
Abstract
Very early onset inflammatory bowel disease (VEOIBD) denotes children with onset of IBD before six years of age. A number of monogenic disorders are associated with VEOIBD including tetratricopeptide repeat domain 7A (TTC7A) deficiency. TTC7A-deficiency is characterized by apoptotic colitis in milder cases with severe intestinal atresia and immunodeficiency in cases with complete loss of protein. We used whole exome sequencing in a VEOIBD patient presenting with colitis characterized by colonic apoptosis and no identified known VEOIBD variants, to identify compound heterozygous deleterious variants in the Ubiquitin protein ligase E3 component N-recognin 5 (UBR5) gene. Functional studies demonstrated that UBR5 co-immunoprecipitates with the TTC7A and the UBR5 variants had reduced interaction between UBR5 and TTC7A. Together this implicates UBR5 in regulating TTC7A signaling in VEOIBD patients with apoptotic colitis.
Similar content being viewed by others


Introduction
Very Early Onset Inflammatory Bowel Disease (VEOIBD) may be associated with monogenic disorders1,2,3,4,5,6,7,8,9,10,11. Recent studies have demonstrated that approximately 3% of Pediatric IBD patients have a monogenic cause for their disease and younger age at diagnosis is a risk factor12.
Previously, we and others identified TTC7A deficiency as a cause of severe intestinal disease1, 13,14,15,16,17,18. Over 50 patients have been identified with pathogenic variants in TTC7A associated with a heterogeneous array of phenotypes involving the intestine and immune system1, 13,14,15,16,17,18,19,20,21,22,23,24,25. VEOIBD patients with TTC7A variants have apoptotic enterocolitis and functional studies show loss of interaction with Phosphatidylinositol 4-kinase Type III Alpha (PI4KIIIα) to be the causative factor1. PI4KIIIα is involved in the production of phosphatidylinositol 4-phosphate (PI4P) at the plasma membrane (PM)26,27 with the help of TTC7A and FAM126A which scaffold PI4KIIIα from endoplasmic reticulum to PM where the complex also interacts with EFR3A/B28,29. We also identified Ubiquitin protein ligase E3 component N-recognin 5 (UBR5) as a Tetratricopeptide Repeat Domain 7A (TTC7A) interacting protein using tandem mass spectrometry1.
UBR5 is a E3 ubiquitin ligase that has been implicated in several cellular process such as the regulation of DNA damage30, metabolism31, transcription32, and apoptosis33. Ubr5-/- mice fail to grow beyond the E10.5 embryonic development stage and knockout of hyd (‘hyperplastic discs’) and UBR5′s homologue in Drosophila melanogaster, results in lethality in the pupal or larval stages34. Recent studies have described UBR5 as an oncogene in colorectal cancer (CRC)35,36,37. UBR5 was also found to be more often mutated in a higher percentage of cases resulting in transitioning from IBD to CRC compared to only CRC cases38.
Here, we identified bi-allelic damaging variants in UBR5 in a VEOIBD patient who presented with severe colonic disease. Functional studies demonstrate that UBR5 co-immunoprecipitates (co-IP) with TTC7A implicating UBR5 in the TTC7A-PI4KIIIα complex signalling.
Results
Patient summary
A boy of Spanish ancestry, born to healthy non-consanguineous parents, presented with diarrhea, rectal bleeding and rectal prolapse at age 2 years and 9 months (Supplementary Figure 1A and Table 1 for blood work analysis). He had no extra intestinal manifestations of disease. Colonoscopy and histopathological examination demonstrated patchy inflammatory cell infiltrates with apoptosis leading to a diagnosis of IBD-Unclassified (Supplementary Figure 1B). He was initially treated with intravenous steroids but eventually required oral tacrolimus due to poor response. His treatment was changed to rectal 5-ASA and azathioprine after response to tacrolimus. At 4 years and 10 months age, his disease flared (Pediatric Ulcerative Colitis Activity Index39,40 [PUCAI] score of 75 points) with no response to intravenous steroids and was switched to infliximab. Currently, he is maintained with 10 mg/kg/4 weeks infliximab (IFX) and rectal 5-ASA (due to a proctitis unresponsive to IFX) and oral 5-ASA with his most recent endoscopy showing only mild proctitis (Supplementary Figure 1C).

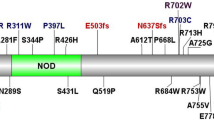
Filtration strategy from Whole Exome Sequencing (WES) for selection of UBR5 as a disease causing variant and genetic analysis of the trio for UBR5 patient. (A) WES of the TRIO identified total of 123,982 variants in the patient. Low-quality variants were removed. Afterwards, common variants with maf < 0.01 from 1000Genomes_phase362 were removed. To isolated potential causative variants, only protein coding variants were included in the inheritance analysis. Finally, variants with CADD > 20 and max maf < 0.01 were identified resulting in various inheritance models such as autosomal recessive, de novo, x-linked, and compound heterozygous. (B) Sanger sequencing of the compound heterozygous variants found in the patient and parents. (C) Pedigree of the affected patient’s family and the inheritance pattern of the mutations in the patient. Aminode analysis for UBR5 from multiple species shows strong conservation of (D) Proline (P) at position 84 and (E) Leucine (L) at position 1405. The red line shows amount of sequence substitution at that amino acid position63. ECR = evolutionarily constrained region. CLUSTALW multiple species sequence alignment for UBR5 by MUSCLE shows strong conservation of (F) Proline at position 84 and (G) Leucine at position 1405. (H) Location of the mutations on the UBR5 domain architecture. Figure adapted from Shearer et al.51. UBA = ubiquitin associated (UBA) domain, UBR = ubiquitin recognin box, NLS = nuclear localization sequences, and PABC = domain homologous to C-terminus of Poly-Adenylation Binding Protein.
