
Abstract
Glehnia littoralis is an important medicinal halophyte—the dried root of which is used as Chinese herbal medicine. However, the use, selection and stability of reference genes are rarely verified in studies of G. littoralis, which hampers investigation of its salt tolerance and metabolism. In this study, we selected 13 candidate reference genes from the transcriptome data of G. littoralis—serine/threonine-protein phosphatase PP2A (PP2A), polyubiquitin 10 (UBQ10), actin (ACT), elongation factor 1-α (EF1-α), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), α-tubulin (α-TUB), β-tubulin (β-TUB), polypyrimidine tract-binding protein 1 (PTBP1), expressed protein 1 (EXP1), expressed protein 2 (EXP2), TIP41-like (TIP41), SAND family (SAND), and cyclophilin 2 (CYP2), and used qRT-PCR to analyse their expression levels in roots of G. littoralis treated with NaCl, polyethylene glycol (PEG), abscisic acid (ABA), and methyl jasmonate (MeJA), as well as in various organs of G. littoralis. The ΔCt, geNorm, NormFinder, and BestKeeper algorithms were used to assess the expression stability of the candidate reference genes and the results were then used to generate a comprehensive rank list with the RankAggreg R package. The most stable reference genes for normalisation were EXP1 and PP2A in response to NaCl, EXP2 and PP2A in response to ABA, CYP2 and α-TUB in response to MeJA, and ACT and EXP1 in the PEG and the organ subsets. GAPDH, β-TUB, and UBQ10 exhibited low stability and so were unsuitable for normalisation. This study is the first systematic analysis of candidate reference genes in G. littoralis and will facilitate further investigation of normalisation of gene expression in G. littoralis.
Similar content being viewed by others


Introduction
Glehnia littoralis Fr. Schmidt ex Miq. is an herbaceous perennial in Family Umbelliferae. It is a medicinal halophyte naturally distributed in coastal areas, such as eastern China, Japan, the Korean Peninsula, Russia, and the United States1. Peeled and dried G. littoralis roots, named Radix Glehniae, are commonly used as a traditional Chinese herbal medicine for moistening the lungs and removing lung-heat; relieving thirst, dry throat and cough; and curing gastrointestinal disorders2,3. Radix Glehniae is rich in coumarins, coumarin glycosides, phospholipids, and polysaccharides, and is a component of anti-aging, anti-inflammation, and health-promoting agents2,3,4. The annual demand for Radix Glehniae is more than 8,000 tons. Due to the destruction of the ecological environment of beaches, G. littoralis-producing regions have shrunk or disappeared and G. littoralis cultivation has moved inland, resulting in an insufficient supply of uneven quality. Therefore, coastal cultivation of G. littoralis must be restored to guarantee the quality of medicinal materials. However, due to long-term inland cultivation and artificial selection, G. littoralis has insufficient salt tolerance and the mechanisms underlying its salt tolerance are unclear. Investigation of the salt-tolerance mechanism of G. littoralis would assist in restoring coastal production. High salinity affects the accumulation of secondary metabolites in plants5,6,7; for example, the contents of polyphenols and carotenoids in buckwheat, flavonoids in barley, and polyphenols in Cakile maritima increased after salt treatment8,9,10. The coastal salt environment of G. littoralis likely impacts the accumulation of secondary metabolites; indeed, accumulation of furocoumarins was increased by salt stress in G. littoralis11. However, the underlying mechanism is unknown. Studies of the salt tolerance of G. littoralis and its effect on the synthesis and accumulation of secondary metabolites are needed to promote production of high-quality medicinal materials, improve the quality of germplasm resources, and facilitate investigation of the function and regulation of genes in plants.
Quantitative real-time PCR (qRT-PCR) is used to analyse the expression patterns of plant genes because of its high sensitivity, quantitative accuracy, low cost, and high throughput12,13,14. To eliminate the influence of different templates and reverse-transcription efficiencies, stable reference genes are used to normalise gene expression values determined by qRT-PCR. However, the expression levels of reference genes differ among environmental conditions, tissues, organs, and developmental stages. They can only remain relatively stable within a range of experimental conditions. The use of a reference gene irrespective of the experimental conditions will affect the reliability of the results. Housekeeping genes such as 18S rRNA, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), actin (ACT), and tubulin (TUB) are commonly used as reference genes in plants15,16,17. Novel reference genes, such as eukaryotic initiation factor 4α (eIF-4α), serine/threonine-protein phosphatase PP2A (PP2A), SAND family (SAND), and TIP41-like (TIP41), have also been used for normalisation18,19,20. However, no reference gene of G. littoralis has been assessed under normal and abnormal conditions, which hampers research into its metabolic pathways and mechanism of salt tolerance.
We previously performed a comprehensive transcriptome analysis of G. littoralis in response to salt stress via RNA sequencing (RNA-seq)2. The RNA-seq results identified a large number of unigenes involved in the response to salt stress in G. littoralis. In the present study, we selected 13 candidate reference genes based on the transcriptome data of G. littoralis and evaluated their expression levels using qRT-PCR in response to salt (NaCl), drought (PEG), abscisic acid (ABA), and methyl jasmonate (MeJA), and in various organs. We also evaluated the expression stability of the candidate reference genes using the statistical algorithms ΔCt21, geNorm22, NormFinder23, and BestKeeper24, and performed a comprehensive stability ranking25. Finally, we validated the candidate reference genes using several stress-related genes from G. littoralis transcriptome data. This work provides the first systematic analysis of candidate reference genes in G. littoralis, and the results will facilitate future studies on gene expression in G. littoralis, as well as in other species of Umbelliferae.
