
Abstract
Perturbations in the intrauterine environment can result in lifelong consequences for metabolic health during postnatal life. Intrahepatic cholestasis of pregnancy (ICP) can predispose offspring to metabolic disease in adulthood, likely due to a combination of the effects of increased bile acids, maternal dyslipidemia and deranged maternal and fetal lipid homeostasis. Whereas ursodeoxycholic acid (UDCA) is a commonly used treatment for ICP, no studies have yet addressed whether it can also prevent the metabolic effects of ICP in the offspring and fetoplacental unit. We therefore analyzed the lipid profile of fetal serum from untreated ICP, UDCA-treated ICP and uncomplicated pregnancies and found that UDCA ameliorates ICP-associated fetal dyslipidemia. We then investigated the effects of UDCA in a mouse model of hypercholanemic pregnancy and showed that it induces hepatoprotective mechanisms in the fetal liver, reduces hepatic fatty acid synthase (Fas) expression and improves glucose tolerance in the adult offspring. Finally, we showed that ICP leads to epigenetic changes in pathways of relevance to the offspring phenotype. We therefore conclude that UDCA can be used as an intervention in pregnancy to reduce features of metabolic disease in the offspring of hypercholanemic mothers.
Similar content being viewed by others


Introduction
The prevalence of non-communicable diseases such as obesity and type 2 diabetes has grown rapidly over the last three decades1. Increasing evidence links gestational, perinatal and early infancy factors to susceptibility of metabolic disease in adulthood2. Type 2 diabetes is more prevalent among people whose mothers experienced malnutrition during the Dutch famine in 1944-453, and a substantive literature suggests that maternal obesity and gestational diabetes mellitus (GDM) are associated with elevated metabolic syndrome markers in childhood and later life4. These observational studies have been strengthened by studies in animal models, which have shown that the maternal nutritional and metabolic status can profoundly influence cardiovascular and metabolic function in young and adult offspring5,6,7.
Intrahepatic cholestasis of pregnancy (ICP) affects 0.5% of pregnancies in UK and up to 2% globally8. Its etiology is complex, with genetic and endocrine components. A subgroup of affected women possess genetic variants in biliary transporters (ABCB4/MDR3 and ABCB11/BSEP) and nuclear receptors (NR1H4/FXR) that perturb bile acid (BA) homeostasis9,10. The phenotype of ICP is unmasked by pathologically elevated levels of reproductive hormone metabolites in genetically predisposed women11,12,13,14. Women with ICP present with pruritus, hepatobiliary injury and hypercholanemia (elevated serum BA concentrations), as well as dyslipidemia2,3,15. Increased rates of GDM have been reported15,16. Maternal BA concentrations ≥40 μmol/L are associated with adverse fetal outcomes, including spontaneous preterm labor, fetal hypoxia and meconium-stained amniotic fluid17,18,19. Fetal BA levels are raised in umbilical cord blood in ICP pregnancies20.
Exposure of the fetus to elevated BA in utero as a result of ICP predisposes the adolescent offspring to metabolic disease. Analysis of the Northern Finland Birth cohort database (1986) revealed that 16-year old children from cholestatic pregnancies had increased body mass index and dyslipidemia, and the males had raised fasting insulin compared to age matched adolescents of uncomplicated pregnancies21. In a mouse model of 0.5% cholic acid (CA) feeding that replicates the hypercholanemia of cholestatic pregnancy, female offspring exposed to increased BAs in utero developed hepatic steatosis, glucose intolerance and insulin resistance when challenged with a Western diet (WD) for six weeks. Human samples and in vivo experiments showed that increased transplacental cholesterol transport and greater fat storage in the fetoplacental unit contribute to this phenotype21. Furthermore, maternal hypercholanemia resulted in altered DNA methylation patterns in the agouti Avy yellow mouse offspring, indicating epigenetic alterations in the ICP-associated offspring phenotype21.
Administration of ursodeoxycholic acid (UDCA) is a treatment for ICP that reduces BA levels and improves symptoms and biochemical abnormalities in some mothers. However, it is not known whether UDCA improves fetal abnormalities and susceptibility of the offspring to metabolic disease later in life22,23.
We hypothesized that treatment with UDCA in cholestatic pregnancy can reverse the fetal metabolic phenotype, as well as placental dyslipidemia. This may also prevent features of metabolic syndrome in the adult offspring. To address our hypothesis, we first compared the fetal and maternal biochemical profile in UDCA-treated ICP pregnancies, untreated ICP and uncomplicated pregnancies. Based on these findings, we then administered UDCA in a mouse model of maternal hypercholanemia to mimic UDCA treatment in ICP21 and we investigated the impact on the metabolic phenotype of the adult offspring. Based on our previous findings in the agouti Avy yellow mouse models and the emerging evidence that epigenetic processes in early life stages are key factors that influence metabolism24,25, we also explored modifications in the methylation patterns of umbilical cord leukocytes and placentas from human ICP using an epigenome-wide association study (EWAS) and global methylation studies.
