
Abstract
Cactoblastis cactorum, a species of moth native to Argentina, feeds on several prickly pear cactus species (Opuntia) and has been successfully used as a biological control of invading Opuntia species in Australia, South Africa and native ruderal Opuntia species in some Caribbean islands. Since its introduction to the Caribbean its spread was uncontrolled, invading successfully Florida, Texas and Louisiana. Despite this long history of invasion, we are still far from understanding the factors determining the patterns of invasion of Cactoblastis in North America. Here, we explored three non-mutually exclusive explanations: a) a stepping stone model of colonization, b) long distance colonization due to hurricanes, and/or c) hitchhiking through previously reported commercial routes. Genetic diversity, genetic structure and the patterns of migration among populations were obtained by analyzing 10 nuclear microsatellite loci. Results revealed the presence of genetic structure among populations of C. cactorum in the invaded region and suggest that both marine commercial trade between the Caribbean islands and continental USA, as well as recurrent transport by hurricanes, explain the observed patterns of colonization. Provided that sanitary regulations avoiding human-mediated dispersal are enforced, hurricanes probably represent the most important agent of dispersal and future invasion to continental areas.
Similar content being viewed by others

Introduction
During the last decades, biological invasion studies have strongly benefited from the use of neutral molecular markers to disentangle routes of invasion1,2,3. This has helped to identify source populations, frequency of invasion events, and the geographical patterns and demographic consequences of invasive species spread4,5. Reconstruction of past events of invasion can help in the recognition of the mechanisms of dispersal to prevent the prevalence of invasion or further spread to additional areas1,6,7,8. However, understanding local dispersal remains a central challenge to prevent and control the economic and biodiversity costs of biological invasions.
Initially, molecular markers were used to identify sources and frequency of dispersal events of invasive species to non-native regions1,2,3,9. Whereas in some cases, these patterns explain the lower genetic variation of invasive species relative to native areas1,10,11,12, combination of migration events from different source populations have sometimes increased genetic variation within invaded regions, potentially increasing the risk of fast adaptation to novel conditions13,14,15,16. Once invasive species are established within non-native areas, understanding further local dispersal is a major challenge to identify potential environmental barriers and recommend control management programs7,17. It is possible to use highly variable molecular markers for understanding local scale dispersal and the entangled nature of the species invasion phenomena.
The cactus moth, Cactoblastis cactorum, is an oligophagous herbivore during its larval stage that consumes the inner tissue of cladodes of plants within the genus Opuntia, negatively affecting the plant survival18. This moth, native to South America, was used in 1924 for biological control purpose against exotic Opuntia species with successful results in Australia. Later, was intentionally introduced to South Africa (1933), New Caledonia (1933) and Hawaii (1950). In 1957 moth larvae, from South Africa and Australia, were introduced in the island of Nevis, seeking to control the exponential growth of native Opuntia populations caused by deforestation and cattle ranging19,20. According to a recent study, this has been the single introduction event of C. cactorum to the Caribbean area21. Since its introduction in Nevis and latter in Saint Kitts in 1957 and until 1987-1989, the moth was detected in other areas as far as Florida22, increasing the risk of extinction of native species23. The entire Florida peninsula is now invaded and the moth has spread through the Gulf of Mexico coasts20. According to the Texas Invasive Species Institute the moth has been detected in Brasoria County (Texas, US) in 2018 (around 800 km from the Northern Mexican border). Following the Atlantic coast of North America, it has dispersed up to Charleston County in South Carolina24,25,26. The rate of dispersal of the moth according to females flight distances was 3-6 km and 16-24 km in 2.5 years in South Africa and Australia respectively27,28. Hence the rapid spread throughout the Caribbean and North America probably involved additional factors. Previous reports suggest that spread among Hawaiian Islands probably occurred through island-hopping29 and tropical storms30. In turn, human-mediated dispersal and climatic events like hurricanes have been proposed as sources of dispersal in the Caribbean21,30,31,32. The threat for North American deserts is that they have the highest diversity of Opuntia cacti species, which may suffer from this oligophagous moth18. Furthermore, domesticated Opuntia in Mexico have a high cultural and economic value as a food resource33. Thus, given the risk of further spread to continental areas and the ecological, social and economic associated costs, the present study examined possible routes of local dispersal in the Caribbean and Florida to better understand the factors that may favor additional introductions of this invasive species to the continent.
