
Abstract
Fungal virulence is regulated by a tight interplay of transcriptional control and chromatin remodelling. Despite compelling evidence that lysine acetylation modulates virulence of pathogenic fungi such as Candida albicans, the underlying mechanisms have remained largely unexplored. We report here that Gcn5, a paradigm lysyl-acetyl transferase (KAT) modifying both histone and non-histone targets, controls fungal morphogenesis – a key virulence factor of C. albicans. Our data show that genetic removal of GCN5 abrogates fungal virulence in mice, suggesting strongly diminished fungal fitness in vivo. This may at least in part arise from increased susceptibility to killing by macrophages, as well as by other phagocytes such as neutrophils or monocytes. Loss of GCN5 also causes hypersensitivity to the fungicidal drug caspofungin. Caspofungin hypersusceptibility requires the master regulator Efg1, working in concert with Gcn5. Moreover, Gcn5 regulates multiple independent pathways, including adhesion, cell wall-mediated MAP kinase signaling, hypersensitivity to host-derived oxidative stress, and regulation of the Fks1 glucan synthase, all of which play critical roles in virulence and antifungal susceptibility. Hence, Gcn5 regulates fungal virulence through multiple mechanisms, suggesting that specific inhibition of Gcn5 could offer new therapeutic strategies to combat invasive fungal infections.
Similar content being viewed by others


Introduction
Invasive fungal infections claim about 1.5 million lives each year1. Candida species (spp) rank among the top three for four causes of nosocomial infectious diseases2,3. While Candida spp are typically normal commensal colonizers of mucosal barriers in healthy individuals4,5, they can cause life-threatening invasive infections in intensive care unit patients, as well as those with impaired immune defence such as particular neutropenia6,7,8. Invasive candidemia is associated with high mortalities of 35–55%7,9 and accounts for up to 10% of nosocomial blood stream infections (BSIs)10. Limited therapeutic options to treat invasive fungal infections, and increased emergence of antifungal drug resistance in related species such as Candida glabrata and Candida auris11,12,13 pose a significant and growing healthcare problem14,15.
Pathogenicity mechanisms of C. albicans include adherence as biofilms, morphogenetic switching, tissue tropism, secretion of hydrolases and metabolic adaptation as well as chromatin remodelling16,17,18. For example, the fungal cell wall, a prime antifungal target19,20, is home to many adhesins and undergoes dynamic remodelling during host stress or immune response to evade detection21,22,23. Moreover, three major MAPK signaling pathways, the Mkc1-mediated cell integrity pathway, the Hog1-dependent high osmolarity pathway and the Cek1-mediated invasion and filamentation pathway, respond to environmental stimuli and thus cooperate in regulating C. albicans virulence24,25,26,27,28. Signaling pathways converge at dedicated downstream transcriptional regulators such as Efg1 and Cph1 and others that control signaling integration to regulate morphogenesis, virulence but also immune evasion29,30,31,32.
Interestingly, most if not all fungal virulence traits are tightly controlled by a dual-layer network that engages transcriptional regulatory networks, whose activity is modulated by specific histone modification enzymes that alter chromatin states. For example, genetic ablation of lysine acetyltransferases and lysine deacetylases (KATs/KDACs) Set3C, Rpd3, Rp31, Hat1, Hst3 and Rtt109 abolishes fungal virulence33,34,35,36,37,38,39. Indeed, KATs and KDACs cooperate with transcriptional regulators in the control of fungal virulence18,40,41 but the molecular mechanisms underlying KATs/KDACs function in fungal pathogenesis remain poorly understood. However, several fungal-specific lysine modifications indicate a potential as valuable therapeutic targets with minimal toxic side effects40,42,43.
As a hallmark fungal lysyl acetyltransferase, Gcn5 (general control nonderepressible-5), is a paradigm KAT and member of the evolutionary conserved Gcn5-related N-acetyltransferase family (GNATs). Yeast Gcn5 is part of large transcriptional multiprotein complexes, including SAGA (Spt-Ada-Gcn5 acetyltransferase), ADA (Ada2-Gcn5-Ada3), HAT-A2 and SLIK (SAGA-like). These evolutionary conserved regulatory complexes recruit the basal transcription machinery and coactivators to specific promoters, control chromatin modification and nucleosome remodelling, as well as retrograde signaling44,45,46,47. For example, Gcn5 is essential for stress response both in fission yeast Schizosaccharomyces pombe and budding yeast Saccharomyces cerevisiae48,49,50. Further, the UmGcn5 homologue in the maize pathogen Ustilago maydis participates in the epigenetic regulation of morphogenesis and pathogenesis51,52. The Aspergillus nidulans GcnE homologue is crucial for inducing genes responsible for conidiation and conidiophore development53. Interestingly, CnGcn5 in Cryptococcus neoformans, the primary cause of meningoencephalitis with highest frequencies in HIV patients, controls stress responses and attenuates virulence54. Gcn5-mediated histone H3 acetylation is required for conidiation, dimorphic transition and virulence of entomopathogenic fungi Beauveria bassiana55. Candida albicans Gcn5 also attenuates pathogenicity and affects morphogenesis56, but the mechanisms of Gcn5-mediated gene regulation, and more importantly, how Gcn5 controls fungal pathogenicity remains largely unknown. Here, we show that Gcn5 controls C. albicans invasive infections by acting downstream of multiple signaling pathways that control cell wall architecture and surface remodeling. Importantly, Gcn5 critically determines susceptibility to killing by innate immune cells, as well as to the fungicidal action by caspofungin. The data establish Gcn5 as drug target which may be suitable for interfering with invasive fungal infections.