Results
Selection of candidate reference genes, primer specificity, and amplification efficiency
Based on the transcriptome data of G. littoralis obtained in our previous study2, thirteen candidate reference genes were selected with reference to the reference genes of Arabidopsis thaliana, which are frequently used in studies based on qRT-PCR18. The candidate reference genes were PP2A, polyubiquitin 10 (UBQ10), ACT, elongation factor 1-α (EF1-α), GAPDH, α-TUB, β-TUB, polypyrimidine tract-binding protein 1 (PTBP1), expressed protein 1 (EXP1), expressed protein 2 (EXP2), TIP41, SAND, and cyclophilin 2 (CYP2). The sequences of the 13 candidate reference genes from RNA-seq are listed in Supplementary File S1 and their characteristics and qRT-PCR primer sequences are shown in Table 1. Primer specificity was evaluated via agarose gel electrophoresis, sequencing, and melting curve analysis. A single DNA band at the correct molecular weight and a single peak in the melting curve for each gene indicated high primer specificity (Supplementary Figs. S1 and S2). The electropherograms of sanger sequencing for qRT-PCR amplicons obtained from 13 reference genes are listed in Supplementary File S2. The amplification efficiency (E) and correlation coefficient (R2) of each candidate reference gene were calculated from calibration curves with R2 values of > 0.99; values of E ranged from 88% to 108% (Table 1).
Expression profiles of the candidate reference genes
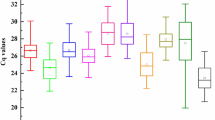
We performed qRT-PCR to evaluate the expression of the 13 candidate reference genes. The expression of the candidate reference genes was determined by calculating the threshold cycle (Ct) values; the higher the Ct value, the lower the expression level. The Ct values of the 13 genes ranged from 17.02 to 29.34, with the majority ranging from 19 to 27 (Fig. 1; Supplementary Dataset 1). EF-1a had the lowest mean Ct value of 19.06 ± 1.47 (standard deviation [SD]), indicating that it was the most highly expressed gene, followed by GAPDH (20.74 ± 1.67), a-TUB (21.35 ± 1.58), β-TUB (21.4 ± 1.52), and ACT (21.42 ± 1.66). UBQ10 (22.52 ± 1.34), PP2A (23.07 ± 1.18), TIP41 (23.93 ± 1.05), CYP2 (24.42 ± 0.86), EXP2 (24.56 ± 1.15), and SAND (24.8 ± 1.25) were moderately expressed, and EXP1 (26.19 ± 0.93) and PTBP1 (25.41 ± 1.32) were expressed at low levels. CYP2 showed the least variation in expression, and GAPDH and ACT showed the greatest variation (Fig. 1).

Threshold cycle (Ct) values of the candidate reference genes. Boxes indicate the 25th and 75th percentiles, horizontal lines represent the medians, spots indicate the means, and whiskers represent the maximum and minimum values.
Analysis of the expression stability of the candidate reference genes
To analyse the stability of the candidate reference genes, the statistical algorithms ΔCt21, geNorm22, NormFinder23, and BestKeeper24 were used.
The ΔCt method involves comparing the relative expression (ΔCt values) of pairs of genes within each test group. The mean SD (mSD) was used to assess the expression stability of the 13 candidate genes (Supplementary Table S1). A low mSD value indicates stable expression. In all samples and in the NaCl subset, PP2A was the most stable gene, whereas GAPDH was the least stable gene. ACT was the most stable gene in PEG subset and CYP2 was the most stable gene in MeJA subset, whereas β-TUB was the least stable gene in both subsets. In the ABA subset, EXP2 and PP2A were the top two stable genes, whereas EF1-α was the least stable gene. Across all organs, EXP1 and ACT were the most stable genes and GAPDH was the least stable gene.
The geNorm algorithm is used to rank the stability of reference genes by generating expression stability values (M-values); a low M-value indicates stable expression (Fig. 2). EXP1 and CYP2 were the most stable genes in all test samples, PP2A and EXP1 were most stable in NaCl subset. ACT and CYP2 were most stable in the PEG subset. Equally, α-TUB and β-TUB in the ABA subset, α-TUB and EXP2 in the MeJA subset, and β-TUB and EXP2 in the organs were the most stable genes. Across all test samples, GAPDH was the least stable gene, as in the NaCl and organ subsets. β-TUB was the least stable gene in the PEG and MeJA subsets. In the ABA subset, EF1-α was the least stable gene.

Expression stability and ranking of the 13 reference genes by geNorm. The least stable genes are on the left, and the most stable on the right.
According to the NormFinder algorithm, genes with the lowest stability values (SV) have the most stable expression. The NormFinder ranked list of the 13 candidate genes was shown in Supplementary Table S2. PP2A was the most stable gene in all test samples, followed by EXP2, CYP2, and PTBP1. In the NaCl subset, PTBP1 was the most stable gene, followed by CYP2, PP2A, and EXP1. In the PEG subset, PP2A ranked first, followed by EXP1, CYP2, and ACT. In the ABA subset, EXP2 was the most stable gene, followed by PP2A, PTBP1 and CYP2. In the MeJA subset, CYP2 was the most stable gene. Across all organs, EXP1 and ACT was the top two stable reference genes.
The Bestkeeper algorithm calculates the stability of candidate reference genes based on the coefficient of variance (CV) and SD values. The reference gene with the lowest CV ± SD was considered the most stable gene. The BestKeeper ranked list of the 13 candidate reference genes can be found in Supplementary Table S3. In all test samples, CYP2 (which had the lowest CV ± SD value) exhibited stability levels similar to that calculated by geNorm. In the NaCl subset, EXP2 was the most stable gene. In the PEG subset, ACT was the most stable gene. In the ABA and MeJA subsets, PP2A was the most stable gene. Across all organs, EF1-α was the reference gene most suitable for normalisation.