Genetic analysis
Analysis of whole exome sequencing of DNA samples from the families did not identify any known monogenic disorder associated with VEOIBD (see Fig. 1A for analysis strategy and Supplementary Table 2 for full list of potential variants). However, we identified biallelic compound heterozygous nonsynonymous variants in the UBR5 gene of the patient (Supplementary Table 2 for further information regarding UBR5 mutation including MAF and damaging scores). A variant in exon 4 of UBR5 [hg38.g.chr8:102360605(G>T); NM_015902:c.250C>A] resulting in a proline to threonine substitution at amino acid position 84 (p.P84T) was inherited from the unaffected mother (Fig. 1B,C). This variant is rare (gnomAD41 minor allele frequency (MAF) = 0.000081) and predicted to be deleterious by bioinformatic algorithms, including a PHRED scaled combined annotation dependent depletion42 (CADD) score (ver 1.3) of 23.5. A variant in trans in exon 33 of UBR5 [hg38.g.chr8:102294091(G>C); NM_015902:c.4213C>G] was inherited from the unaffected father (Fig. 1B,C) resulting in a leucine to valine substitution at amino acid position 1405 (p.L1405V). This variant has a CADD score of 24.6 and is not present in gnomAD. Both affected residues, P84 and L1405, are highly conserved across species (Fig. 1D–G respectively). While the p.P84T variant is not located in a known domain, the p.L1405V variant is predicted to affect the second nuclear localization sequence of UBR543 (Fig. 1H). A number of VEOIBD databases were searched for other patients with potential UBR5 variants but none were identified.
Functional studies
Immunohistochemistry
As Hemotoxylin and Eosin staining of the patient’s colonic biopsies (Supplementary Figure 1B) demonstrated increased apoptosis, we further examined apoptosis using immunohistochemistry (IHC) staining for cleaved (Cl) Caspase 3. Dual labeling of colon sections in a healthy control and patient with IBD and with known variants, we observed minimal cleaved(cl)-Caspase 3 positive cells (Fig. 2A,B). In contrast, patients with UBR5 or TTC7A variants had increased cleaved-Caspase 3 positive cells (Fig. 2C,D). In our patient with UBR5 variants, we observed increased cleaved-Caspase 3 positive cells in the lamina propria (Fig. 2D). In contrast, in the TTC7A patient section (Fig. 2C), the cleaved-Caspase 3 positive cells are only in the epithelium. There is no observed difference in the intensity and architecture for β-catenin between UBR5 patient section (Fig. 2D) and control sections (healthy control + IBD patient sections) (Fig. 2A,B). However, there is a disruption in epithelial layer architecture for β-catenin in TTC7A patient section (Fig. 2C).

Elevated caspase-3 activity in the UBR5 patient. Immunohistochemistry (IHC) of Formalin-Fixed Paraffin Embedded (FFPE) colon sections from (A) healthy control, (B) IBD patient without mutations, (C) TTC7A patient, and (D) UBR5 patient. Cleaved (Cl) caspase-3 is shown in green, β-catenin in red and nuclear counterstaining in blue (RedDot2).
IHC staining for UBR5 and TTC7A in a healthy control showed that in the colon, UBR5 localized mainly in the nuclei of immune cells with minimal epithelial expression (Fig. 3A). In colonic sections from an IBD patient without UBR5 or TTC7A variants and a TTC7A-deficiency patient (previously described44), we observed upregulation of UBR5 in epithelial cells of the colon (Fig. 3B,C). No colocalization of UBR5 and TTC7A labeling could be observed in the TTC7A patient (Fig. 3E and Supplementary Table 3). Interestingly, colonic sections from our UBR5 variant patient demonstrated a different pattern of localization in the patient, as compared to healthy controls and IBD patients with strong signal intensity for UBR5 aggregated in the epithelial indicative of intra-epithelial lymphocytes (Fig. 3D,E).

IHC of FFPE colon sections from (A) a healthy individual, (B) an IBD patient without mutations, (C) a patient with TTC7A variants and (D) a patient with compound heterozygous variants in UBR5. UBR5 is shown in green, TTC7A in red and nuclear counterstaining in blue (RedDot2). (E) Shows percentage of cells showing colocalization between UBR5 and TTC7A in 100 cells stained positive for both, UBR5 and TTC7A, in selected areas.