Methods
Human samples
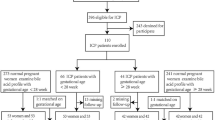
Women with untreated ICP (n = 16), UDCA-treated ICP (n = 17) and controls (n = 18) were recruited from Queen Charlotte’s and Chelsea and St Thomas’ Hospitals, London. ICP diagnosis was based on presentation with gestational pruritus and serum BA over 10 µmol/L and no additional identifiable cause of liver dysfunction. Exclusion criteria included other causes of hepatic dysfunction (including pre-eclampsia, hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome, acute fatty liver of pregnancy, primary biliary cholangitis, active viral hepatitis, and any ultrasound abnormality that may result in biliary obstruction) and multi-fetal pregnancy. Controls were women with uncomplicated pregnancies. The decision to treat with UDCA was made by the individual specialist clinician looking after each woman with ICP. Maternal serum and umbilical cord blood were collected and prepared as previously described20.
Animal handling and maintenance
Age-matched (6–8-week-old) female and male C57BL/6 inbred mice were purchased from Harlan Laboratories (Derbyshire, UK) and maintained in a 24-hour light-dark cycle (12 h/12 h) with free access to normal chow diet (RM3; SDS, Essex, UK) and water. Mice were allowed to acclimatize for one week before any experiment. For all studies, the second litters were used as previously described21. A week before mating for the second litter, females were randomly assigned to one of the three dietary groups: RM3 control diet (normal chow-NC; n = 6), RM3 diet supplemented with 0.5% CA (CA diet; n = 6) or RM3 diet supplemented with 0.5% CA plus 0.5% UDCA (CA + UDCA diet; n = 4). All diets were purchased from LBS Serving Biotechnology Ltd, Horley, UK. To address the effects of UDCA in gestational cholestasis, pregnant mothers were sacrificed on day 18 of pregnancy after a 4-hour fast. Maternal and fetal liver and blood as well as placentas were collected. Maternal body weight, liver weight, litter number, fetal weight and placental weight were monitored.
To investigate the effects of UDCA on long-term health of the offspring, a different set of mice were placed on an identical maternal dietary regime and were allowed to give birth to offspring (n = 4–6 mothers per dietary regimen). Litters were reduced to four mice per mother on post-natal day 2; 2 females and 2 males from each mother were kept when possible. Only female offspring were analyzed in this study, as it was previously shown that females from this dietary regime develop a more extreme phenotype21. At 12 weeks of age, one female mouse from each litter was challenged with a western diet for six weeks (LBS Serving Biotechnology Ltd), whereas female littermates continued on a NC diet(n = 4–6 per dietary regimen from each different diet mothers). A glucose tolerance test was performed two days before sacrifice of the animals at 18 weeks of life as previously described (intraperitoneal injection, 2 g/kg)21.
Biochemical measurements
Serum and tissue biochemical parameters (total, LDL- and HDL-cholesterol, triglycerides (TG) and FFA) were measured using an LX20 autoanalyzer (Beckman Coulter) as previously described21. BA measurements were performed by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) with the use of internal standards for extraction and quantification as previously reported11. The same techniques were used for both human and animal studies.
Epigenome wide association studies(EWAS)
Venous umbilical cord samples were collected from pregnancies affected by ICP (n = 43; 22 of which received treatment with UDCA) and uncomplicated pregnancies (n = 17). Samples were collected in plain vacutainers and centrifuged at 3500 rpm for 10 minutes. The serum was stored at −80 °C until use. EDTA blood collection tubes were used to store whole blood at −80 °C. DNA was extracted using the Qiagen QIAamp Blood mini kit, according to the manufacturers instructions (Qiagen, Skelton House, Manchester, UK). Following elution of DNA into AE buffer, samples were returned to storage at −80 °C until use. 1 µg of the genomic DNA was treated with Sodium Bisulfite using Zymo EZ DNA Methylation-Gold kit (ZymoResearch, Irvine, California, USA, D5007) and processing of the HumanMethylationEPIC (Illumina, Inc. CA, USA) platform was carried out by the Centre for Molecular Medicine and Therapeutics (CMMT) (http://www.cmmt.ubc.ca).
Raw image array files were processed using the R statistical software (R version 3.5.0). A total of 866,836 probes for 60 samples were analysed with minfi (version 1.26) and probe signals normalised using the stratified quantile normalisation algorithm26. We identified probes that were detected (P > 0.01) in one or more samples (n = 863,110) and were not at the sites of single-nucleotide polymorphisms (SNPs) (minor allele frequency – maf = 0)26 (n = 833,907) or cross-reactive (identified for the EPIC dataset27). After excluding probes that did not meet all these criteria, a total of 792,068 probes, including those on sex chromosomes, were maintained for subsequent analyses.
Statistical analyses for the methylation data were performed using the logit transformed M-values for methylation to avoid bias that would have incurred if beta values were selected28. Differentially methylated CpGs (dmCpGs) were assigned using Limma (version 3.36.1)29 and adjusted for status, gender and ethnicity as confounders as well as treatment, gender and ethnicity in the second model. A 5% Benjamini-Hochberg False Discovery Rate (FDR)30 correction for multiple testing was applied. All dmCpGs were assigned to their corresponding genomic context and location using the R/Bioconductor package IlluminaHumaMethylationEPICanno.ilm10b2.hg19 (version 0.6.0)31.