Results
Genetic ablation of the Gcn5 histone acetyltransferase impairs filamentation
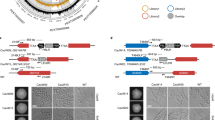
First, we asked if the type A KAT, Gcn5, is involved in fungal morphogenesis. We created homozygous deletion (gcn5∆/∆) mutants using the recyclable NAT1 flipper method57 in a SC5314 wild type (wt) strain background, including a restored gcn5∆/∆::GCN5 strain. While this work was in progress, Chang et al. reported that Gcn5 is required for the invasive growth, hyphal elongation in hyphal-inducing conditions56. Consistent with this notion, we found that lack of Gcn5 led to a smooth colony morphology on filament-inducing media, indicating a severe morphogenesis defect when compared to wild type control and the reconstituted gcn5∆/∆::GCN5 strain (Fig. 1A). Mutant cells also displayed a morphology defect as indicated by the pseudo-hyphal morphology on complete YPD medium, and by the aberrant chitin deposition as visualized by calcofluor white (CFW) staining (Fig. 1B). In full agreement with a previous report56, we show that filamentation of gcn5∆/∆ cells was severely impaired, since true hyphae were absent. Moreover, daughter cells often remained attached to mother cells, indicating a separation defect (Fig. 1C).

Lack of GCN5 impairs bud separation, hyphae formation and agar invasion. (A) Logarithmically growing cells of SC5314 wild-type (wt), homozygous deletion (gcn5Δ/Δ) and restored (gcn5Δ/Δ::GCN5) Candida strains were plated on YPD plates supplemented with 10% fetal calf serum. Colony morphology was analysed after incubating for 3 days at 37 °C. Photographs were taken using a Discovery V12 Stereoscope equipped with an Axiocam MR5 camera (Zeiss). Scale bar = 1 mm. (B) Representative confocal microscopy image showing fungal cells stained with calcofluor white (CFW) to detect cell wall chitin. Cells were fixed in 4% p-formaldehyde for 2 hours, washed and stained with CFW 1 mg/ml for 5 min. Differential Interference Contrast (DIC) and UV light images (UV) of the same cells are shown at 60x magnification. Scale bar = 5 µM. (C). Representative confocal DIC images showing the hyphal morphology of strains at indicated temperatures. Logarithmically growing cells in YPD supplemented with 10% FCS were fixed in 4% p-formaldehyde for 2 hours and washed and images were taken with an LSM 700 Zeiss Confocal microscope at 60x magnification. Scale bar = 5 µM.
Since morphological defects often associate with altered drug susceptibilities, we also tested cell growth on solid and liquid media supplemented with various stress agents including caffeine, the cell wall stressor SDS and Congo Red, and specific antifungal agents, as well as different carbon sources (Supplementary Fig. S1). Comprehensive phenotyping showed an altered sensitivity to several stress conditions, including increased susceptibility to antifungal agent caspofungin (CSP), albeit azole sensitivity was not significantly altered (Supplementary Fig. S1A,B). The gcn5∆/∆ mutant showed slightly increased resistance to peroxide stress (Supplementary Fig. S1B). Cells lacking GCN5 showed growth defects in media containing citric acid, ethanol and sodium acetate used as sole carbon sources (Supplementary Fig. S1C,D), but consumed glucose and glycerol like wild type cells. The elevated sensitivity to CSP and SDS was also confirmed by MIC50 assays in liquid media, showing that SDS and CSP sensitivities were 12 and ~3-fold elevated in gcn5∆/∆, respectively, while caffeine sensitivity was essentially unchanged (Supplementary Fig. S1E). Taken together, these data suggest that Gcn5 is required for normal morphogenesis and cell separation, stress adaptation, carbon source utilization, as well as antifungal susceptibility and cell wall homeostasis.