A previous study using a mitochondrial gene (COI) reported that commercial transportation of ornamental cacti from Dominican Republic and Puerto Rico was the most likely route of invasion of C. cactorum to Florida31. However, a more recent study with the same marker and a more extended population survey suggested that hurricanes may have also contributed to the dispersal of the moth within the region32. Whereas commercial transportation of ornamental cacti has been controlled ever since the detection of this invasive species18, further introductions within the continent may occur via climatic events. Accordingly, the goal of this study was to further advance in the comprehension of the patterns of dispersal of C. cactorum by using nuclear microsatellites and computational Bayesian approaches. This approach will allow us to add a new piece of evidence to determine whether dispersal through trade, climatic events (hurricanes) or both, better account for the current genetic structure of C. cactorum in North America.
Material and Methods
Sample collection
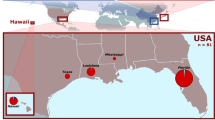
From 2011 to 2012, a total of 229 larvae of the cactus moth, C. cactorum, were collected from 10 invasive sites within the Caribbean region and Florida (USA) (N = 185) (Fig. 1), and two native ones in Argentina (N = 44) from where the moth was initially collected and transported for biological control of ruderal Opuntia species in Australia in 1924 (Table 1). Within each collecting site, only one larva was selected per individual host of Opuntia spp. to avoid over-representation of genotypes belonging to the same genetic family. All samples were stored in 95% ethanol until DNA extraction.

Geographic location of the invading Caribbean populations of Cactoblastis cactorum. The location is on the conductance map for the Caribbean and Atlantic sea obtained with the CIRCUITSCAPE program based on hurricane incidence values, the darkest areas show those with the highest probability of flow. This conductance map was built using the isopletes shown at Lugo et al.48 and available in NOAA (https://www.nhc.noaa.gov/).
DNA extraction and microsatellite analyses
We performed total DNA extraction with DNEasy blood and tissue kits (Qiagen, MD, USA) following the manufacturer protocol. Population genetic variation was determined using nuclear microsatellites developed by Genetic Marker Services (http://www.geneticmarkerservices.com). We removed loci that did not amplify among the 29 primers pairs tested. We chose 14 potential polymorphic loci (Table 2) and labeled with fluorophores (Applied Biosystems) for fragment analysis. Each multiplex PCR mixture (5 µL) contained 2.5 µL RadyTaq (Qiagen cat. 206143) and 1 µL DNA template (20 ng) 5pmol for each fluorescent labeled primers. PCR were performed through touchdown reaction starting with initial heat activation at 95 °C for 10 min followed by 6 denaturation cycles of 94 °C for 1 min, annealing for 1 min and 1 min of extension at 72 °C; annealing cycle temperature began at 60 °C or 57 °C and decreased 1 °C every cycle. The PCR reaction ended with two stages of 12 cycles each (57° and 56°, respectively) and final elongation of 72 °C for 5 min. The PCR product was diluted and run on an ABI 3730xl automated capillary sequencer. The allele size was manually scored using a Liz 600 size standard (Applied Biosystems) in GeneMarker (V2.2.0) (Soft Genetics LLC, State College, Pennsylvania, USA).
Basic population estimators of genetic variation
Genetic variation within sampled populations was characterized using the mean number of alleles per locus (NA), the allelic richness (AR), the mean expected- (He)34 and observed heterozygositiy (Ho), the Fixation Index (Fis) and estimation of FST35 between each pair of sampling site using FSTAT 2.9.3.36. Mean allelic richness (AR) was calculated using the rarefaction method of Leberg37. Exact test for deviation from Hardy-Weinberg equilibrium were performed with GENEPOP38. We used FreeNA to determine the frequency of null alleles using EM algorithm39. Pairwise population differentiation was tested using only those loci that were in H-W equilibrium in more than 50% of the populations.
Genetic clustering of populations was examined using STRUCTURE40 (v 2.3.3). We chose the admixture model with correlated allele frequencies. Two analyses were performed, one including all native and invaded population samples, and one including only populations from the invaded area. The first analysis examined whether genetic differentiation has occurred after the human-mediated journey of C. cactorum from its native Argentina to the Caribbean (1957). The second analysis was performed to explore genetic structuring within the invaded Caribbean only. Because groups of larvae were collected from discrete sites, sampling locations were used as prior information41. Each run consisted of a burn-in period of 105 Markov Chain Monte Carlo (MCMC) iterations, followed by 106 iterations. 20 replicated runs were carried out for each value of the potential number of clusters (K) set, between 1 and 9. STRUCTURE HARVESTER42 (available at http://taylor0.biology.ucla.edu/structureHarvester/) was used to collating output results from STRUCTURE and to determine the uppermost level of structure using the method of Evanno et al.43. Because STRUCTURE assumes the absence of null alleles and H-W equilibrium for all loci, analyses were performed using 10 out of 14 microsatellites that fulfilled the required assumptions.