The use of different algorithms resulted in different gene expression ranks. Therefore, the ranks of the candidate reference genes were determined using the R package RankAggreg25. RankAggreg aggregated the ranks determined by ΔCt, geNorm, NormFinder, and BestKeeper to produce a comprehensive ranking list. For all test samples, the stability of the candidate reference genes was in the order CYP2 > PP2A > EXP1 > PTBP1 > EXP2 > SAND > β-TUB > ACT > EF1-α > UBQ10 > α-TUB > TIP41 > GAPDH (Fig. 3a). EXP1 comprehensively ranked first in the NaCl and organ subsets, and ACT and EXP1 were ranked as the top two stable genes in the PEG subset (Fig. 3b,c,f). In the ABA and MeJA subsets, EXP2 and CYP2 were the most stably expressed genes, respectively (Fig. 3d,e). The expression of GAPDH was most unstable in the NaCl and organ subsets (Fig. 3b,f). The expression of β-TUB was most unstable in response to PEG and MeJA (Fig. 3c,e). For the ABA subset, EF1-α was the least stable gene (Fig. 3d).

Rank aggregation of the 13 candidate reference genes in the six subsets. The RankAggreg package was loaded into R software. The ΔCt, BestKeeper, NormFinder, and geNorm ranks are represented as grey lines. The black line represents the mean rank of each gene according to each method. The red line indicates the result of the Cross-Entropy algorithm.
Selection of the optimal combination of PCR reference genes
Using the geNorm algorithm, the pairwise variation Vn/Vn+1 was also automatically calculated by geNorm between two consecutive normalisation factors across subsets to determine the optimal number of reference genes22. A value of Vn/Vn+1 of <0.15 indicated that n reference genes were the optimal combination compared with n + 1 reference genes. Figure 4 shows that all pairwise variations of the PEG, ABA, and MeJA subsets were <0.15, suggesting that the use of two reference genes was the optimal combination for normalising gene expression. In all test samples, and in the NaCl and organ subsets, the V3/V4 values were <0.15; thus, in these subsets, the use of three reference genes was the optimal combination for normalisation (Fig. 4). For example, according to the comprehensive ranking list (Fig. 3), “ACT + EXP1”, “EXP2 + PP2A” and “CYP2 + α-TUB” could be regarded as the optimal combinations of qRT-PCR reference genes for the PEG, ABA and MeJA subsets, respectively. Likewise, “CYP2 + EXP1 + PP2A” could be regarded as the optimal combination of qRT-PCR reference genes for all test samples and the NaCl subset. “EXP1 + ACT + β-TUB” was regarded as the optimal combination for the organ subset.

Pairwise variation (Vn/Vn+1) values calculated by geNorm to determine the optimal number of reference genes. The cut-off value to determine the optimal number of reference genes for normalisation using qRT-PCR was 0.15.
Validation of reference genes
To validate the stability of the candidate reference genes, we first compared their expression in the presence and absence of NaCl stress based on the RNA-seq analysis of G. littoralis in response to salt stress2. Gene expression levels are reported as FPKM values, and the variation in expression levels as CVs. A lower CV indicates more stable expression. PP2A, EXP1, CYP2, and β-TUB had lower CVs than the other candidate reference genes under NaCl treatment, whereas UBQ10, α-TUB, GAPDH, and ACT had higher CVs (Fig. 5). The stability of these genes in the RNA-seq data was mostly consistent with the comprehensive list in the NaCl-treatment subset (Fig. 3b).

Expression and stability of the candidate reference genes based on RNA-seq in the salt (NaCl) treatment. FPKM values indicate gene expression. Lower coefficient of variation (CV) values indicate more stable expression. FPKM, fragments per kilobase of exon model per million mapped reads.
To further validate the reference genes, the expression pattern of a stress-related target gene, PYL (Pyrabactin resistance 1-like gene), in the NaCl, PEG, ABA, MeJA, and organ subsets was examined using qRT-PCR (Fig. 6). The two most (rank1 and 2) and least (rank 12 and 13) stable reference genes in Fig. 3 were used as the reference genes for each subset. Different letters indicate statistically significant difference in each condition (P < 0.05, Duncan’s multiple range test). PYL expression increased significantly at 24 h after NaCl treatment when normalised to the four candidate reference genes, but the expression was markedly higher when normalised to α-TUB. At 6 h, the PYL expression showed the opposite trend and significant difference when normalised to GAPDH and α-TUB (least stable) compared to PP2A and EXP1 (most stable) (Fig. 6a). In the PEG subset, the expression levels of PYL after PEG treatment were upregulated when normalised to GAPDH and β-TUB (least stable) or to EXP1 and ACT (most stable). At 6 h, PYL expression was significantly upregulated when normalised to GAPDH and β-TUB compared to EXP1 and ACT, and PYL expression showed the opposite trend at 24 h after PEG treatment when normalised to GAPDH compared to the other three candidate reference genes (Fig. 6b). In the ABA subset, PYL expression was upregulated at 6 h and decreased at 24 h when normalised to EF1-α, TIP41 (both least stable), PP2A, and EXP2 (both most stable) (Fig. 6c). However, the PYL expression showed no statistical significant compared with that at time 0 h when normalised to EF1-α. In the MeJA subset, the variation in PYL expression normalised to CYP2, α-TUB (both most stable), β-TUB, and TIP41 (both least stable) increased at 6 h and 24 h after treatment, and the expression level of PYL was more variable when normalised to β-TUB (Fig. 6d). The expression level of PYL was highest in leaves followed by roots when normalised to the most stable (EXP1 and ACT) and least stable (GAPDH and PTBP1) genes (Fig. 6e). Therefore, when the most stable reference gene was used for normalisation, the expression level of target gene was more reliable and reproducible.