Lack of Gcn5 differentially regulates MAPK signaling
External stimuli trigger MAPK signaling pathways to transmit signals to dedicated downstream transcription factors that control morphogenesis24,58,59. To test how Gcn5 affects MAPK signaling, we immunoblotted for both activated and total levels of Mkc1, Cek1 and Hog1 (Fig. 2). Interestingly, activated Mkc1-P were 2-fold lower, whereas Cek1-P and Hog1-P showed 5-fold higher basal levels in gcn5∆/∆ mutant cells when compared to the total non-phosphorylated Mkc1, Cek1 and Hog1 controls (Fig. 2A,B), which remained unchanged in gcn5∆/∆ cells (Fig. 2A). No significant changes were observed in the restored gcn5∆/∆::GCN5 strain (Fig. 2A,B). This differential activation of Mkc1, Cek1 and Hog1 in gcn5∆/∆ mutants implies a possible cross-talk between these MAPKs pathways that may, at least in part, account for altered cell wall phenotypes.

Lack of Gcn5 differentially regulates MAPK signaling and cell wall components. (A) Logarithmically growing cultures of SC5314 wild-type (wt), homozygous deletion (gcn5Δ/Δ) and restored (gcn5Δ/Δ::GCN5) Candida strains were used to prepare cell free extracts using the TCA protocol as described in materials and methods. Extracts corresponding to 1 OD600 (1 × 107 cells) were fractionated by SDS-Page and blotted for proteins as indicated. Signals from the same whole cell extracts were detected using antibodies for total and phosphorylated MAP kinases. The commercial antibodies recognized Mkc1 and Cek1 (p44/42 MAPK Erk1/2, Cell Signaling), and Hog1 (y-215, Santa Cruz), and phosphorylated Mkc1-P and Cek1-P (Phospho-p44/42 MAPK (Erk1/2), Cell Signaling) and Hog1-P (Phospho-p38, Cell Signaling). Reprobing with PSTAIR antibody (Sigma) recognizing Cdc28 served as a loading control. (B) Densitometry analysis was done by using image studio software (LI-COR). Data are expressed as fold-change normalized to the PSTAIR (Cdc28) loading control from three independent biological samples (±SEM, **p ≤ 0.005, ***p ≤ 0.0005 ****p < 0.0001). (C) Flow cytometry-based quantification of cell wall components in Candida albicans. Logarithmically growing cultures of SC5314 wild-type (wt), homozygous deletion (gcn5Δ/Δ) and restored (gcn5Δ/Δ::GCN5) Candida strains were washed and triple-stained to decorate cell wall components before quantification of β-D-glucan (FITC), chitin (BV421) and mannan (Texas Red) in suitable laser Channels. Data represents the mean fluorescence intensity (±SEM, *p < 0.05, **p < 0.01) from three independent experiments. (D) For qPCR quantification, RNA was extracted using the Trizol extraction method from wt, gcn5Δ/Δ and gcn5Δ/Δ::GCN5 cultures as described in materials and methods. FKS1, FKS2 and FKS3 transcript levels were measured. Gene associated with Topoisomerase II (PAT1) mRNA was used as a normalization control and data represent mean relative expression to PAT1 from three t < six different experiments (±SEM, *p < 0.05, **p < 0.01, ***p<0.0005).
The fungicidal caspofungin (CSP) inhibits the Fks1-dependent synthesis and deposition of β-1, 3-D-glucan into the cell wall14. Notably, CSP treatment also elevates intracellular reactive oxygen species (ROS) and increases DNA damage. Vitamin C is an antioxidant that can quench ROS and protect cells from oxidative damage39. However, caspofungin hypersusceptibility was not due to elevated intracellular ROS, since gcn5∆/∆ cells actually produced less ROS upon caspofungin (Supplementary Fig. S2A,B).
GCN5 deletion alters cell surface components and FKS expression
We next analysed in more detail the effect of Gcn5 loss on cell wall architecture by quantifying all cell wall components (chitin, β1,3-glucan, branched β1,6-glucan and mannoproteins) using a FACS-based triple staining assay60. Both β-D-glucan and chitin levels were about 2-fold elevated in gcn5∆/∆ cells when compared to the wild type control or the restored strain. Outer mannoprotein levels were unchanged, indicating that gcn5∆/∆ causes specific cell wall defects rather than gross effects (Fig. 2C). We hypothesized that the increased β-D-glucan content in the gcn5∆/∆ mutant may be due to increased expression of glucan synthases such as FKS, which synthesises β-1, 3-D-glucan. To test this, we quantified mRNA expression levels of all three FKS genes, FKS1, FKS2 and FKS3, encoded in the C. albicans genome. Unexpectedly, we found that expression of the CSP target Fks1 was significantly reduced upon removal of Gcn5, whereas both FKS2 and FKS3 were significantly upregulated (Fig. 2D). This implies a compensatory inverse regulation of FKS genes caused by the loss of Gcn5. Thus, we believe that the increased expression of FKS1 upon CSP treatment in wt cells explains the CSP tolerance, which in turn is impaired when Gcn5 is ablated. In other words, the FKS2/3-triggered compensatory increase in glucan is not sufficient to restore CSP tolerance when GCN5 is deleted. We cannot exclude a distinct glucan distribution or an aberrant integration of newly synthesized glucan into the functional cell wall architecture.