Identification of TTC7A as a binding partner of UBR5
Previously, tandem mass spectrometry (MS) using TTC7A WT and the E71K variants as bait identified UBR5 as a potential interactor of TTC7A1. To further validate these finding, we used co-immunoprecipitation (co-IP) studies in HEK 293 T cells and showed that UBR5 did co-IP with TTC7A (Fig. 4A) and TTC7A with UBR5 (Fig. 4B). TTC7A showed reduced co-IP with the identified UBR5 L1405V mutant protein, as compared with UBR5 WT (Fig. 5A,B). However, TTC7A showed increased co-IP with a catalytically dead UBR5 E3 ligase C2768A mutant in the HECT domain. Further analysis of the previously identified TTC7A VEOIBD mutants (E71K, Q526X, and A832T)1 also showed that UBR5 had reduced co-IP to the missense variants but increased co-IP with the Q526X truncation (Fig. 6A,B). These results validate the previously identified interaction between TTC7A and UBR5 and indicate a role of UBR5 in TTC7A signaling1. Full uncropped blots are available in Supplementary Figures 2–5.


UBR5 L1405V mutant shows significantly reduced binding to TTC7A in HEK 293 T cells. (A) TTC7A co-IPs with differential binding affinities to UBR5 mutants. UBR5 plasmids were tagged with FLAG and the His tag removed. Lysate and IP samples were derived from the same experiment but ran on different blots. (B) UBR5 L1405V and C2768A show reduced and increased binding to TTC7A, respectively. Densitometry of western blot experiments from (A) were quantified and the values from all samples were made relative to the samples transfected with TTC7A and UBR5. Error bars indicate SD. *Denotes p < 0.05, ns = non-significant (student’s t-test). n = 4. Full length blots for (A) are presented in Supplementary Figure 5.

UBR5′s differential binding affinities to TTC7A VEO-IBD mutants. (A) UBR5 co-IPs with differential binding affinities to TTC7A VEO-IBD mutants. HEK 293 T cells were transfected with His/EGFP tagged UBR5 and Myc/FLAG tagged TTC7A WT, the mutants (A832T, Q526X, or E71K) or the tagged backbone vector only. (B) Densitometry of western blot from (A) was quantified and the values from all samples were made relative to the samples transfected with TTC7A and UBR5. Error bars indicate SD. **Denotes p < 0.01. n = 4. Full length blots for (A) are presented in Supplementary Figure 6.
Discussion
We have recently demonstrated that 3% of pediatric IBD patients have monogenic forms of IBD12. Like the case presented here, many of these patients present at a very early age with severe disease that is difficult to treat12. The young age of our patient, the severity of disease requiring biologic therapy, and the presence of apoptosis on biopsy made this patient a strong candidate for genetic analysis to determine a potential monogenic cause of disease. We first screened for variants in genes known to be associated with VEOIBD, and as this patient was male we focused on X-linked genes associated with intestinal epithelial apoptosis including FOXP3 and XIAP, and also autosomal recessive genes including LRBA, ARPC1B, and TTC7A (see 12,45 for a complete list of genes associated with apoptosis). However, neither variants in these genes nor other genes associated with VEOIBD were identified. Therefore, we examined novel candidate genes and prioritized variant that were rare and damaging based on known biological function, animal models, and known interaction with previously identified VEOIBD genes.
UBR5 was selected as a potential gene candidate based on evidence from our previous study utilizing tandem mass spectrometry to identify potential binding partners for TTC7A1,18. Bi-allelic deleterious variants in TTC7A were identified as a causal gene for severe intestinal and immune disease with high penetrance1,13,14,15,16,18,21,22,23,24,46,47,48,49. Many TTC7A-deficient patients present with clinical features associated with monogenic IBD or VEOIBD1,14,15,18,21,22,47. A key pathological feature of TTC7A deficiency is increased intestinal epithelial cell apoptosis1,13,18,47 that is not commonly found in typical non-genetic forms of IBD. Although, our patient did not have the severe immunodeficiency and intestinal stricturing disease observed in severe loss-of-function TTC7A mutations, he did have apoptotic colonic disease associated with the less severe form of the disease caused by hypomorphic TTC7A mutations.
With any potential novel VEOIBD variants, functional studies are required to demonstrate a potentially causative defect. Here we used co-IP experiments to validate our genetic studies and showed that variants in both TTC7A and UBR5 reduce co-IP. Previous studies of Ubr5-/- mice demonstrated widespread apoptosis by E9.5 embryonic development stage50. UBR5 is a HECT E3 ubiquitin ligase that is found to be mutated or amplified in various cancer types (including colorectal cancer51), to inhibit intestinal apoptosis35 and UBR5 knockdown resulted in increased apoptosis in ovarian cells52. Pathological examination of biopsies from patients with both TTC7A and UBR5 variants demonstrate that UBR5 has low expression in the healthy gut but is highly upregulated in TTC7A-deficiency patient indicating a possible compensatory mechanism. Also, in our VEOIBD patient with UBR5 variants, there was increased apoptosis in both epithelial and immune cells. As TTC7A is expressed in both epithelial and immune cells, this may be due to dysregulation of TTC7A-PI4K signalling but as UBR5 has a role in DUBA signalling in T-cells, this may contribute to disease progression53. It is interesting to speculate that our patient may benefit from treatment with Leflunomide as it was recently shown in a preclinical study as a potential therapy for TTC7A-deficiency54.