Relative expression levels of PYL normalised to four reference genes for NaCl, PEG, ABA, MeJA and organs subsets. Error bars indicate means ± SD. Different letters indicate statistically significant difference in each condition (P < 0.05, Duncan’s multiple range test).
We then selected eight potentially stress-related genes from G. littoralis RNA-seq data for expression analysis using the top two stable reference genes of each stress treatment. The eight genes included stress-responding protein kinases, transporter proteins, a transcription factor and a synthetase (Fig. 7; Supplementary Table S4). Reference genes EXP1, ACT, EXP2, and CYP2 were used for NaCl, PEG, ABA, and MeJA treatments respectively in Fig. 7a–d,i–l. PP2A, EXP1, PP2A, and α-TUB were also used for each treatment respectively in Fig. 7e–h,m–p. Different letters above each bar indicate statistically significant difference in each treatment (P < 0.05, Duncan’s multiple range test). The eight selected genes showed differential expression under NaCl, PEG, ABA and MeJA treatments by qRT-PCR; most were induced by these treatments (Fig. 7). The expression levels of comp35393_c0_seq6, comp33363_c0_seq1, and comp34770_c0_seq12 were significantly increased after NaCl treatment when normalised to both stable reference genes. The expression of comp35862_c0_seq13, comp30905_c0_seq3, comp35199_c0_seq4, comp35393_c0_seq6, comp37685_c0_seq1, and comp25557_c0_seq2 showed significant differences at 24 h after PEG treatment compared with that at 0 h. The expression of comp30905_c0_seq3 and comp35199_c0_seq4 were significantly increased after ABA treatment. The expression of comp35862_c0_seq13, comp35393_c0_seq6, and comp34770_c0_seq12 showed significant differences at 24 h after MeJA treatment compared with that at 0 h, as well as comp30905_c0_seq3 and comp25557_c0_seq2 at 6 h. Although the fold changes in relative expression normalised to the top two stable reference genes did not match perfectly, the trends in expression of these genes were generally consistent, confirming the reproducibility of the qRT-PCR results.

Expression patterns of eight stress-related genes normalised to the top two stable reference genes for NaCl, PEG, ABA, MeJA treatment. EXP1, ACT, EXP2, and CYP2 were used for NaCl, PEG, ABA, MeJA treatments respectively in (a–d,i–l). PP2A, EXP1, PP2A, and α-TUB were used for each treatment respectively in (e–h,m–p). (a,e) comp35862_c0_seq13/MAPK; (b,f) comp34770_c0_seq12/CDPK; (c,g) comp30905_c0_seq3/SOS1; (d,h) comp35199_c0_seq4/CIPK; (i,m) comp35393_c0_seq6/TPC1; (j,n) comp25557_c0_seq2/WRKY; (k,o) comp33363_c0_seq1/P5CS; (l, p) comp37685_c0_seq1/SnRK2. Error bars indicate means ± SD. Different letters indicate statistically significant difference in each treatment (P < 0.05, Duncan’s multiple range test).
Discussion
Glehnia littoralis is an important medicinal halophyte used in studies of the salt-tolerance mechanism, breeding, synthesis of active ingredients, and the relationship between high salinity and metabolism in G. littoralis. Additionally, qRT-PCR is a highly sensitive, accurate, and high-throughput gene expression analysis technology26. However, the accuracy of qRT-PCR depends largely on the reference genes used. It is particularly important to select appropriate reference genes for the tissue, organ, and experimental conditions27. At present, the use and selection of reference genes, and the stability of these genes are rarely verified in studies of G. littoralis, which hampers research. We used the RNA-seq data of G. littoralis to screen 13 candidate reference genes. The expression levels and stability of the 13 candidate reference genes were assessed in roots of G. littoralis subjected to NaCl, PEG, ABA, and MeJA treatments, as well as in the major organs. G. littoralis is a halophyte and its leaves have the structure typical of most mesophytes28. Plant hormones regulate all aspects of plant growth and development. MeJA, a derivative of jasmonate, is an important signalling molecule in plant secondary metabolism29,30,31. We ranked the stability of the 13 candidate reference genes under a variety of conditions using the ΔCt, geNorm, NormFinder, and BestKeeper algorithms. Because of their different principles, the rank lists generated by the four algorithms were non-identical (Fig. 2; Supplementary Tables S1–S3)32. For example, PP2A was ranked first by geNorm and ΔCt in the NaCl subset, but it was ranked third by Normfinder and fifth by BestKeeper. The geNorm algorithm calculates the SD of variation between two reference genes after logarithmic transformation as the M-value and indicates the optimal number of reference genes necessary for accurate normalisation22. Normfinder works on a similar principle as geNorm, but it considers both the inter- and intra-group variation in expression and outputs only the most stable gene23. BestKeeper, an Excel-based tool, identifies stable reference genes using pair-wise correlation and can analyse up to 10 target genes24,33,34. The ΔCt method determines the stability of reference genes by comparing the relative expression (Ct value) of pairs of genes within each sample based on geNorm data21. In the present study, in the NaCl and PEG subsets, the gene ranked first by geNorm was identical to that ranked by the ΔCt method. For all samples and in the ABA, MeJA and organ subsets, the gene ranked first by Normfinder was identical to that ranked by the ΔCt method. Although the stability ranks of the candidate reference genes differed among the four methods, the RankAggreg R package or a geometric mean analysis can be used to combine the rank lists into an optimal list of reference genes25,35,36. Several previous studies have suggested that the use of a combination of reference genes is better than a single reference gene36,37,38. The geNorm algorithm also determined the optimal number of reference genes for accurate normalisation by pairwise variation between the normalisation factors. For different cases, an optimal combination of reference genes can be used for comprehensive normalisation to acquire the most reasonable results. In the present study, the use of two reference genes was the optimal combination for qRT-PCR analysis under PEG, ABA and MeJA treatments, and the use of three reference genes was the best for all test samples, NaCl and organ subsets.