Top 1000 dmCpGs, with a p value <1.74E-04 and FDR < 13%, were selected from the disease (treated and untreated) vs control probe-wise differential methylation analyses to perform IPA pathway analyses (QIAGEN Inc., version 01–07). The increase in FDR was adjusted to maintain a significant number of CpGs32, including 18 passing FDR at 5%.
Differentially methylated regions were identified using the DMRcate package (version 1.16.0)33. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were performed with R/Bioconductor package missMethyl (version 1.14.0) using the gometh function34 and the MethylationEPIC genes as background. Further pathway analyses were completed with Ingenuity IPA (QIAGEN Inc., version 01–07) utilising the Ingenuity knowledge base reference gene set. An IPA search of individual genes was also run to establish their biological significance.
Global DNA methylation studies
DNA extraction of placentas was carried out with a DNeasy Blood and Tissue Kit (Qiagen) as per supplier’s instructions. DNA was eluted in buffer AE, which guarantees optimal DNA stability, and stored at −20 °C for further use. Quantification of DNA methylation was performed using the Epigentek MethylFlash Methylated DNA Kit (colorimetric assay; Insight Biotechnology Ltd, Wembley, UK), according to manufacturer’s instructions. 200 ng of sample DNA as well as positive and negative controls were included. The light absorbance was quantified in a microplate reader at 450 nm (Pherastar FS, BMG Labtech, Bucks, UK). Final amounts were calculated as percentages based on the controls’ standard curve as instructed by the manufacturer.
Quantitative real-time PCR
1 μg of total RNA (Qiagen, Manchester UK) from mouse tissues and human placentas was processed as previously described21. Primer sequences (Sigma-Aldrich, Poole, UK) are given in Supplementary Table 1.
Statistical analysis
Data are presented as means ± sem unless otherwise stated. Statistical analysis for multiple comparisons was performed by repeated measures of ANOVA and Newman–Keuls post hoc testing with the GraphPad Prism 7.00 software (GraphPad Software Inc., San Diego, CA, USA). The significance cutoff was set at p ≤ 0.05.
Study approval
Written informed consent was received from participants prior to inclusion in the study, which conformed to the 1975 Declaration of Helsinki guidelines. Biochemical studies were approved by the local ethics committee of Hammersmith Hospital NHS Trust (11/L0/0396). Epigenome-wide studies were also approved by the ethics committees of Hammersmith Hospitals NHS Trust, London (97/5197 and 08/H0707/21), and King’s College Hospitals NHS Trust, London (03WH06).
All experimental procedures were approved by the ethical committee for animal welfare at King’s College London, and all animal studies were compliant with the UK Animals (Scientific Procedures) Act of 1986 and the guidelines from the biological sciences unit at King’s College London.
Results
UDCA improves fetal dyslipidemic features associated with ICP
ICP is associated with umbilical cord blood dyslipidemia and metabolic syndrome in 16-year-old children21. Analysis of umbilical cord blood lipid profiles in fetal cord blood from ICP + UDCA treatment showed an increase in cholesterol and FFA in females exposed to ICP, whereas FFA and TG were increased in exposed males. These features were reversed by UDCA treatment (Fig. 1).

Serum biochemical profile of umbilical cord blood from uncomplicated pregnancies, untreated ICP and UDCA-treated ICP. n = 18, 16 and 17, respectively. Data are presented as mean and standard error of the mean (SEM) and were analyzed with multiple measures of ANOVA followed by Neuman Keul’s post-hoc testing. (a) p < 0.05 for comparison ICP vs Control in females; (b) p < 0.05 for comparison ICP vs ICP + UDCA; (c) p < 0.05 for comparison ICP vs Control in males; (d) p < 0.05 for comparison ICP vs ICP + UDCA males; ICP: intrahepatic cholestasis of pregnancy; UDCA: ursodeoxycholic acid; FFA: free fatty acids; TG: triglycerides.
UDCA improves elevated maternal BA concentrations associated with CA feeding and induces fetal hepatoprotective mechanisms in mouse hypercholanemic pregnancy
Due to the positive effects of UDCA on ICP-induced fetal dyslipidemia and the limited options for in vivo human fetal studies in ICP, we expanded our studies into a mouse model in which a 0.5% (CA) diet fed to pregnant mice mimicked the hypercholanemia of ICP, as previously described21. We first investigated whether dietary supplementation with UDCA is sufficient to reverse the CA diet-induced increase in maternal and fetal BA concentrations (CA + UDCA group). UDCA prevented the elevation of taurocholic acid (TCA) and taurodeoxycholic acid (TDCA) in maternal serum and liver (Fig. 2A). However, UDCA did not affect the altered mRNA expression of CA diet-induced Fxr targets such as Shp, Cyp7a1 and Bsep (Fig. 2B). Unlike maternal BA levels, UDCA did not significantly reverse the CA diet-induced increase in fetal serum BAs (Fig. 2C). UDCA did not alter fetal hepatic Cyp7a1 and Shp mRNA expression (Fig. 2D).