Isolation genetic and connectivity in the invaded region
The hypothesis that genetic differentiation between a pair of populations is a linear function of the geographic distance between them was examined through a Mantel test that correlates genetic and geographic distance matrices. Paired genetic distances were estimated as [FST/ (1-FST)] and geographic distances were estimated as the logarithm of the Euclidean distances (blog) using GenAlEx44 (v.6.4). Isolation was also tested considering that hurricanes may also shaped the genetic structure of populations in this area. To explore the relationship between genetic distances and the frequency of hurricanes incidence across pairs of populations, a resistance matrix was constructed using the CIRCUITSCAPE program45 (Fig. 1). Based on a previous subdivision of the geographic area using a grid with the probability of incidence of hurricanes (categories 1 to 5) following NOAA46,47,48. Pairs of populations connected by cells with high incidence of hurricanes were assigned a high conductance value (i.e., low resistance to migration). This rationale was applied to all pairs of populations to construct a resistance matrix (indicated by light areas in Fig. 1). To correlate the resistance matrix against the genetic distance matrix controlling for the geographic distance among populations, a partial Mantel test with 10,000 permutations was implemented.
Approximate Bayesian Computational analysis
To provide a quantitative evaluation of the dispersal routes of the cactus moth into Florida (USA), an Approximate Bayesian Computational Analysis was performed with DIYABC49. Analyses were performed with ten microsatellites that were at Hardy-Weinberg equilibrium. This approach uses genetic information provided by microsatellites data and historical data given by dates of first observation within invaded regions. Three hypothetical invasion scenarios were examined (Fig. 2), and parameter values were drawn from prior distributions (Table 3). In all three cases we used the native Argentinian population of Ayuí as the initial source of invasion. This population was genetically close in average with the invasive populations (Paired FST = 0.225). Since moths were taken from Argentina to Australia, and then to South Africa, the analysis considered a “ghost” population representing a non-sampled population that may represent this step. Given the location and date of introduction to the Caribbean, the sampled population of Antigua was considered the source population that initiated the invasion in the region. Antigua was preferred over Nevis because of the larger number of individual larvae in our collection, the high genetic similarity between both islands (Paired FST = 0.012, N.S.) and the short difference in time of introduction between them (1 year). The following step in the invasion process was characterized by the population La Romana (Dominican Republic), given that the moth was detected in the island of Desecheo, located between Puerto Rico and Dominican Republic in 1963. After this step, hypotheses differ in how the cactus moth reached Florida. In the first hypothesis, Cuba, represented by the sampled population of Santiago, is the source of the Floridian invasive population (Fig. 2, Scenario 1). Due to US embargo to Cuba, any dispersal of the moth from Cuba to Florida is more likely related to environmental agents (such as hurricanes) rather than commercial transportation. The second hypothesis considers that moths belonging to La Romana (Dominican Republic) directly invaded Florida (Highlands) (Fig. 2, Scenario 2). This route constitutes the recorded trajectory of commercial traffic to Florida of ornamental cacti infested with C. cactorum from Dominican Republic and Puerto Rico detected in 198950. However, we cannot rule out the possibility that hurricanes also contributed to dispersal among these areas. The third hypothesis considers that the population of Highlands (Florida) was the result of the admixture of genotypes belonging to Cuba and Dominican Republic (Fig. 2, Scenario3).

Scheme of the three competing scenarios evaluated with the ABC model. The prior distribution values of parameters are described in Table 3 (0 = year 2012, assuming two generations per year back in time). The scenarios was built and evaluated with DIYABC (V. 2). The edition was made in the Inskape program.
Given that C. cactorum usually has two generations per year27,28,51, this generation time was used to scale coalescent time in all scenarios. Dates of introduction to Australia (Tg) and Antigua (T2) were given as fixed values based on historical data of the intentional introduction events18. In the other areas (T1 (Argentina), T3-5 (Dominican Republic, Cuba and USA); Table 3), a minimum and maximum value was assigned to each time parameter, with the dates of first observation of the insect as the lower boundary (Table 3). We simulated three million microsatellite data sets (one million for each scenario). Subsequently, the probability for each scenario was inferred by polychotomous logistic regression on the 1% of the simulated data sets closest to the observed data set49. The selected scenario was chosen as that with the highest posterior probability value. To evaluate the robustness of the analysis, we used pseudo-observed simulated data sets to quantify the type I error rate (risk to exclude the focal scenario when it is the true one) and the type II error rate (risk to select the focal scenario when it is false)49. All simulations and ABC analyses were carried out in DIYABC (v.2) sofware52.
