2232–2235 (2020).
Gao, Y. et al. Effect of telmisartan on preventing learning and memory deficits via peroxisome proliferator-activated receptor-gamma in vascular dementia spontaneously hypertensive rats. J. Stroke Cerebrovasc. Dis. 27(2), 277–285 (2018).
Chaitanya, G. V., Steven, A. J. & Babu, P. P. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 8, 31 (2010).
Candelario-Jalil, E., Yang, Y. & Rosenberg, G. A. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 158(3), 983–994 (2009).
Mandler, R. N. et al. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in cerebrospinal fluid differ in multiple sclerosis and Devic’s neuromyelitis optica. Brain 124(Pt 3), 493–498 (2001).
Nicolescu, A. C. et al. Inhibition of matrix metalloproteinase-2 by PARP inhibitors. Biochem. Biophys. Res. Commun. 387(4), 646–650 (2009).
Christensen, J. & Shastri, V. P. Matrix-metalloproteinase-9 is cleaved and activated by cathepsin K. BMC Res. Notes 8, 322 (2015).
Bi, J. et al. Critical role of matrix metallopeptidase 9 in postoperative cognitive dysfunction and age-dependent cognitive decline. Oncotarget 8(31), 51817–51829 (2017).
Wang, H. et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285(5425), 248–251 (1999).
Klune, J. R. et al. HMGB1: endogenous danger signaling. Mol. Med. 14(7–8), 476–484 (2008).
Gao, H. M. et al. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 31(3), 1081–1092 (2011).
Paudel, Y. N. et al. HMGB1: a common biomarker and potential target for TBI, neuroinflammation, epilepsy, and cognitive dysfunction. Front Neurosci. 12, 628 (2018).
Famakin, B. M. et al. HMGB1 is a potential mediator of astrocytic TLR4 signaling activation following acute and chronic focal cerebral ischemia. Neurol. Res. Int. 2020, 3929438 (2020).
Singh, V. et al. HMGB1 as a key mediator of immune mechanisms in ischemic stroke. Antioxid Redox Signal 24(12), 635–651 (2016).
Acknowledgements
The authors would like to thank the core imaging facility for small animals (CIFSA) of the Georgia Cancer Center and Augusta University animal facility for performing the imaging and for help in performing analysis. We would also like to thank the Electron Microscope and Histology Core at Augusta University for sectioning the brain tissues. Special thanks to members of Dr. Adviye Ergul’s lab for providing technical assistance during animal sacrifice. We would like to thank Mir Adil for his help in figure preparation.
Funding
This study was supported by National Institute of Health (NIH) R01 NS104573 to Adviye Ergul and Susan C. Fagan; R21-NS088016 and Veterans Affairs (VA) Merit Review (BX000891) to Susan C. Fagan; R01 NS083559, VA Merit Review (BX000347), VA Senior Research Career Scientist Award (IK BX004471) to Adviye Ergul.
Author information
Authors and Affiliations
Contributions
M.A.S. was responsible for overall study design, data collection and analysis as well as performing behavioral testing, microscopy studies and writing the manuscript. W.E. performed the stroke surgeries. M.A., B.P. and W.A. helped in data collection and analysis for molecular and behavioral experiments. M.H.J. was responsible for the statistical analysis. A.S.A. supported the study with the MRI data. A.E. interpreted the data and reviewed the manuscript. S.C.F. designed the study, managed the experiments, interpreted the data, and reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sayed, M.A., Eldahshan, W., Abdelbary, M. et al. Stroke promotes the development of brain atrophy and delayed cell death in hypertensive rats. Sci Rep 10, 20233 (2020). https://doi.org/10.1038/s41598-020-75450-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-75450-6
This article is cited by
-
Experimental Ischemic Stroke Induces Secondary Bihemispheric White Matter Degeneration and Long-Term Cognitive Impairment
Translational Stroke Research (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