Our studies suggest that UBR5 may be associated with VEOIBD; however, there are a number of important limitations. First, despite searching a number of VEOIBD databases, we were unable to identify a second patient with bi-allelic variants in UBR5. UBR5 does not have any homozygous loss-of-function variants on gnomad (https://gnomad.broadinstitute.org/) suggesting that loss of function is detrimental and often it takes time to identify additional patients. Second, our functional studies using tandem mass-spec and co-immunoprecipitation experiments have shown that TTC7A and UBR5 appear to interact; however, we did not determine the precise role of UBR5 in TTC7A signaling. Therefore, further study into the function of this putative interaction and the relevant cell type(s) will be critical in our understanding of the disease pathogenesis.
Methods
Next-generation sequencing and data analysis
WES was performed in collaboration with the Regeneron Genetics Center (RGC) on this proband and his unaffected parents who were enrolled and consented in our NEOPICS partnership (https://www.neopics.org/). Exome capture was carried out using the NimbleGen VCRome 2.1 and sequencing was done using an Illumina HiSeq 2500 platform with paired-end 75 bp reads. Sequencing reads were aligned to human reference genome (GRCh38). Variants were called using the Genome Analysis Toolkit (GATK) (pmid:20644199) and the generated VCF files were subsequently annotated with snpEff (pmid:22728672). Polymorphisms reported in public databases with Minor Allele Frequency (MAF) > 1% and synonymous variants were filtered out. Potential pathogenicity protein-coding variants were prioritized using evolutionary conservation and various prediction tools (SIFT, PolyPhen2, Mutation Taster) from dbNSFP55. Inheritance modeling was carried out using GEMINI software56 (https://gemini.readthedocs.io/en/latest/) to identify variants that fit autosomal recessive, de novo, and X-linked inheritance patterns.
Patient data availability
The identified UBR5 variants of our patient will be submitted to the ClinVar57 database (https://www.ncbi.nlm.nih.gov/clinvar/) upon publication. Information on the raw whole-exome sequencing data will not be published to protect research participant privacy.
Sanger sequencing
Sanger sequencing was performed in the patient and parents to validate the compound heterozygous variants identified by WES. The genetic details for the variants are listed below:
NM_015902 (Homo sapiens ubiquitin protein ligase E3 component n-recognin 5 (UBR5), transcript variant 1, mRNA).
P84T: c.250c>a; dbSNP rs143719892; GRCh37 8:103372833 G>T; exon4.
L1405V: c.4213c>g; not in dbSNP; GRCh37 8:103306319 G>C; exon33.
The following primers were used to sequence P84T: forward TGGTAGAGTTTGCAGGATTGG (sense), and reverse TGATAACTGACTCCTCTGCTACT (anti-sense). The following primers were used to sequence L1405V: forward CCAAGGACTGTGGGACAAA (sense), and reverse CTCTTGCCACTGAACGTAGAA (anti-sense).
Plasmid constructs
pCMV-Tag2B EDD and C2768A were a gift from Darren Saunders & Charles Watts (Addgene plasmid # 37188 and 37189 respectively)43. These plasmids were modified by deleting the His tag on the C-terminus of the cDNA for the experiment in Fig. 5. UBR5 mutant plasmids were created from the pCMV-Tag2B EDD WT plasmid by ACGT Corporation (Toronto). pEGFP-C1 EDD was also a gift from Darren Saunders & Charles Watts (Addgene plasmid # 37190)43. TTC7A WT and mutant plasmids (E71K, Q526X and A832T) with myc-DDK tags were previously generated1. Another TTC7A plasmid was constructed with pLJM1-EGFP entry vector with the deletion of the EGFP from the vector. A N-terminus HA-tagged TTC7A cDNA was subcloned into pLJM1 entry vector. pLJM1-EGFP backbone vector (or GFP vector) was used as a control for HA tagged TTC7A plasmids.
Co-immunoprecipitation assay
HEK 293 T cells were grown on 10 cm plates and transfected with various combinations of plasmids using PolyJet (SignaGen Laboratories) according to standard protocols. 48 h post-transfection, cells were lysed with lysis buffer (150 mM NaCl, 50 mM HEPES, 1% Triton-X, 10% glycerol, 1.5 mM MgCl2 and 1.0 mM EGTA) supplemented with protease inhibitors (1 mM PMSF, 1 mM P2714, 2 mM Na3VO4 and 5 mM NaF). Lysates were not precleared before beginning the immunoprecipitation except for Fig. 5A experiment. 1 mg of lysate was immunoprecipitated with anti-FLAG beads (Sigma Aldrich or BioLegend) or anti-GFP beads (BioLegend) for 2 h at 4 °C. Negative control included lysates of all samples pooled with protein G beads (BioLegend) and 2 μg of mouse IgG antibody. Beads were washed 3 times with the same lysis buffer with protease inhibitors used to lyse the cells. Bound proteins were eluted using 35 μL of 2X SDS protein sample buffer (40% glycerol, 240 mM Tris/HCl, 8% SDS, 0.4% bromophenol blue, 5% beta-mercaptoethanol). 50 μg of lysate with 1-2X sample buffer and 30 μL of IP sample (15 μL for experiments in Fig. 4A,B) was loaded into 8–10% SDS-PAGE and subject to western blot analysis. Semi-dry transfer was performed using a Bio-Rad machine and nitrocellulose membrane. All experiments were performed in triplicate unless otherwise stated.