Effects of UDCA on mouse bile acid homeostasis. (A) UDCA supplementation in 0.5% CA-fed mice reversed increased taurine-conjugated bile acids in the maternal serum and liver. (B) UDCA did not affect maternal hepatic mRNA expression levels of Fxr target genes. (C) UDCA supplementation in CA feeding did not affect taurine-conjugated bile acids in the fetal serum (D) UDCA supplementation in CA feeding did not affect Shp or Cyp7a1 mRNA expression levels in the fetal liver. Data are presented as mean and standard error of the mean (SEM) and were analyzed with multiple measures of ANOVA followed by Neuman Keul’s post-hoc testing. *p < 0.05 between comparisons connected by lines; NC n = 6, C A n = 6, CA + UDCA n = 4. NC, CA and CA + UDCA represent maternal diet.
As UDCA did not alter CA diet-induced serum BAs and hepatic Fxr targets in the fetus, we investigated whether UDCA can induce fetal hepatoprotective mechanisms. To address this, we studied the gene expression profile of BA transporters in the fetal liver affected by hypercholanemia. UDCA increased Mrp2 and Oatp1a1 mRNA, whereas it reversed the Bsep mRNA upregulation caused by gestational hypercholanemia in the fetal liver (Fig. 3). No statistically significant changes were observed in gene expression of fetal hepatic Oatp1b2 and Mrp3 mRNA.

Effects of UDCA on fetal hepatic bile acid transporters. Gene expression profile of the BA transport genes Mrp2, Mrp3, Bsep, Oatp1a1 and Oatp1b2 mRNA in mouse fetal liver. Data are presented as mean and standard error of the mean (sem) and were analyzed with multiple measures of ANOVA followed by Neuman Keul’s post-hoc testing. *p < 0.05 between comparisons connected by lines, NC n = 6, CA n = 6, CA + UDCA n = 4. NC, CA and CA + UDCA represent maternal diet.
We previously showed that maternal hypercholanemia in the mouse increased liver to body weight ratio, as well as placental weight. UDCA reversed the increase in placental weight, but it did not affect the maternal liver to body weight ratio (Supplementary Figure 1).
UDCA improves hypercholanemia-induced maternal and fetal dyslipidemia in the mouse
We previously established that, in line with ICP, mouse hypercholanemic pregnancy results in maternal and fetoplacental dyslipidemia15,21. As shown in Fig. 4A, UDCA prevented the CA diet-induced changes in total, HDL- and LDL-cholesterol in maternal serum. It also reduced hepatic TG and cholesterol levels. In the fetal serum, UDCA reversed the decrease in HDL cholesterol and the increase in TG levels, (Fig. 4B). Also, UDCA supplementation in hypercholanemic pregnancy reversed the increase of cholesterol and TG levels in the placenta (Fig. 4C). However, UDCA did not affect the dyslipidemic phenotype of the fetal liver (Fig. 4B).

Effects of UDCA on lipid profile in mouse hypercholanemic pregnancy. (A) UDCA improved maternal serum cholesterol and LDL-cholesterol as well as hepatic cholesterol increase in gestational cholestasis. (B) UDCA reversed cholestasis induced changes in HDL and TG in the fetal serum, but it did not affect cholesterol and TG raised levels in the fetal liver. (C) UDCA reversed cholesterol and TG elevated levels associated with maternal cholestasis in placenta. Data are presented as mean and standard error of the mean (sem) and were analyzed with multiple measures of ANOVA followed by Neuman Keul’s post-hoc testing. *p < 0.05 between comparisons connected by lines. NC n = 6, CA n = 6, CA + UDCA n = 4. NC, CA and CA + UDCA represent maternal diet. FFA: free fatty acids; HDL: high-density lipoprotein; LDL: low-density lipoprotein; TG: triglyceride.
To further investigate whether maternal and fetal serum lipid profiles are associated with changes in hepatic metabolism, we looked at the transcription of hepatic rate-limiting cholesterol and fatty acid synthesis genes, HMG-CoA reductase (Hmgcr) and fatty acid synthase (Fas) mRNA. No effects were observed in the hepatic Hmgcr mRNA either in the mother or fetus in response to UDCA (Fig. 5A,C). In contrast, UDCA prevented the CA-associated increase of Fas mRNA in the fetal liver (Fig. 5D) but not in the maternal liver (Fig. 5B).

Effects of UDCA on lipid gene expression profile in mouse hypercholanemic pregnancy. (A) Hmgcr mRNA expression in maternal liver. (B) Fas mRNA expression in maternal liver (C) Hmgcr mRNA expression in fetal liver (D) Fas mRNA expression in fetal liver. Data are presented as mean and standard error of the mean (sem) and were analyzed with multiple measures of ANOVA followed by Neuman Keul’s post-hoc testing. NC n = 6, CA n = 6, CA + UDCA n = 4. *p < 0.05 between comparisons connected by lines. NC: normal chow; CA: diet supplemented with cholic acid; CA + UDCA: diet supplemented with cholic acid + ursodeoxycholic acid; Hmcr: HMG-coA reductase; Fas: fatty acid synthase.