Results
No evidence of linkage disequilibrium was detected for all 14 microsatellite loci. Four loci were excluded from the analysis because they had more than 20% of null alleles and were not at Hardy-Weinberg equilibrium (i.e., cc7b, cc16, cc65, cc6b). With the remaining ten microsatellites loci we found that the native population from Yuquerí (Argentina) had on average the highest number of alleles (3.8), while the lowest average number of alleles (1.6) was observed in one of the most recent invasive population (Lee Co., USA) (Table 1). Allelic richness (AR) was higher in native range (2.016–2.185) than invaded populations (1.453–1.992; Table 1). A deficit of heterozygotes was detected in one of the native populations (Ayuí), and in three of the Caribbean invaded populations (Antigua, Nevis and Santiago; Table 1). On average, lower levels of heterozygosity were recorded for invaded than native populations (Table 1).
Pairwise comparisons indicated significant differentiation between the native Argentinian populations and all invaded populations in the Caribbean and Florida (mean FST = 0.229, range = 0.128–0.49). Within the invaded region, genetic differentiation ranged from almost no differences to rather high levels of genetic differentiation (Table 4). The average level of differentiation within the invaded region was FST = 0.185. In general, early invaded populations (Nevis and Antigua) presented lower levels of differentiation with the rest of the populations in the region than recently invaded populations. The Jamaican population (Palisadores) presented the greatest level of genetic differentiation within the Caribbean (mean FST = 0.310; Table 4), whereas Florida populations had a low level of differentiation with population La Romana (Dominican Republic) and low to moderate differentiation with two of the closest Cuban populations (Pinar del Río and Trinidad; Table 4).
Clustering genetic analyses using STRUCTURE on the whole dataset revealed the presence of two main groups (K = 2, with the delta K method) distinguishing native from all invasive populations (Fig. 3B), and tend to confirm a single introduction event. When analyzing only the group of invasive populations, K = 2 is also selected (Fig. 3A). While individuals from Puerto Rico, Palisadores and Pinar del Rio belong to a well-defined cluster, the rest of the populations showed an admixed pattern. Analyses of isolation by distance and connectivity including only the invading populations indicated a low but almost significant level of isolation by distance (r = 0.165; P = 0.05). On the other hand, the isolation by resistance hypothesis based on hurricane and wind currents was significantly supported (r = 0.435; P = 0.008).

Ancestry estimation based on the Bayesian clustering method STRUCTURE in the Cactoblastis cactorum samples. (A) Genetic clustering of the 10 introduced populations, assuming two population clusters (K = 2). (B) Genetic clustering of the 10 introduced populations and the 2 native populations, assuming two population clusters (K = 2). Note: each vertical line corresponds to an individual and the shades of gray represent the probability of belonging to a group. Individuals are grouped by population sample.
Results from Approximate Bayesian Analyses (ABC) support quiet well the hypothesis described in the third scenario (Fig. 2), due to (1) a higher consistency between simulated and sampled data (i.e. the highest posterior probability; P = 0.7798), (2) non-overlapping confidence intervals, and (3) low Type I and mean Type II errors (0.038 and 0.000; respectively). This dispersal scenario considers that the invasion to Florida (represented by the Highlands population) was funded by insects belonging to at least two sources of dispersal, one from Dominican Republic and the other from Cuba (represented in the analysis by the Santiago population; Fig. 2). The posterior probability estimation of parameters of model three assigned a median admixture rate of R = 0.4.
Discussion
During the 1981–1991 decade, North American customs authorities intercepted Opuntia plants infested with C. cactorum. Seizures occurred in plant shipments sent from the Dominican Republic to Miami and in luggage to Dallas Texas International Airport53. Commercial trade of ornamental cacti from the Dominican Republic to the United States of America coupled with the constant influx of American tourists to the Caribbean islands lead to the proposal that human-mediated dispersal has been the major agent of migration to the continent. Similarly, the comparison of the mitochondrial haplotypes from eastern populations of C. cactorum of Florida and those of Dominican Republic supports this idea21,31,32. Despite other potential mechanisms of dispersal as island-hopping and tropical storms or hurricanes30, the present study support the genetic similarity and likelihood of migration between La Romana (Dominican Republic) and Highlands (Florida) populations. However, the distribution of genetic variation and ABC analyses of invasion routes within the Caribbean region also suggest the presence of other nonexclusive routes of dispersal.