Housekeeping genes with stable constitutive expression, such as ACT, TUB, 18S rRNA, UBQ, CYP, GAPDH, and EF1-α, are typically used as reference genes. However, the expression levels of these reference genes are not constant in some species and cases16,18,39,40,41,42. GAPDH has good stability in Arabidopsis pumilain under drought- and heat-stress and in Kentucky bluegrass roots under cold- and drought-stress but was one of the most unstable reference genes in Baphicacanthus cusia43 as well as in G. littoralis in this study. Similarly, β-TUB was the least stable reference gene in the roots of G. littoralis exposed to PEG and MeJA, as well as in Metasequoia exposed to ABA36. However, β-TUB showed good stability in Syntrichia caninervis under abiotic stresses and desiccation/rehydration44, and in a variety of tissues of Corchorus capsularis32. In this study, CYP2 was highly stable in G. littoralis under salt stress but was less stable in orchardgrass45. Novel reference genes, such as PP2A, EXP1, TIP41, and SAND, exhibited better stability in specific cases18,19,46,47. For example, PP2A and SAND were highly stable in Caragana intermedia under salt-, drought-, and heat-stress47, and EXP1 was the most stable gene in Lycoris aurea under salt-, heat-, and ABA-stress and in various tissues35. In this study, PP2A, EXP1, EXP2, and CYP2 were the four most stable genes in G. littoralis in most subsets. In Peucedanum praeruptorum Dunn, a species in Umbelliferae, SAND, ACT2, UBC9, PP2A, and PTBP1 were the most suitable genes for normalisation of qRT-PCR data48.
In our previous RNA-seq analysis of G. littoralis, we also assessed differentially expressed genes (DEGs) under salt stress. The results of the previous study were compared against the stability assessment in the present study for the NaCl treatment to confirm the stability and reliability of the candidate reference genes (Figs. 3 and 5). To validate the stability of these reference genes, PYL was first used as a test gene (Fig. 6). PYL is a core regulatory component of the ABA signaling pathway, and its expression is induced by various stresses49. In the transcriptome analysis of G. littoralis in response to salt stress2, PYL expression was significantly upregulated by salt stress. In our study, the two most and least stable reference genes were used as the reference genes for each subset. The more stable the reference gene, the more reliable the expression trend of the target gene. For example, a clearly opposite trend in PYL expression was observed at 24 h when GAPDH (unstable reference gene) was used as the reference gene compared to the most stable reference gene for PEG treatment, as well as when it was used as the reference gene for NaCl treatment, which can lead to misinterpretation of results, as some previous studies have indicated50,51,52. For further validation, we analysed and compared the relative expression levels of other stress-related genes from G. littoralis RNA-seq data under different stress conditions with the top two stable reference genes of each subset. The qRT-PCR results revealed similar trends in the expression patterns of these genes when the two selected reference genes were used for normalisation. These genes include Ca2+-dependent protein kinase (CDPK), CBL-interacting protein kinase (CIPK), mitogen-activated protein kinase (MAPK), and the ser/thr protein kinase OPENSTOMATA 1 (OST1)/SnRK2.6/SnRK2E, all of which play pivotal roles in plant responses to multiple stresses53,54,55,56; salt overly sensitive (SOS) signaling pathway gene SOS1 and vacuolar ion channel TWO PORE CHANNEL1 (TPC1), which show positive responses to salt stress57,58; ∆1-pyrroline-5-carboxylate synthetase (P5CS), which resists drought stress59; and the transcription factor WRKY60. Most of these genes display stress-induced expression in G. littoralis as well as in other plants. All of the above results provide the evidence for validation of the stability of the recommended reference genes. Therefore, the use of appropriate reference genes or gene combinations is essential for normalisation of the expression of target genes in G. littoralis. Our systematic analysis of candidate reference genes will facilitate future studies on gene expression in G. littoralis, as well as in other species of Umbelliferae.
Materials and methods
Plant materials and stress treatments
Glehnia littoralis seeds were originally collected from Tannanwan Beach, Pingtan, Fujian Province, China (25°26′1.86″N, 119°45′14.4″E) under the permission of competent authority and cultivated in the Germplasm Garden of Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing, China2. The G. littoralis was identified and conserved by the Herbarium of Institute of Botany, Jiangsu Province and Chinese Academy of Sciences (voucher specimen: No.0631502). The experimental research on G. littoralis is legal and no field experiments are involved. In this study, seedlings of G. littoralis with a root length of 4–6 cm were transplanted from Germplasm Garden into flowerpots containing washed sand. The seedlings were grown in a chamber under a 14-h-light (26 °C)/10-h-dark (22 °C) photoperiod and irrigated with Hoagland nutrient solution. After 3 months of growth, the seedlings were subjected to salt (200 mM NaCl), drought (20% PEG 6000), or hormone (100 μM ABA or 100 μM MeJA) treatment for 0, 6, or 24 h, as described previously35. The underground part of G. littoralis is used as herbal medicine, and so G. littoralis roots were sampled separately at different time points for each treatment. For organ samples, leaves and flowers of G. littoralis were collected in July during the same year. Plants grown under normal conditions were collected as controls. Samples were harvested from three replicate plants, immediately frozen, and stored at −80 °C for RNA extraction.