UDCA in mouse hypercholanemic pregnancy improves glucose tolerance in female offspring
We previously established that female adult offspring exposed to CA in utero develop glucose intolerance when fed a WD for six weeks21. We therefore tested whether exposure of the offspring to UDCA in pregnancy can ameliorate this phenotype. Although UDCA did not affect the liver to body weight ratio (Fig. 6A), it did significantly improve glucose tolerance in 18-week old female offspring (Fig. 6B). Remarkably, whereas offspring fed a NC diet responded to intraperitoneal glucose injection within 15′ regardless of the maternal dietary regime during pregnancy (Fig. 6B, left panel), offspring fed a WD diet who were exposed to UDCA in utero did not develop glucose intolerance (Fig. 6B, right panel). Intriguingly, 30′ after the intraperitoneal glucose injection, blood glucose levels were significantly decreased in WD-fed females exposed to UDCA in gestation compared to WD-fed females from NC- or CA-fed mothers (Fig. 6B).

Effects of UDCA on liver to body weight ratio and glucose tolerance in offspring of mouse hypercholanemic pregnancy. (A) Effects of UDCA on liver to body weight ratio (B) Effects of UDCA on glucose tolerance in NC-fed and WD-fed female offspring of maternal hypercholanemia. Data are presented as mean and standard error of the mean (sem) and were analyzed with multiple measures of ANOVA followed by Neuman Keul’s post-hoc testing. $p < 0.05 0′ vs 15′, *p < 0.05 15′ CA WD vs 15′ NC/CA + UDCA WD, #p < 0.05 30′ CA WD vs 30′ NC/CA + UDCA WD, NC n = 6, CA n = 6, CA + UDCA n = 4. NC/CA/CA + UDCA represent maternal diet; NC/WD represent offspring diet. LW/BW: liver weight/body weight; NC: normal chow; WD: western diet; GTT: glucose tolerance test.
ICP alters DNA methylation profiles of relevance to offspring phenotype
Eighteen CpGs were differentially methylated (with a 5% Benjamini-Hochberg FDR) when all cases of ICP (treated and untreated, labelled as disease group) were compared to controls (control versus disease comparison, Table 1). Specifically, these dmCpGs were located mainly in the gene body (TEX29, TBX1, MAMDC2, WDFY4, AHNAK, NFIA and WDR70); within the 5′ untranslated region, between the TSS and the ATG start site (H6PD, ILR21, DFNB59) and one was located in the 3′UTR, between the stop codon and poly A signal (PYCR2) (Table 1). An IPA search of the genes associated with the DMCpGs was then performed to analyse their biological significance. H6PD is related to carbohydrate metabolism biological processes, whilst AHNAK is related to impaired glucose tolerance. When analyses were run to include the treatment as a factor in the model, both the control versus untreated and also the treated versus untreated comparisons identified two significantly differentially methylated CpGs, all assigned to the PRR25 gene (located within the CpG island, Table 1), which does not have any known direct relevance to the phenotype.
To further analyse the biological importance of the methylation differences in each comparison, canonical pathways were identified using IPA, GO and KEGG tools through the MissMethyl R package. In the control versus disease analysis, IPA identified 45 pathways that were differentially methylated, including one related to fatty acid oxidation (Fig. 7). Additional pathways were identified with the R gometh function in the MissMethyl package by GO and KEGG (Table 2). IPA analysis, using the Ingenuity knowledge base reference gene set, also highlighted the presence of gene promoters that were differentially methylated between controls and disease (Fig. 8B). Similarly, several disease processes were revealed for the same comparison, including endocrine system disorders, lipid metabolism, carbohydrate metabolism and metabolic disease (Fig. 8A).

Canonical pathways identified by IPA in the control versus disease comparison (treated and untreated ICP). Bars represent –log (p value). Continuous line represents p value threshold equivalent to 0.05. Lines with squares represent the number of molecules in the dataset that are part of the pathway/number of molecules in the pathway (ratio).

Disease processes and differentially methylated molecules identified by IPA in the control versus disease comparison (treated and untreated ICP). (A) Biological processes. Bars represent –log (p value). Continuous line represents p value threshold equivalent to 0.05. (B) Molecules Hypomethylated in disease (C) Molecules hypermethylated in disease.
ICP affects the methylation profile of human placentas
The placenta plays an important role in transferring maternal environmental cues to the fetus, thereby potentially influencing metabolic programming35. Placentas from ICP pregnancies had a significant increase in global methylation when compared to placentas from uncomplicated pregnancies (Fig. 9).

Global DNA methylation patterns in placentas from pregnancies affected by ICP and controls. Data are presented as mean and standard error of the mean (sem). 5-mC%: percentage of 5-methylcytosie; ICP: Intrahepatic cholestasis of pregnancy. *p < 0.05.
Discussion
In this study we demonstrated for the first time that UDCA has a protective role against the development of ICP-associated fetal dyslipidemia. If persistent, these effects may protect offspring from the development of metabolic disease in later life. Using a model of mouse gestational hypercholanemia that was also administered UDCA, we showed a similar effect in the mother and fetus. In addition, UDCA prevented the development of glucose intolerance in the obese offspring of these mice. We also showed for the first time that ICP induces epigenomic alterations in fetal cord blood. Similarly, maternal hypercholanemia altered global methylation patterns in human placentas.