In the Caribbean and North Atlantic region, hurricanes are a ubiquitous temporary phenomenon with consistent wind current patterns. Studies of genetic variation have shown that hurricanes and marine currents influence the patterns of dispersal and constitute a source of variation in ecological and demographic processes54. Wind has been identified as one of the most important factors promoting the spread and long-distance dispersal of invasive insect species7,55 and to enhance distance and/or speed of dispersion as in the case of the monarch butterfly56, the cattle egret57 or the mealy bug, Oracella aculata58. Also, the grasshopper Eumetopina flavipes, vector of the sugarcane virus Ramu, entered Australia from New Zealand favored by wind currents59,60. Wind is also responsible for long-distance dispersal of the wasp Megastigmus schimitscheki in France7. Although previously suggested, the role of hurricanes on local dispersal of C. cactorum within the Caribbean was not examined until recently32. Our previous results using mitochondrial COI32 coupled with the findings of this study using nuclear microsatellites, are consistent and provide new pieces of evidence supporting the role of hurricanes on the spread of C. cactorum toward mainland North America. In comparison with the isolation by distance model, the isolation by resistance migration approach was a much better predictor of the current pattern of genetic variation of C. cactorum in the Caribbean and Florida. This finding suggests that hurricanes were one of the main drivers of dispersal from the Caribbean to mainland North America as previously shown using a more conserved mitochondrial marker32. In addition, ABC analyses of the invasion routes supported our hypothesis that moths entered Florida both through commercial transportation from the Dominican Republic and from Cuba as a consequence of climatic events (hurricanes); whereas trade with Cuba was suspended as a result of the embargo. Moreover, this finding suggests that hurricanes may have also dispersed C. cactorum from its initial introduction in Nevis, Antigua and Montserrat to other islands onto the Caribbean region. Historical records of hurricanes in the region (https://www.nhc.noaa.gov/) further support this statement. After the initial human-mediated introduction of C. cactorum, three hurricanes connected the Lesser Antilles and the Dominican Republic between 1963 and 1967, four impacted the south of Cuba and the Dominican Republic between 1975 and 1980, and two passed from Cuba and impacted the USA in 1985 and 1987. Nevertheless, the presence of the moth in the lower Bahamas in 198361, and the possibility of island hopping, as recorded in the Hawaiian archipelago, can also represent a possible route of dispersal towards Florida. During the last decades at least three hurricanes impacted the Bahamas before reaching the Atlantic coast of Florida (Mitch in 1988; Andrew in 1992, and Katrina in 2005). Hence a more extended survey of populations in this area of the Caribbean will provide further insight on the dispersion routes across the region.
The process of invasion is generally associated with the loss of genetic variation due to successive demographic bottlenecks as the invasion of new locations proceeds62. This process can be counteracted if repeated invasion events occur in a given location, thus increasing the amount of genetic variation to equivalent, or larger levels of variation, than that observed on the native region17,63. Our analyses revealed that populations of C. cactorum from the Caribbean and Florida, sustained less genetic variation than in the native region, likely as a consequence of genetic drift. Given that present populations of C. cactorum outside the Caribbean region are located more that 8,000 km away in other biogeographical areas and continents, the genetic structure results support previous analyses using mitochondrial markers suggesting that the insect entered the Caribbean probably once in 195721. Similar to the Lepidopteran tomato pest, Tuta absoluta, in Africa and the Mediterranean Basin62, the occurrence of just one introduction of the cactus moth was enough to allow its spread throughout the Caribbean despite the lower genetic variability than the source population from South America. Unlike the tomato pest and other invasive insect species1,9 significant genetic differentiation was detected in the invaded region suggesting again a possible effect of drift. A more complete survey of invaded populations in Australia, South Africa and in other continents will help to decipher whether the magnitude of reduction in genetic variation will affect the adaptive potential of the moth into other continental areas with different climatic conditions as those of the Caribbean and Florida.
The combined effect of hurricanes and genetic drift reducing genetic variation through space will likely affect the evolutionary potential of the moth during future introductions to the continent. Hence, monitoring programs of climatic events like hurricanes in the region and environmental genomics surveys will add new insight to better understand possible barriers to the expansion of the moth into continental areas.
References
Coisi, M. et al. Invasion of Europe by the western corn rootworm, Diabrotica virgifera virgifera: multiple transatlantic introductions with various reductions of genetic diversity. Mol. Ecol. 17, 3614–3627, https://doi.org/10.1111/j.1365-294X.2008.03866.x (2008).