RNA extraction, cDNA synthesis, and qRT-PCR
RNA from G. littoralis was extracted using RNAiso Plus reagent (TaKaRa, Dalian, China) according to the manufacturer’s instructions. The RNA concentration was measured using a Nanodrop ND-2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). RNA samples with a ratio of absorbance at 260 nm (A260) to absorbance at 280 nm (A280) of 1.8–2.2 and an A260/A230 ratio of > 1.8 were used for cDNA synthesis. For qRT-PCR, 600 ng of RNA was used for reverse transcription in a 20 μL reaction volume using the Prime Script™ RT Reagent kit with gDNA Eraser (TaKaRa).
The qRT-PCR amplification was performed in 96-well plates using a qTOWER 2.2 Real-Time PCR system (Analytik Jena AG, Jena, Germany) with TB Green Premix Ex Taq™ II (Tli RNaseH Plus; TaKaRa). Reactions were performed in a total volume of 20 μL containing 2.0 μL of 10-fold diluted cDNA, 0.8 μL each of forward and reverse primer (10 μM), 10 μL of TB Green Premix Ex Taq™ II, and 6.4 μL of ddH2O. A reaction without template was used as the negative control. The qRT-PCR conditions were as follows: 95 °C for 5 min followed by 40 cycles of 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 25 s. After completion of amplification, a melting curve analysis was performed. Three biological replicates and three technical replicates were performed.
Selection of candidate reference genes and design of primers
We previously performed a comprehensive transcriptome analysis of G. littoralis in response to salt stress via Illumina 2000 sequencing2. A number of unigenes were identified and annotated, and their expression was analysed. Based on commonly used reference genes in Arabidopsis, we identified 13 candidate reference genes from the G.littoralis transcriptome data. Primers for qRT-PCR were designed based on the sequences of the 13 candidate reference genes (Supplementary File S1) using Primer3 Tools (http://primer3.ut.ee/) and the following criteria: primer size 18–23 bp, GC content 45–65%, melting temperature 57–62 °C, and product size 100–250 bp (Table 1). The specificity of the primer pairs was assessed via qRT-PCR and a melting curve analysis. The G. littoralis cDNA template was diluted five-fold with nuclease-free water to establish a calibration curve. The PCR amplification efficiency (E) and the correlation coefficient (R2) were calculated using the slope of the calibration curve according to the equation 10−1/slope – 161. Finally, the amplification products were verified via 1% agarose gel electrophoresis and sequencing.
Analysis of gene stability
The stability of the 13 candidate reference genes was evaluated using four statistical algorithms—the ΔCt method21, geNorm22, NormFinder23, and BestKeeper24. The ΔCT method involved comparison of the relative expression of pairs of genes within each sample. NormFinder, geNorm, and BestKeeper are Excel-based tools, and the Ct values obtained were converted into input files according to the developers’ instructions35,36,44. The geNorm algorithms also recommended the optimal combination number of qRT-PCR reference genes by comparing pairwise variation (Vn/n+1) values and the cut-off value was <0.1522. Using the above four algorithms, ranked lists of gene stability were generated, and the R package RankAggreg was used to generate a list of the overall stability of the 13 candidate reference genes25. R software v. 3.6.0 (R × 64) was used to load the RankAggreg v. 0.6.5 package (http://cran.r-project.org/web/packages/RankAggreg/). The details can be accessed at https://cran.r-project.org/web/packages/RankAggreg/RankAggreg.pdf. The RankAggreg package contains a Cross-Entropy Monte Carlo algorithm and a Genetic algorithm for evaluating rank aggregation.
Validation of the reference genes
Several stress-related genes were selected from the G. littoralis transcriptome data to test the stability of the candidate reference genes. The details of them were shown in Supplementary Table S4. Primers for qRT-PCR were shown in Supplementary Table S5. Relative gene expression was analysed using the 2−ΔΔCT method62.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Information.
References
Peng, Y. et al. Growth and physiological characteristics of Glehnia littoralis in response to salt stress. Jiangsu J. Agr. Sci. 30, 1273–1278 (2014).
Li, L., Li, M., Qi, X., Tang, X. & Zhou, Y. De novo transcriptome sequencing and analysis of genes related to salt stress response in Glehnia littoralis. PeerJ 6, e5681 (2018).
Yuan, Z., Tezuka, Y., Fan, W., Kadota, S. & Li, X. Constituents of the underground parts of Glehnia littoralis. Chem. Pharm. Bull. 50, 73–77 (2002).
Tomshich, S. V. et al. Biologically active polysaccharides from medicinal plants of the Far East. Chem. Nat. Compd. 33, 146–149 (1997).
Ramakrishna, A. & Ravishankar, G. A. Influence of abiotic stress signals on secondary metabolites in plants. Plant Signal. & Behav. 6, 1720–1731 (2011).
Selmar, D. Potential of salt and drought stress to increase pharmaceutical significant secondary compounds in plants. Landbauforschung Volkenrode 58, 139–144 (2008).
Selmar, D. & Kleinwachter, M. Stress enhances the synthesis of secondary plant products: the impact of stress-related over-reduction on the accumulation of natural products. Plant & Cell Physiol. 54, 817–826 (2013).
Ali, R. M. & Abbas, H. M. Response of salt stressed barley seedlings to phenylurea. Plant Soil & Environ 49, 158–162 (2003).