Statistical analysis
Co-IP: Odyssey FC (LI-COR Biosciences), a chemiluminescence scanner, was used for imaging of the western blot. Densitometry of the western blot was obtained by ImageStudioLite (LI-COR Biosciences) and quantified by making the ratio of the IP band for a protein in a sample relative to the lysate band in the same sample. For each sample, the IP/lysate ratio obtained for TTC7A for each sample was divided with IP/lysate ratio for UBR5 from the same sample. For each sample, the values obtained for TTC7A/UBR5 ratios of IP/lysate were made relative to TTC7A WT + UBR5 transfected sample. Vice versa was done for Fig. 5A to generate Fig. 5B. GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA) was used to create the graph and student’s t-test was performed for statistical analysis of the variables of interest. All experiments were n = 3 unless otherwise stated.
IF histochemical staining on formalin-fixed paraffin-embedded (FFPE) sections
Colon-sigmoid mucosa tissue samples were retrieved from the Division of Pathology, The Hospital for Sick Children. These samples include healthy control, IBD control with normal GI histology and without validated variants from infants. Patient’s biopsies from Spain (UBR5 patient) and BC (TTC7A patient44) infants were obtained with Ethics approval and informed consent as previously summarized58.
Dual immunofluorescent histochemical staining
The details for IF staining on FFPE section procedure are published58. Briefly, as a first step, paraffin was removed using Xylene, and afterwards rehydrated with different percentages of ethanol. Antigen retrieval was performed with high-pressure cooking in EDTA–borax buffer (1 mM EDTA, 10 mM borax (sodium tetraborate, Sigma, St Louis, MI, USA), 10 mM boric acid (Sigma) with 0.001% Proclin 300 (Supleco, Bellefonte, PA, USA) at pH 8.5. To block non-specific staining, the slides were incubated for 1 h at room temperature in 4% BSA in 1X phosphate-buffered saline (PBS, Multi Cell) 20% normal donkey serum. A properly diluted primary antibody, for example, rabbit anti-TTC7A (see Table 1 for detail) polyclonal antibody and anti-cyto-structure mouse monoclonal antibody (Abcam Inc. Toronto, Ontario, see Table 1) incubation was performed overnight at 4 °C. On the following day, stained slides were washed three times for 5 min with 1X PBS. Secondary antibody, donkey anti-rabbit IgG Fab2 fragment-Rhodamine conjugate mixed with donkey anti-mouse IgG Fab2 fragment-FITC conjugate (Jackson Immuno Research Lab, West Grove, PA) incubation was performed at room temperature in darkness for 2 h, and slides were washed afterwards three times for 10 min in darkness. As a nuclear counterstain reagent, RedDot2 far red fluorescence (Biotium Inc. Fremont CA) was used at a dilution of 1:200. Finally, sections were mounted overnight with Vector shield fluorescence mounting medium (Vector Labs, Burlington, ON).
Confocal microscopy
Double/triple-immunostained sections were imaged using a Leica confocal laser scanning microscope (model TCS-SP8) and LAS-AF software (Leica Microsystems, Wetzlar, Germany), as previously reported59. The variable excitation wavelengths of the krypton/argon laser were 488 nm for fluorescein isothiocyanate conjugate, 568 nm for Texas Red complex, and 695 nm for Alexa Fluor 680 conjugate/RedDot 2 (nuclear counterstaining). Image processing, including color resolution, color separation, and merging of fields, were carried out using Adobe PhotoShop CS5 software (Adobe Systems Incorporated, San Jose, CA, USA).
Morphometric and colocalization analysis
The NIH ImageJ software was used with appropriate algorithms to analyze the degree of co-occurrence and correlation for TTC7A and UBR5 in the images. A total of 100 cells were selected that showed positive staining for both, TTC7A and UBR5, from 5 different areas within each slide. The JACoP plugin60 for ImageJ was applied in these cells for obtaining Pearson and Mander’s coefficients. Previously reported algorithms61 were used to determine co-localization and the associated statistics are reported in Supplementary Table 3. Linear regression/correlation and the t-test were used for the statistical/correction analysis to report on colocalization.
Helsinki guidelines
All human experiments followed the Helsinki Guidelines.
Informed consent
Informed consent was obtained from the participant’s parents and the study had local ethics board approval at Hospital Regional Universitario de Málaga, Málaga, Spain and Hospital for Sick Children, Toronto, Canada (Research Ethics Board: REB1000024905).
References
Avitzur, Y. et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology 146, 1028–1039. https://doi.org/10.1053/j.gastro.2014.01.015 (2014).