Estoup, A. & Guillemaud, T. Reconstructing routes of invasion using genetic data: why, how and so what? Mol. Ecol. 19, 4113–4130, https://doi.org/10.1111/j.1365-294X.2010.04773.x (2010).
Estoup, A. et al. Estimation of demo-genetic model probabilities with Approximate Bayesian Computation using linear discriminant analysis on summary statistics. Mol. Ecol. Resour. 12, 846–855, https://doi.org/10.1111/j.1755-0998.2012.03153.x (2012).
Lombaert, E. et al. Bridgehead effect in the worldwide Invasion of the biocontrol harlequin ladybird. PLOS ONE 5, e9743, https://doi.org/10.1371/journal.pone.0009743 (2010).
Kerdelhué, C., Boivin, T. & Burban, C. Contrasted invasion processes imprint the genetic structure of an invasive scale insect across southern Europe. Heredity 113, 390–400 (2014).
Keller, S. R. & Taylor, D. R. History, chance and adaptation during biological invasion: separating stochastic phenotypic evolution from response to selection. Ecol. Lett. 11, 852–866 (2008).
Lander, T. A. et al. Reconstruction of a windborne insect invasion using a particle dispersal model, historical wind data, and Bayesian analysis of genetic data. Ecol. Evol. 4, 4609–4625, https://doi.org/10.1002/ece3.1206 (2014).
Simberloff, D. et al. Impact of biological invasions: what’s what and the way forward. Trends Ecol. Evol. 28, 58–66 (2013).
Lombaert, E. et al. Rapid increase in dispersal during range expansion in the invasive ladybird Harmonia axyridis. J. Evol. Biol. 27, 508–517 (2014).
Puillandre, N. et al. Genetic bottleneck in invasive species: the potato tuber moth adds to the list. Biol. Invasions 10, 319–333 (2008).
Rubinoff, D., Holland, B. S., Shibata, A., Messing, R. H. & Wright, M. G. Rapid invasion despite lack of genetic variation in the Erythrina gall wasp (Quadrastichus erythrinae Kim) 1. Pac. Sci. 64, 23–31 (2010).
Tsutsui, N. D., Suarez, A. V., Holway, D. A. & Case, T. J. Reduced genetic variation and the success of an invasive species. PNAS 97, 5948–5953, https://doi.org/10.1073/pnas.100110397 (2000).
Fraimount, A. et al. Deciphering the routes of invasion of Drosophila suzukii by Means of ABC Random Forest. Mol. Biol. Evol. 34, 980–996 (2017).
Guillemaud, T., Beaumont, M. A., Ciosi, M., Cornuet, J. M. & Estoup, A. Inferring introduction routes of invasive species using approximate Bayesian computation on microsatellite data. Heredity 104, 88–99, https://doi.org/10.1038/hdy.2009.92 (2010).
Holland, B. S. Invasion without a bottleneck: microsatellite variation in natural and invasive populations of the brown mussel Perna perna (L.). Mar. Biotechnol. 3, 407–415 (2001).
Stepien, C. A., Taylor, C. D. & Dabrowska, K. A. Genetic variability and phylogeographical patterns of a nonindigenous species invasion: a comparison of exotic vs. native zebra and quagga mussel populations. J. Evolution. Biol. 15, 314–328 (2002).
Dlugosch, K. M. & Parker, I. M. Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 17, 431–449, https://doi.org/10.1111/j.1365-294X.2007.03538.x (2008).
Zimmermann, H. G., Bloem, S. & Klein, H. Cactoblastis cactorum. The biology, history, threat, surveillance and control of the cactus moth. FAO/IAEA Division of Nuclear Techniques in Food and Agriculture (2007).
Julien, M. H. & Griffiths, M. W. Biological control of weeds: a world catalogue of agents and their target weeds, 4th edn. CABI Publishing, Wallingford (1998).
Zimmermann, H. G. & Pérez-Sandi, M. The consequences of introducing the cactus moth Cactoblastis cactorum to the Caribbean and beyond (2006).
Marsico, T. D. et al. Geographic patterns of genetic diversity from the native range of Cactoblastis cactorum (Berg) support the documented history of invasion and multiple introductions for invasive populations. Biol. Invasions 13, 857–868, https://doi.org/10.1007/s10530-010-9874-9 (2011).
Dickel, T. S. Cactoblastis cactorum in Florida (Lepidoptera: Pyralidae Phycitinae). Trop. Lepidoptera 2, 117–118 (1991).
Jezorek, H., Baker, A. J. & Stiling, P. D. Effects of Cactoblastis cactorum on the survival and growth of North American Opuntia. Biol. Invasions 14, 2355–2367 (2012).