A limited number of studies have assessed effects of UDCA on the fetoplacental unit and whether it may have a beneficial role for the long-term health of the offspring. One previous study revealed raised numbers of syncytial knots in ICP placentas, indicating that elevated BA concentrations in ICP are associated with increased apoptosis36. These observations were consistent with a study in a rat model of gestational hypercholanemia that demonstrated increased apoptosis and oxidative stress in placentas exposed to excessive BA37. Another study reported a UDCA-induced reversal of transient latent cholestasis in 21-day old offspring from a rat model of hypercholanemic pregnancy38. In ICP, UDCA has also been shown to reduce BA levels in umbilical cord blood and amniotic fluid39,40.
While in our mouse model UDCA successfully reversed maternal gestational hypercholanemia, it did not reverse increased BA concentrations in the fetal serum. However, we showed UDCA-associated induction of fetal hepatic BA transporters, and this may reduce fetal BA toxicity. It is likely that the upregulation of Mrp2 and Oatp1a1 in the fetal liver protects the fetus from hepatocellular accumulation of potentially toxic biliary constituents. In support of this, an increase of Mrp2 mRNA has been reported in rat cholestasis41.
Interestingly, we not only showed that UDCA improved fetal and maternal dyslipidemia in our animal model, but also that it had an impact on FFA synthesis pathways in the fetus. Unlike in the maternal liver, fetal hepatic Fas mRNA expression returned to baseline when hypercholanemic mothers were administered UDCA. This finding is of interest as FFA accumulation in the hepatocyte has been implicated in the development of hepatosteatosis33, which is one of the long-term effects of maternal hypercholanemia in the offspring of this model21. Emerging evidence suggests that hepatosteatosis precedes the metabolic syndrome and is associated with insulin resistance42. We previously established in this model that mild hepatosteatosis precedes progression of glucose intolerance that is unmasked when the offspring are fed an obesogenic diet. Herein, we report that WD-fed female offspring exposed to UDCA in utero were protected from impaired glucose tolerance.
This concurs with previous studies in rodents that have successfully improved obesity-induced hepatosteatosis with UDCA administration43,44. A permanent effect of UDCA on the pathogenesis of non-alcoholic fatty liver disease (NAFLD) and associated impairment of metabolic health is therefore plausible, and might be attributable to the restoration of a healthy FFA metabolism.
UDCA reduced hepatic TG in hypercholanemic mothers, as well as in the fetal serum and placenta. It also reversed the hypercholesterolemic phenotype of the mother and placentas, and the CA-induced reduction in HDL levels in the fetal serum. Curiously, these results were not accompanied by a reduction in mRNA expression of maternal and fetal hepatic cholesterol de novo biosynthesis gene Hmgcr, suggesting that UDCA mediates these effects via alternative mechanisms.
DNA methylation is a typical chromatin modification that can occur when the mother is exposed to an environmental stimulus. We previously established that female offspring of the Avy mouse model, when born to mothers fed a CA diet, have reduced methylation of the IAP promoter21, suggesting that maternal hypercholanemia can trigger epigenetic changes in the offspring. By performing an EWAS in fetal umbilical cord blood, we found that ICP also induces methylation changes in pathways that could contribute to the offspring phenotype. When ICP cases (both treated and untreated) were compared with controls (uncomplicated pregnancies), a total of eighteen differentially methylated CpGs were found. IPA pathway analysis showed that these methylated CpGs are involved in fatty acid oxidation pathways, as well as pathways potentially related to glucose metabolism such as glucose-6-phosphate dehydrogenase activity and glucose 1-dehydrogenase [NAD(P)] activity. Interestingly, such pathways are involved in diseases related to endocrine system, cardiovascular disease, lipid metabolism, carbohydrate metabolism, and metabolic disease, all of which are consistent with our previous findings.
The control versus untreated and the treated versus untreated comparisons yielded limited results and did not identify any loci directly relevant to the phenotype studied. The relatively small number of samples precluded detailed investigation of whether beneficial effects of UDCA stem from epigenetic changes or not. Another limitation of the analysis is that the samples from UDCA-treated and untreated ICP cases were not matched for disease severity as they would be in a randomised, placebo-controlled trial.
While the use of surrogate tissues for human EWAS offers valuable insights into the epigenome, it also presents a challenge for interpretation due to possible tissue-specific epigenetic alterations. However, alterations in DNA methylation in the placenta have been previously reported36, suggesting that placental epigenetic marks may be of relevance to metabolic homeostasis in later life.
To gain further insight into the epigenetic component of hypercholenemic pregnancy, we also performed a global DNA methylation analysis of human placentas. As suggested by the EWAS results, ICP altered the human placental epigenome.
Conclusion
UDCA treatment improves ICP-associated fetal dyslipidaemia. This finding is consistent in mouse hypercholanemic pregnancy. In addition, maternal UDCA administration improved the lipid profile of mothers and placentas and triggered hepatoprotective mechanisms in the fetal liver in mouse gestational hypercholanemia. In adult offspring, UDCA prevented the development of glucose intolerance. Our data support an epigenetic component to the impact of ICP upon offspring metabolic phenotypes. Larger sample sizes will enable more detailed assessment of the effects of UDCA on the epigenome, and investigation of sex-specific alterations. Human cohort studies will be necessary to investigate whether such UDCA effects can persist through adulthood. These data indicate that UDCA may be used as an effective intervention in ICP to prevent susceptibility of the adult offspring to metabolic disease.