Ksouri, R. et al. Salinity effects on polyphenol content and antioxidant activities in leaves of the halophyte Cakile maritima. Plant Physiol. & Biochem. 45, 244–249 (2007).
Lim, J.-H., Park, K.-J., Kim, B.-K., Jeong, J.-W. & Kim, H.-J. Effect of salinity stress on phenolic compounds and carotenoids in buckwheat (Fagopyrum esculentum M.) sprout. Food Chem. 135, 1065–1070 (2012).
Shu, X., Li, N., Tang, X. & Zhou, Y. Effects of NaCl stress on photosynthetic physiology and active component of different Glehnia littoralis provenance. Jiangsu J. Agr. Sci. 35, 790–797 (2019).
Bustin, S. A. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR) trends and problems. J. Mol. Endocrinol. 29, 23–39 (2002).
Radonic, A. et al. Guideline to reference gene selection for quantitative real-time PCR. Biochem. Biophys. Res. Commun. 313, 856–862 (2004).
VanGuilder, H. D., Vrana, K. E. & Freeman, W. M. Twenty-five years of quantitative PCR for gene expression analysis. BioTechniques 44, 619–626 (2008).
Glare, E. M., Divjak, M., Bailey, M. J. & Walters, E. H. beta-Actin and GAPDH housekeeping gene expression in asthmatic airways is variable and not suitable for normalising mRNA levels. Thorax 57, 765–770 (2002).
Thellin, O. et al. Housekeeping genes as internal standards: use and limits. J. Biotechnol. 75, 291–295 (1999).
Dheda, K. et al. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. BioTechniques 37, 112–119 (2004).
Czechowski, T., Stitt, M., Altmann, T., Udvardi, M. K. & Scheible, W.-R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 139, 5–17 (2005).
Libault, M. et al. Identification of four soybean reference genes for gene expression normalization. Plant Genome 1, 44–54 (2008).
Ling, H., Wu, Q., Guo, J., Xu, L. & Que, Y. Comprehensive selection of reference genes for gene expression normalization in sugarcane by real time quantitative RT-PCR. Plos One 9, e97469 (2014).
Silver, N., Best, S., Jiang, J. & Thein, S. L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 7, 33 (2006).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, research0034.1 (2002).
Andersen, C. L., Jensen, J. L. & Orntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250 (2004).
Pfaffl, M. W., Tichopad, A., Prgomet, C. & Neuvians, T. P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515 (2004).
Pihur, V., Datta, S. & Datta, S. RankAggreg, an R package for weighted rank aggregation. BMC Bioinformatics 10, 62 (2009).
Gutierrez, L., Mauriat, M., Pelloux, J., Bellini, C. & Van Wuytswinkel, O. Towards a systematic validation of references in real-time RT-PCR. Plant Cell 20, 1734–1735 (2008).
Dheda, K. et al. The implications of using an inappropriate reference gene for real-time reverse transcription PCR data normalization. Anal. Biochem. 344, 141–143 (2005).
Voronkova, N. M., Burkovskaya, E. V., Bezdeleva, T. A. & Burundukova, O. L. Morphological and biological features of plants related to their adaptation to coastal habitats. Russ. J. Ecol. 39, 1–7 (2011).
Wasternack, C. Jasmonates: an update on biosynthesis, signal transduction and action in plant stress response, growth and development. Ann. Bot. 100, 681–697 (2007).
De Geyter, N., Gholami, A., Goormachtig, S. & Goossens, A. Transcriptional machineries in jasmonate-elicited plant secondary metabolism. Trends Plant Sci. 17, 349–359 (2012).
Wasternack, C. & Hause, B. Jasmonates: biosynthesis, perception, signal transduction and action in plant stress response, growth and development. Ann. Bot. 111, 1021–1058 (2013).
Niu, X. et al. Selection of reliable reference genes for quantitative real-time PCR gene expression analysis in Jute (Corchorus capsularis) under stress treatments. Front. Plant Sci. 6, 848 (2015).
Chang, E. et al. Selection of reference genes for quantitative gene expression studies in Platycladus orientalis (Cupressaceae) using real-time PCR. Plos One 7, e33278 (2012).
Xiao, X. et al. Validation of suitable reference genes for gene expression analysis in the halophyte Salicornia europaea by real-time quantitative PCR. Front. Plant Sci. 5, 788 (2015).
Ma, R., Xu, S., Zhao, Y., Xia, B. & Wang, R. Selection and validation of appropriate reference genes for quantitative real-time PCR analysis of gene expression in Lycoris aurea. Front. Plant Sci. 7, 536 (2016).
Wang, J. J., Han, S., Yin, W., Xia, X. & Liu, C. Comparison of reliable reference genes following different hormone treatments by various algorithms for qRT-PCR analysis of Metasequoia. IJMS 20, 34 (2018).
Huang, N. et al. Identification and evaluation of PCR reference genes for host and pathogen in sugarcane-Sporisorium scitamineum interaction system. BMC Genomics 19, 479 (2018).
Ribeiro, P. R. et al. Identification of reference genes for gene expression studies during seed germination and seedling establishment in Ricinus communis L. Seed Sci. Res. 24, 341–352 (2014).
Nicot, N., Hausman, J. F., Hoffmann, L. & Evers, D. Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J. Exp. Bot. 56, 2907–2914 (2005).
Jian, B. et al. Validation of internal control for gene expression study in soybean by quantitative real-time PCR. BMC Mol. Boil. 9, 59 (2008).
Yang, Y. et al. Characterization of reference genes for quantitative real-time PCR analysis in various tissues of Salvia miltiorrhiza. Mol. Boil. Rep. 37, 507–513 (2010).