Bloem, S., Hight, S. D., Carpenter, J. E. & Bloem, K. A. Developing the most effective trap to monitor the geographical expansion of the cactus moth Cactoblastis cactorum (Lepidoptera:Pyraildae). Fla. Entomol. 88, 300–306 (2005).
Johnson, D. M. & Stiling, P. D. Distribution and dispersal of Cactoblastis cactorum (Lepidoptera: Pyralidae), an exotic opuntia-feeding moth. Fla. Entomol. 81, 12–22 (1998).
Hight, S. D., Bloem, S., Bloem, K. A. & Carpenter, J. E. Cactoblastis cactorum (Lepidoptera: Pyralidae): Observations of courtship and mating behaviors at two locations on the gulf coast of Florida. Fla. Entomol. 86, 400–407 (2003).
Dodd, A. P. The biological campaign against prickly pear. Commonwealth Prickly Pear Board. Brisbane, Australia (1940).
Pettey, F. W. The biological control of prickly pear in South Africa. Sci. Bull., Dept. of Agric. of the Union of South Africa 271, 1–163 (1948).
Tuduri, J. C. G., Martorell, L. F. & Gaud, S. M. Geographical distribution and host plants of the cactus moth, Cactoblastis cactorum (Berg), in Puerto Rico and the United States Virgin Islands. J. Agr. U. Puerto Rico 58, 130–134 (1971).
Stiling, P. Potential Non-target Effects of a Biological Control Agent, Prickly Pear Moth, Cactoblastis cactorum (Berg) (Lepidoptera: Pyralidae), in North America, and Possible Management Actions. Biol Invasions 4, 273–281, https://doi.org/10.1023/A:1020988922746 (2002).
Simonsen, T. J., Brown, R. L. & Sperling, F. A. H. Tracing an Invasion: Phylogeography of Cactoblastis cactorum (Lepidoptera: Pyralidae) in the United States based on mitochondrial DNA. Ann. Entomol. Soc. Am. 101, 899–905, https://doi.org/10.1093/aesa/101.5.899 (2008).
Andraca-Gómez, G. et al. potential invasion route of Cactoblastis cactorum within the Caribbean region matches historical hurricane trajectories. Biol. Invasions 17, 1397–1406, https://doi.org/10.1007/s10530-014-0802-2 (2015).
Soberon, J., Golubov, J. & Sarukhán, J. The Importance of Opuntia in Mexico and Routes of Invasion and Impact of Cactoblastis cactorum (Lepidoptera: Pyralidae). Fla. Entomol. 84, 486–492, https://doi.org/10.2307/3496376 (2001).
Nei, M. Genetic distance between populations. Am. Nat. 106, 283–292 (1972).
Goudet, J. Fstat ver. 2.9.3.2. Institute of Ecology, Biology Building, UNIL, CH-1015, Lausanne, Switzerland (2002).
Wright, S. Coefficients of inbreeding and relationship. Am. Nat. 56, 330–338 (1922).
Leberg, P. L. Estimating allelic richness: effects of sample size and bottlenecks. Mol. Ecol. 11, 2445–2449 (2002).
Raymond, M. & Rousset, F. GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 86, 248–249 (1995).
Chapuis, M. P. & Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 24, 621–631 (2007).
Pritchard, J. K, Wen, X. & Falush, D. Documentation for structure software: version 2.2. University of Chicago, Chicago, pp 1–36 (2007).
Hubisz, M. J., Falush, D., Stephens, M. & Pritchard, J. K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332 (2009).
Earl, D. A. & von Holdt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361, https://doi.org/10.1007/s12686-011-9548-7 (2012).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Peakall, R. & Smouse, P. E. Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295, https://doi.org/10.1111/j.1471-8286.2005.01155.x (2006).
McRae, B. H. & Shah, V. B. Circuitscape user’s guide. The University of California, Santa Barbara (2009).
Landsea, C. W. A climatology of intense (or major) Atlantic hurricanes. Mon. Weather Rev. 121, 1703–1713 (1993).
Landsea, C. W., Nicholls, N., Gray, W. M. & Avila, L. A. Downward trends in the frequency of intense at Atlantic Hurricanes during the past five decades. Geophys. Res. Lett. 23, 1697–1700, https://doi.org/10.1029/96GL01029 (1996).
Lugo, A. E., Rogers, C. S. & Nixon, S. W. Hurricanes, Coral Reefs and Rainforests: Resistance, Ruin and Recovery in the Caribbean. AMBIO 29, 106–114, https://doi.org/10.1579/0044-7447-29.2.106 (2000).