References
Gluckman, P. D. et al. Towards a new developmental synthesis: adaptive developmental plasticity and human disease. Lancet. 373, 1654–1657 (2009).
van Abeelen, A. F. M. et al. Famine exposure in the young and the risk of type 2 diabetes in adulthood. Diabetes. 61, 2255–60 (2012).
Poston., L., Harthoorn, L. F. & van der Beek, E. M. Obesity in Pregnancy: Implications for the Mother and Lifelong Health of the Child. A Consensus Statement. Pediatr. Res. 69, 175–180 (2011).
Burns, S. P. et al. Gluconeogenesis, glucose handling, and structural changes in livers of the adult offspring of rats partially deprived of protein during pregnancy and lactation. J. Clin. Invest. 100, 1768–74 (1997).
Mouralidarane, A. et al. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology. 58, 128–138 (2013).
Samuelsson, A. M. et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertens. 51, 383–92 (2008).
Liang, X. et al. Maternal high-fat diet during lactation impairs thermogenic function of brown adipose tissue in offspring mice. Sci. Rep. 6, 343–45 (2016).
Pataia, V., Dixon, P. H. & Williamson, C. Pregnancy and bile acid disorders. Am. J. Physiol. Liver Physiol. 313, G1–G6 (2017).
Dixon, P. H. et al. A Comprehensive Analysis of Common Genetic Variation Around Six Candidate Loci for Intrahepatic Cholestasis of Pregnancy. Am. J. Gastroenterol. 109, 76–84 (2014).
Dixon, P. H. et al. An expanded role for heterozygous mutations of ABCB4, ABCB11, ATP8B1, ABCC2 and TJP2 in intrahepatic cholestasis of pregnancy. Sci. Rep. 7, 1–8 (2017).
Abu-Hayyeh, S. et al. Prognostic and mechanistic potential of progesterone sulfates in intrahepatic cholestasis of pregnancy and pruritus gravidarum. Hepatology. 63, 1287–1298 (2016).
Abu-Hayyeh, S. et al. Intrahepatic cholestasis of pregnancy levels of sulfated progesterone metabolites inhibit farnesoid X receptor resulting in a cholestatic phenotype. Hepatology. 57, 716–726 (2013).
Chen, Y. et al. Estrogen and Estrogen Receptor-α-Mediated Transrepression of Bile Salt Export Pump. Mol. Endocrinol. 29, 613–626 (2015).
Song, X. et al. Transcriptional dynamics of bile salt export pump during pregnancy: mechanisms and implications in intrahepatic cholestasis of pregnancy. Hepatology. 60, 1993–2007 (2014).
Martineau, M. G. et al. The metabolic profile of intrahepatic cholestasis of pregnancy is associated with impaired glucose tolerance, dyslipidemia, and increased fetal growth. Diabetes Care. 38, 243–8 (2015).
Martineau, M., Raker, C., Powrie, R. & Williamson, C. Intrahepatic cholestasis of pregnancy is associated with an increased risk of gestational diabetes. Eur. J. Obstet. Gynecol. Reprod. Biol. 176, 80–85 (2014).
Glantz, A., Marschall, H. U. & Mattsson, L. Å. Intrahepatic cholestasis of pregnancy: Relationships between bile acid levels and fetal complication rates. Hepatology. 40, 467–474 (2004).
Geenes., V. et al. Association of severe intrahepatic cholestasis of pregnancy with adverse pregnancy outcomes: A prospective population-based case-control study. Hepatology. 59, 1482–1491 (2014).
Ovadia, C. et al. Association of adverse perinatal outcomes of intrahepatic cholestasis of pregnancy with biochemical markers: results of aggregate and individual patient data meta-analyses. Lancet. 393, 899–909 (2019).
Geenes, V. et al. The reversed feto-maternal bile acid gradient in intrahepatic cholestasis of pregnancy is corrected by ursodeoxycholic acid. PLoS One. 9, e83828, https://doi.org/10.1371/journal.pone.0083828 (2014).
Papacleovoulou, G. et al. Maternal cholestasis during pregnancy programs metabolic disease in offspring. J. Clin. Invest. 123, 3172–3181 (2013).
Bacq, Y. et al. Efficacy of Ursodeoxycholic Acid in Treating Intrahepatic Cholestasis of Pregnancy: A Meta-analysis. Gastroenterology. 143, 1492–1501 (2012).
Chappell, L. C. et al. Ursodeoxycholic acid versus placebo, and early term delivery versus expectant management, in women with intrahepatic cholestasis of pregnancy: semifactorial randomised clinical trial. BMJ. 344, e3799, https://doi.org/10.1136/bmj.e3799 (2012).
Burdge, G. C., Slater-jefferies, J., Torrens, C. & Phillips, E. S. Dietary protein restriction of pregnant rats in the F 0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F 1 and F 2 generations. Br. J. Nutr. 97, 435–439 (2007).
Lin, X. et al. Developmental pathways to adiposity begin before birth and are influenced by genotype, prenatal environment and epigenome. BMC Med. 15, 1–18 (2017).