Janecke, A. R. et al. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum. Mol. Genet. 24, 6614–6623. https://doi.org/10.1093/hmg/ddv367 (2015).
Elkadri, A. et al. Mutations in plasmalemma vesicle associated protein result in sieving protein-losing enteropathy characterized by hypoproteinemia, hypoalbuminemia, and hypertriglyceridemia. Cell Mol. Gastroenterol. Hepatol. 1, 381.e387-394.e387. https://doi.org/10.1016/j.jcmgh.2015.05.001 (2015).
Li, Q. et al. Variants in TRIM22 that affect NOD2 signaling are associated with very-early-onset inflammatory bowel disease. Gastroenterology 150, 1196–1207. https://doi.org/10.1053/j.gastro.2016.01.031 (2016).
Kahr, W. H. et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat. Commun. 8, 14816. https://doi.org/10.1038/ncomms14816 (2017).
Parlato, M. et al. Human ALPI deficiency causes inflammatory bowel disease and highlights a key mechanism of gut homeostasis. EMBO Mol. Med. 10, e8483 (2018).
Kotlarz, D. et al. Human TGF-beta1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nat. Genet. 50, 344–348. https://doi.org/10.1038/s41588-018-0063-6 (2018).
Lehle, A. S. et al. Intestinal inflammation and dysregulated immunity in patients with inherited caspase-8 deficiency. Gastroenterology 156, 275–278. https://doi.org/10.1053/j.gastro.2018.09.041 (2019).
vandeGeer, A. , et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J. Clin. Investig. 128, 3957–3975. https://doi.org/10.1172/JCI97116 (2018).
Li, Y. et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 116, 970–975. https://doi.org/10.1073/pnas.1813582116 (2019).
van Haaften-Visser, D. Y. et al. Ankyrin repeat and zinc-finger domain-containing 1 mutations are associated with infantile-onset inflammatory bowel disease. J. Biol. Chem. 292, 7904–7920. https://doi.org/10.1074/jbc.M116.772038 (2017).
Crowley, E. et al. Prevalence and clinical features of inflammatory bowel diseases associated with monogenic variants, identified by whole-exome sequencing in 1000 children at a single center. Gastroenterology https://doi.org/10.1053/j.gastro.2020.02.023 (2020).
Bigorgne, A. E. et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J. Clin. Invest. https://doi.org/10.1172/JCI71471 (2013).
Chen, R. et al. Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J. Allergy Clin. Immunol. 132(656), e617-664.e617. https://doi.org/10.1016/j.jaci.2013.06.013 (2013).
Samuels, M. E. et al. Exome sequencing identifies mutations in the gene TTC7A in French-Canadian cases with hereditary multiple intestinal atresia. J. Med. Genet. https://doi.org/10.1136/jmedgenet-2012-101483 (2013).
Agarwal, N. S. et al. Tetratricopeptide repeat domain 7A (TTC7A) mutation in a newborn with multiple intestinal atresia and combined immunodeficiency. J. Clin. Immunol. 34, 607–610. https://doi.org/10.1007/s10875-014-0067-7 (2014).
Fernandez, I. et al. Multiple intestinal atresia with combined immune deficiency related to TTC7A defect is a multiorgan pathology: study of a French-Canadian-based cohort. Medicine 93, e327. https://doi.org/10.1097/MD.0000000000000327 (2014).
Lemoine, R. et al. Immune deficiency-related enteropathy-lymphocytopenia-alopecia syndrome results from tetratricopeptide repeat domain 7A deficiency. J. Allergy Clin. Immunol. 134, 1354. https://doi.org/10.1016/j.jaci.2014.07.019 (2014).
Lawless, D. et al. Bialellic mutations in tetratricopeptide repeat domain 7A (TTC7A) cause common variable immunodeficiency-like phenotype with enteropathy. J. Clin. Immunol. 37, 617–622. https://doi.org/10.1007/s10875-017-0427-1 (2017).
Leclerc-Mercier, S. et al. Ichthyosis as the dermatological phenotype associated with TTC7A mutations. Br. J. Dermatol. 175, 1061–1064. https://doi.org/10.1111/bjd.14644 (2016).
Neves, J. F. et al. Missense mutation of TTC7A mimicking tricho-hepato-enteric (SD/THE) syndrome in a patient with very-early onset inflammatory bowel disease. Eur. J. Med. Genet. 61(4), 185–188 (2017).
Lien, R. et al. Novel mutations of the tetratricopeptide repeat domain 7A gene and phenotype/genotype comparison. Front. Immunol. 8, 1066. https://doi.org/10.3389/fimmu.2017.01066 (2017).
Kammermeier, J. et al. Stem cell transplantation for tetratricopeptide repeat domain 7A deficiency: long-term follow-up. Blood 128, 1306–1308. https://doi.org/10.1182/blood-2016-01-696385 (2016).
Yang, W. et al. Compound heterozygous mutations in TTC7A cause familial multiple intestinal atresias and severe combined immunodeficiency. Clin. Genet. 88, 542–549. https://doi.org/10.1111/cge.12553 (2015).