Lilly, S. T., Drummond, R., Pearson, M. & Macdiarmid, R. Identification and validation of reference genes for normalization of transcripts from virus-infected Arabidopsis thaliana. Mol. Plant Microbe. Interact. 24, 294–304 (2011).
Huang, Y. et al. Stable internal reference genes for normalizing real-time quantitative PCR in Baphicacanthus cusia under hormonal stimuli and UV irradiation and in different plant organs. Front. Plant Sci. 8, 668 (2017).
Li, X. et al. Characterization of reference genes for RT-qPCR in the desert moss Syntrichia caninervis in response to abiotic stress and desiccation/rehydration. Front. Plant Sci. 6, 38 (2015).
Huang, L. et al. Reference gene selection for quantitative real-time reverse-transcriptase PCR in orchardgrass subjected to various abiotic stresses. Gene 553, 158–165 (2014).
Han, B., Yang, Z., Samma, M. K., Wang, R. & Shen, W. Systematic validation of candidate reference genes for qRT-PCR normalization under iron deficiency in Arabidopsis. BioMetals 26, 403–413 (2013).
Zhu, J. et al. Reference gene selection for quantitative real-time PCR normalization in Caragana intermedia under different abiotic stress conditions. Plos One 8, e53196 (2013).
Zhao, Y. et al. Selection of reference genes for gene expression normalization in Peucedanum praeruptorum dunn under abiotic stresses hormone treatments and different tissues. Plos One 11, e0152356 (2016).
Zhao, Y. et al. ABA receptor PYL9 promotes drought resistance and leaf senescence. Proc. Natl. Acad. Sci. USA 113, 1949–1954 (2016).
Chen, Y. et al. Selection and validation of reference genes for target gene analysis with quantitative RT-PCR in leaves and roots of bermudagrass under four different abiotic stresses. Physiol. Plant 155, 138–148 (2014).
Chen, Y. et al. Selection of reference genes for quantitative real-time PCR normalization in creeping bentgrass involved in four abiotic stresses. Plant Cell Rep. 34, 1825–1834 (2015).
Wang, H. et al. Identification and validation of reference genes for Populus euphratica gene expression analysis during abiotic stresses by quantitative real-time PCR. Physiol. Plantarum 152, 529–545 (2014).
Yu, L. et al. Phosphatidic acid mediates salt stress response by regulation of MPK6 in Arabidopsis thaliana. New Phytol. 188, 762–773 (2010).
Ding, Y. et al. OST1 kinase modulates freezing tolerance by enhancing ICE1 stability in Arabidopsis. Dev. Cell 32, 278–289 (2015).
Yang, T., Chaudhuri, S., Yang, L., Du, L. & Poovaiah, B. A calcium/calmodulin-regulated member of the receptor-like kinase family confers cold tolerance in plants. J. Biol. Chem. 285, 7119–7126 (2010).
Mustilli, A.-C., Merlot, S., Vavasseur, A., Fenzi, F. & Giraudat, J. Arabidopsis OST1 protein kinase mediates the regulation of stomatal aperture by abscisic acid and acts upstream of reactive oxygen species production. Plant Cell 14, 3089–3099 (2002).
Qiu, Q., Guo, Y., Dietrich, M. A., Schumaker, K. S. & Zhu, J. Regulation of SOS1, a plasma membrane Na /H exchanger in Arabidopsis thaliana, by SOS2 and SOS3. Proc. Natl. Acad. Sci. USA 99, 8436–8441 (2002).
Evans, M. J., Choi, W.-G., Gilroy, S. & Morris, R. J. A ROS-assisted calcium wave dependent on the AtRBOHD NADPH oxidase and TPC1 cation channel propagates the systemic response to salt stress. Plant Physiol. 171, 1771–1784 (2016).
Bandurska, H. et al. Regulation of proline biosynthesis and resistance to drought stress in two barley (Hordeum vulgare L.) genotypes of different origin. Plant Physiol. Bioch. 118, 427–437 (2017).
Rushton, P. J., Somssich, I. E., Ringler, P. & Shen, Q. J. WRKY transcription factors. Trends Plant Sci. 15, 247–258 (2010).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 (2009).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 25, 402–408 (2001).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 31800272) and grant from Jiangsu Key Laboratory for the Research and Utilization of Plant Resources (JSPKLB201812).
Author information
Authors and Affiliations
Contributions
L.L. and Z.Y.F. designed the experiments. L.L. performed the experiments and analysed the data. L.N.W., F.H.L. and Q.X.U. provided the materials and technical supports. L.L. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Li, N., Fang, H. et al. Selection and Validation of Reference Genes for Normalisation of Gene Expression in Glehnia littoralis. Sci Rep 10, 7374 (2020). https://doi.org/10.1038/s41598-020-63917-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-63917-5
This article is cited by
-
Autophagy formation, microtubule disorientation, and alteration of ATG8 and tubulin gene expression under simulated microgravity in Arabidopsis thaliana
npj Microgravity (2024)
-
Sporogenesis, gametophyte development and embryogenesis in Glehnia littoralis
BMC Plant Biology (2023)
-
Screening and validating of endogenous reference genes in Chlorella sp. TLD 6B under abiotic stress
Scientific Reports (2023)
-
Selection and validation of reliable reference genes for quantitative real-time PCR in Barnyard millet (Echinochloa spp.) under varied abiotic stress conditions
Scientific Reports (2023)
-
Selection and validation of appropriate reference genes for RT–qPCR analysis of Nitraria sibirica under various abiotic stresses
BMC Plant Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