Aryee, M. J. et al. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 30, 1363–1369 (2014).
McCartney, D. L. et al. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genomics Data. 9, 22–24 (2016).
Du, P. et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 11, 587 (2010).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47, https://doi.org/10.1093/nar/gkv007 (2015).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple. Testing. J. R. Stat. Soc. Ser. B. 57, 289–300 (1995).
Hansen, K. D. IlluminaHumanMethylationEPICanno.ilm10b2.hg19: Annotation for Illumina’s EPIC methylation arrays. https://doi.org/10.18129/B9.bioc.IlluminaHumanMethylationEPICanno.ilm10b2.hg19 (2016)
Zhang, X. et al. DNA methylation signatures of illicit drug injection and hepatitis C are associated with HIV frailty. Nat. Commun. 8, 2243, https://doi.org/10.1038/s41467-017-02326-1 (2017).
Peters, T. J. et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 8, 6, https://doi.org/10.1186/1756-8935-8-6 (2015).
Phipson, B., Maksimovic, J. & Oshlack, A. missMethyl: an R package for analyzing data from Illumina’s HumanMethylation450 platform. Bioinformatics. 32, 560 (2015).
Godfrey, K. M., Costello, P. M. & Lillycrop, K. A. The developmental environment, epigenetic biomarkers and long-term health. J. Dev. Orig. Health Dis. 6, 399–406 (2015).
Geenes, V. L. et al. A placental phenotype for intrahepatic cholestasis of pregnancy. Placenta. 32, 1026–1032 (2011).
Perez, M. J., Macias, R. I. R. & Marin, J. J. G. Maternal cholestasis induces placental oxidative stress and apoptosis. Protective effect of ursodeoxycholic acid. Placenta. 27, 34–41 (2006).
Macias, R. I. et al. Long-term effect of treating pregnant rats with ursodeoxycholic acid on the congenital impairment of bile secretion induced in the pups by maternal cholestasis. J Pharmacol Exp Ther. 312, 751–758 (2005).
Brites., D. Intrahepatic cholestasis of pregnancy: Changes in maternal-fetal bile acid balance and improvement by ursodeoxycholic acid. Ann. Hepatol. 1, 20–28 (2002).
Mazzella, G. et al. Ursodeoxycholic acid administration in patients with cholestasis of pregnancy: effects on primary bile acids in babies and mothers. Hepatology. 33, 504–508 (2001).
Fickert, P. et al. Effects of Ursodeoxycholic and Cholic Acid Feeding on Hepatocellular Transporter Expression in Mouse Liver. Gastroenterology. 121, 170–183 (2001).
Lonardo, A., Ballestri, S., Marchesini, G., Angulo, P. & Loria, P. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. 47, 181–190 (2015).
Pathil, A., Mueller, J., Warth, A., Chamulitrat, W. & Stremmel, W. Ursodeoxycholyl lysophosphatidylethanolamide improves steatosis and inflammation in murine models of nonalcoholic fatty liver disease. Hepatology. 55, 1369–1378 (2012).
Nie, B. et al. Specific bile acids inhibit hepatic fatty acid uptake in mice. Hepatology. 56, 1300–1310 (2012).
Acknowledgements
We would like to acknowledge the support of all members of the Maternal and Fetal Disease Group, School of Life Course Sciences, King’s College London. The study was funded by the Wellcome Trust (Grant P30874), ICP Support, Genesis Research Trust and the National Institute for Health Research Biomedical Research Centers at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London and Imperial College Healthcare NHS Trust. KMG is supported by the UK Medical Research Council (MC_UU_12011/4), the National Institute for Health Research (NIHR Senior Investigator (NF-SI-0515-10042) and the NIHR Southampton Biomedical Research Centre) and the European Union (Erasmus+ Programme Early Nutrition eAcademy Southeast Asia-573651-EPP-1-2016-1-DE-EPPKA2-CBHE-JP). The views expressed are those of the authors and not necessarily those of the NHS, NIHR or the Department of Health. No conflicts of interest declared.
Author information
Authors and Affiliations
Contributions
G.P., L.B.M., F.F., V.P., A.A.Q., S.A.H., S.M., E.J., A.W. conducted scientific experiments. J.C. and M.V.-L. identified cases and contributed to phenotyping. G.P., K.L., E.J., N.K., H.U.M., K.L., K.M.G. and P.D. analyzed data. G.P. and C.W. conceived and designed the study. All authors contributed to the final draft of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Borges Manna, L., Papacleovoulou, G., Flaviani, F. et al. Ursodeoxycholic acid improves feto-placental and offspring metabolic outcomes in hypercholanemic pregnancy. Sci Rep 10, 10361 (2020). https://doi.org/10.1038/s41598-020-67301-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67301-1
This article is cited by
-
Perinatal exposure to UDCA prevents neonatal cholestasis in Cyp2c70-/- mice with human-like bile acids
Pediatric Research (2023)
-
A multi-centered trial investigating gestational treatment with ursodeoxycholic acid compared to metformin to reduce effects of diabetes mellitus (GUARD): a randomized controlled trial protocol
Trials (2022)
-
GWAS meta-analysis of intrahepatic cholestasis of pregnancy implicates multiple hepatic genes and regulatory elements
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