Jardine, S., Dhingani, N. & Muise, A. M. TTC7A: steward of intestinal health. Cell Mol. Gastroenterol. Hepatol. 7, 555–570. https://doi.org/10.1016/j.jcmgh.2018.12.001 (2019).
Balla, A. et al. Maintenance of hormone-sensitive phosphoinositide pools in the plasma membrane requires phosphatidylinositol 4-kinase III alpha. Mol. Biol. Cell 19, 711–721. https://doi.org/10.1091/mbc.E07-07-0713 (2008).
Tan, J. L., Oh, K., Burgess, J., Hipfner, D. R. & Brill, J. A. Pl4Klll alpha is required for cortical integrity and cell polarity during Drosophila oogenesis. J. Cell Sci. 127, 2601–2601. https://doi.org/10.1242/jcs.154898 (2014).
Baskin, J. M. et al. The leukodystrophy protein FAM126A (hyccin) regulates PtdIns(4)P synthesis at the plasma membrane. Nat. Cell Biol. 18, 132–138. https://doi.org/10.1038/ncb3271 (2016).
Lees, J. A. et al. Architecture of the human PI4KIIIalpha lipid kinase complex. Proc. Natl. Acad. Sci. USA 114, 13720–13725. https://doi.org/10.1073/pnas.1718471115 (2017).
Zhang, T., Cronshaw, J., Kanu, N., Snijders, A. P. & Behrens, A. UBR5-mediated ubiquitination of ATMIN is required for ionizing radiation-induced ATM signaling and function. Proc. Natl. Acad. Sci. 111, 12091–12096. https://doi.org/10.1073/pnas.1400230111 (2014).
Jiang, W. et al. Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase. Mol. Cell 43, 33–44. https://doi.org/10.1016/j.molcel.2011.04.028 (2011).
Ong, S. S. et al. Stability of the human pregnane X receptor is regulated by E3 ligase UBR5 and serine/threonine kinase DYRK2. Biochem. J. 459, 193–203. https://doi.org/10.1042/BJ20130558 (2014).
Henderson, M. J. et al. EDD mediates DNA damage-induced activation of CHK2. J. Biol. Chem. 281, 39990–40000 (2006).
Mansfield, E., Hersperger, E., Biggs, J. & Shearn, A. Genetic and molecular analysis of hyperplastic disks, a gene whose product is required for regulation of cell-proliferation in Drosophila-Melanogaster imaginal disks and germ-cells. Dev. Biol. 165, 507–526. https://doi.org/10.1006/dbio.1994.1271 (1994).
Ji, S. Q., Zhang, Y. X. & Yang, B. H. UBR5 promotes cell proliferation and inhibits apoptosis in colon cancer by destablizing P21. Die Pharmazie Int. J. Pharm. Sci. 72, 408–413. https://doi.org/10.1691/ph.2017.7433 (2017).
Xie, Z. et al. Significance of the E3 ubiquitin protein UBR5 as an oncogene and a prognostic biomarker in colorectal cancer. Oncotarget 8, 108079–108092. https://doi.org/10.18632/oncotarget.22531 (2017).
Wang, J., Zhao, X., Jin, L., Wu, G. & Yang, Y. UBR5 contributes to colorectal cancer progression by destabilizing the tumor suppressor ECRG4. Digest. Dis. Sci. 62, 2781–2789. https://doi.org/10.1007/s10620-017-4732-6 (2017).
Robles, A. I. et al. Whole-exome sequencing analyses of inflammatory bowel disease-associated colorectal cancers. Gastroenterology 150, 931–943. https://doi.org/10.1053/j.gastro.2015.12.036 (2016).
Turner, D. et al. Development, validation, and evaluation of a pediatric ulcerative colitis activity index: a prospective multicenter study. Gastroenterology 133, 423–432. https://doi.org/10.1053/j.gastro.2007.05.029 (2007).
Turner, D. et al. Appraisal of the pediatric ulcerative colitis activity index (PUCAI). Inflam. Bowel Dis. 15, 1218–1223. https://doi.org/10.1002/ibd.20867 (2009).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285. https://doi.org/10.1038/nature19057 (2016).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucl. Acids Res. 47, D886–D894. https://doi.org/10.1093/nar/gky1016 (2018).
Henderson, M. J. et al. EDD, the human hyperplastic discs protein, has a role in progesterone receptor coactivation and potential involvement in DNA damage response. J. Biol. Chem. 277, 26468–26478. https://doi.org/10.1074/jbc.M203527200 (2002).
Saunders, J. R. et al. Novel exonic deletions in TTC7A in a newborn with multiple intestinal atresia and combined immunodeficiency. J. Clin. Immunol. https://doi.org/10.1007/s10875-019-00669-6 (2019).
Uhlig, H. H. et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 147(990), e1003-1007.e1003. https://doi.org/10.1053/j.gastro.2014.07.023 (2014).
Guana, R. et al. The complex surgical management of the first case of severe combined immunodeficiency and multiple intestinal atresias surviving after the fourth year of life. Pediatr. Gastroenterol. Hepatol. Nutr. 17, 257–262. https://doi.org/10.5223/pghn.2014.17.4.257 (2014).