Cornuet, J. M. et al. Inferring population history with DIYABC: a user-friendly approach to approximate Bayesian computiation. Bioinformatics 24, 2713–2719 (2007).
Habeck, D. H. & Bennett, F. D. Cactoblastis cactorum Berg (Lepidoptera:Pyralidae), a phycitine new to Florida. Fla. Department Agric. & Consumer Services, Division of Plant Industry. (1990).
Mann, J. Cacti naturalized in Australia and their control. Department of Lands, Brisbane. 1969.
Cornuet, J.-M. et al. DIYABC v2. 0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30, 1187–1189 (2014).
Pemberton, R. W. Cactoblastis cactorum (Lepidoptera: Pyralidae) in the United States. An immigrant biological control agent or an introduction of the nursery trade? Am. Entomol. 41, 230–232 (1995).
Cisneros de la Cruz, D. et al. Short barriers affect genetic variability of Rhizophora mangle L. in the Yucatan peninsula. Ecol. Evol. 8, 11083–11099, https://doi.org/10.1002/ece3.4575 (2018).
Liebhold, A. M. & Tobin, P. C. Population ecology of insect Invasions and their management. Annu. Rev. Entomol. 53, 387–408, https://doi.org/10.1146/annurev.ento.52.110405.091401 (2008).
Clarke, A. R. & Zalucki, M. P. Monarchs in Australia: on the winds of a storm? Biol. Invasions 6, 123–127, https://doi.org/10.1023/B:BINV.0000010120.29634.db (2004).
Botkin, D. B. The naturalness of biological invasions. West. N. Am. Naturalist 61, 261–266 JSTOR, www.jstor.org/stable/41717172 (2001).
Chen, Y., Vasseur, L. & You, M. Potential distribution of the invasive loblolly pine mealybug, Oracella acuta (Hemiptera: Pseudococcidae), in Asia under future climate change scenarios. Climatic Change 141, 719–732, https://doi.org/10.1007/s10584-017-1917-0 (2017).
Anderson, C. D. et al. Considering spatial and temporal scale in landscape-genetic studies of gene flow. Mol. Ecol. 19, 3565–3575, https://doi.org/10.1111/j.1365-294X.2010.04757.x (2010).
New, T. R. Alien species and insect conservation. Springer International Publishing, Cham. (2016).
Starmer, W. T., et al. The yeast community associated with decaying Opuntia stricta (Haworth) in Florida with regard to the moth Cactoblastis cactorum (Berg). Fla. Scientist 51, 7–11, JSTOR, www.jstor.org/stable/24319982 (1988).
Guillemaud, T. et al. The tomato borer, Tuta absoluta, invading the Med.terranean Basin, originates from a single introduction from Central Chile. Sci. Rep. 5, 8371, https://doi.org/10.1038/srep08371 (2015).
Lavrinienko, A. et al. First record of the invasive pest Drosophila suzukii in Ukraine indicates multiple sources of invasion. J. Pest. Sci. 90, 421–429, https://doi.org/10.1007/s10340-016-0810-3 (2017).
Acknowledgements
We thank Daniel Piñero, Antonio González, Jordan Golubov for helpful comments during the elaboration of the project and three reviewers for their constructive criticism. Ana Wegier and Matias Baranzelli provided useful comments that improved the manuscript. Sivael Morales, Mikel Cañizares, Jacqueline Pérez-Camacho and Sofía Ochoa offered invaluable help during field collection of sampling in Puerto Rico, Cuba and Jamaica. This study was financed by Comisión Nacional para el Uso y Conocimiento de la Biodiversidad (CONABIO-JE002) granted to JF, KB and CAD. GA-G thanks CONACYT and Posgrado en Ciencias Biológicas (UNAM) for the scholarship for graduate studies during her PhD project.
Author information
Authors and Affiliations
Contributions
G.A.G., J.F. conceived broad ideas. G.A.G., M.O., R.P.I., K.B., C.D., J.F. realized field collections. G.A.G., E.L. performed genetic analyses. G.A.G., J.F. wrote the manuscript. All authors contributed substantially to the final version and approved it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Andraca-Gómez, G., Lombaert, E., Ordano, M. et al. Local dispersal pathways during the invasion of the cactus moth, Cactoblastis cactorum, within North America and the Caribbean. Sci Rep 10, 11012 (2020). https://doi.org/10.1038/s41598-020-66864-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66864-3
This article is cited by
-
Meta-analysis reveals less sensitivity of non-native animals than natives to extreme weather worldwide
Nature Ecology & Evolution (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