Woutsas, S. et al. Hypomorphic mutation in TTC7A causes combined immunodeficiency with mild structural intestinal defects. Blood 125, 1674–1676 (2015).
Mandiá, N., Perez-Muñuzuri, A. & Lopez-Suarez, O. Congenital intestinal atresias with multiple episodes of sepsis. Medicine 97, e10939 (2018).
Fullerton, B. S., Velazco, C. S., Hong, C. R., Carey, A. N. & Jaksic, T. High rates of positive severe combined immunodeficiency screening among newborns with severe intestinal failure. JPEN J. Parenter. Enter. Nutr. 42, 239–246. https://doi.org/10.1002/jpen.1013 (2018).
Saunders, D. N. et al. Edd, the murine hyperplastic disc gene, is essential for yolk sac vascularization and chorioallantoic fusion. Mol. Cell. Biol. 24, 7225–7234. https://doi.org/10.1128/MCB.24.16.7225-7234.2004 (2004).
Shearer, R. F., Iconomou, M., Watts, C. K. & Saunders, D. N. Functional roles of the E3 ubiquitin ligase UBR5 in cancer. Mol. Cancer Res. MCR 13, 1523–1532. https://doi.org/10.1158/1541-7786.MCR-15-0383 (2015).
Bradley, A. et al. EDD enhances cell survival and cisplatin resistance and is a therapeutic target for epithelial ovarian cancer. Carcinogenesis 35, 1100–1109. https://doi.org/10.1093/carcin/bgt489 (2014).
Rutz, S. et al. Deubiquitinase DUBA is a post-translational brake on interleukin-17 production in T cells. Nature 518, 417–421. https://doi.org/10.1038/nature13979 (2015).
Jardine, S. et al. Drug screen identifies leflunomide for treatment of inflammatory bowel diseases caused by TTC7A deficiency. Gastroenterology https://doi.org/10.1053/j.gastro.2019.11.019 (2019).
Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 24, 2125–2137. https://doi.org/10.1093/hmg/ddu733 (2015).
Paila, U., Chapman, B. A., Kirchner, R. & Quinlan, A. R. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput. Biol. 9, e1003153. https://doi.org/10.1371/journal.pcbi.1003153 (2013).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucl. Acids Res. 46, D1062–D1067. https://doi.org/10.1093/nar/gkx1153 (2017).
Pan, J., Thoeni, C., Muise, A., Yeger, H. & Cutz, E. Multilabel immunofluorescence and antigen reprobing on formalin-fixed paraffin-embedded sections: novel applications for precision pathology diagnosis. Mod. Pathol. 29(6), 557–569 (2016).
Pan, J., Yeger, H. & Cutz, E. Innervation of pulmonary neuroendocrine cells and neuroepithelial bodies in developing rabbit lung. J. Histochem. Cytochem. 52, 379–389. https://doi.org/10.1177/002215540405200309 (2004).
Bolte, S. & Cordelières, F. P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232. https://doi.org/10.1111/j.1365-2818.2006.01706.x (2006).
Villalta, J. I. et al. New algorithm to determine true colocalization in combination with image restoration and time-lapse confocal microscopy to map kinases in mitochondria. PLoS ONE 6, e19031. https://doi.org/10.1371/journal.pone.0019031 (2011).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74. https://doi.org/10.1038/nature15393 (2015).
Chang, K. T., Guo, J., di Ronza, A. & Sardiello, M. Aminode: identification of evolutionary constraints in the human proteome. Sci. Rep. 8, 1357–1357. https://doi.org/10.1038/s41598-018-19744-w (2018).
Acknowledgements
AMM is funded by a Canada Research Chair (Tier 1) in Pediatric IBD, CIHR Foundation Grant and NIDDK (RC2DK118640) Grant. AMM, SBS, CK, DK are supported by the Leona M. and Harry B. Helmsley Charitable Trust. CK and DK are supported by the Collaborative Research Consortium SFB1054 project A05. Special thanks to (1) Dr. Julie Brill and Dr. Andras Kapus for their project advice, (2) The Regeneron Genetics Center who performed whole exome sequencing for the patient and parents, and (3) ACGT Corporation for modifying the UBR5 plasmids and creating UBR5 mutant plasmids.
Author information
Authors and Affiliations
Contributions
N.D., C.G., N.W., Q.L., J.P., S.J., G.L., and A.M.M. conceived the experiment(s) and/or provided input into the experimental design. V.M.N.L. provided patient information and critical discussion on disease pathogenesis. N.D. and J.P. conducted the experiments. N.D., C.G., J.P., Q.L., and A.M.M. analyzed the results. N.D. and A.M.M. with S.B.S., D.K., C.K., C.J.G., and V.M.N.L. analyzed the data and wrote the manuscript with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
CGJ is a full-time employee of the Regeneron Genetics Center from Regeneron Pharmaceuticals, Inc. and receives stock options as part of compensation. All other authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dhingani, N., Guo, C., Pan, J. et al. The E3 ubiquitin ligase UBR5 interacts with TTC7A and may be associated with very early onset inflammatory bowel disease. Sci Rep 10, 18648 (2020). https://doi.org/10.1038/s41598-020-73482-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-73482-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