To test whether loss of Gcn5 alters cell membrane permeability, which may explain the altered drug sensitivity, we measured the kinetics of fluorescein diacetate (FDA) uptake, whose linear uptake is solely a function of lipid membrane permeability61 and thus independent of transporters (Supplementary Fig. S2C). Indeed, CSP increased FDA uptake in the wild type and restored strains, whereas basal FDA uptake in gcn5∆/∆ cells was already higher, indicating altered membrane lipid permeability that was not further increased by CSP (Supplementary Fig. S2C). To conclude, these results show that genetic ablation of GCN5 alters the cell wall composition, FKS expression, and membrane lipid permeability.
Transcriptional response of wild type and gcn5Δ/Δ cells upon caspofungin stress
To dissect the Gcn5-dependent regulatory networks that may be underlying the observed phenotypes, we performed RNA-seq of gcn5∆/∆ cells and wild type in the presence and absence of CSP (Fig. 3). Growth curves and CSP sensitivity in liquid culture suggested a concentration of 10 ng/ml as appropriate, since this concentration exerted significant toxicity but still allowed for growth of all cells (Fig. 3A). The gcn5∆/∆ mutant showed sensitivity to 10 ng/ml CSP but retained more than 85% viability after 45 min but not at 60 min, where 30% cells were inviable (Fig. 3A; data not shown). Hence, we considered the time points 15- and 45-min post CSP treatment as optimal for RNA-seq. Differentially regulated genes were included when expression experienced a ≥ or ≤log21.5-fold change, with an adjusted P-value cut-off ≤ 0.05 (Fig. 3, Supplementary Table S4).

Transcriptional response of wt and gcn5Δ/Δ cells upon caspofungin stress. Total RNA was isolated from cultures of SC5314 wild-type (wt) and homozygous deletion strain (gcn5Δ/Δ) grown in the absence or presence of caspofungin and subjected to RNA-seq analysis. The bioinformatics pipeline for data analysis and hierarchial clustering is described in materials and methods. (A) Genetic removal of GCN5 increases caspofungin susceptibility. Growth of indicated strains in YPD containing or lacking 10 ng/ml caspofungin (CSP10) at 30 °C. Data represent the means from three independent growth analyses (±SEM). Solid and dotted lines indicate untreated and caspofungin-treated cultures, respectively. (B) Heat map of hierarchical clustered and differentially expressed genes in wt and gcn5Δ/Δ cells in response to caspofungin treatment (CSP). Lanes 1 and 2 compare 15 min and 45 min CSP-treated wt cultures with untreated YPD-grown cells, respectively. Lanes 3 and 4 compare 15 min and 45 min CSP-treated gcn5Δ/Δ cultures with untreated YPD-grown cells, respectively. Lane 5 compares gcn5Δ/Δ with wt cells. Lanes 6 and 7 compare 15 min and 45 min CSP-treated gcn5Δ/Δ cultures with CSP-treated wt cells. Log-scaled expression values are colour-coded according to the legend on the bottom. (C) Venn diagrams depicting the overlap between up- (upper panel) and down-regulated (lower panel) genes after 0 min, 15 min and 45 min CSP-treatment of gcn5Δ/Δ cells compared to respective CSP-treated wt cells.
We first compared transcript levels of YPD-grown gcn5∆/∆ to YPD-grown wild type cells and found 336 differentially regulated genes (Fig. 3B, lane 5). Out of these, 235 genes were induced, and 101 genes were repressed (Fig. 3C). The distribution of genes responding to GCN5 deletion classified according to gene ontology (GO)-slim biological processes included major-enriched (cell adhesion, oxidation-reduction, transmembrane transport and pathogenesis) and minor-enriched (arginine biosynthesis, ergosterol and hydrogen sulphide biosynthesis, heme transport and zinc sequestration) as depicted in Supplementary Fig. S3A. Notably, genes implicated in pathogenesis were exclusively induced, except for the alpha-1,3-mannosyltransferase MNN14. Three-quarters of the differentially regulated genes were up-regulated in gcn5∆/∆ cells, implying that Gcn5 target genes are primarily negatively regulated. Together, our genome-wide analysis of gcn5∆/∆ cells suggests that GCN5 deletion changes the landscape of several C. albicans transcriptional networks by regulating various target gene sets.
To determine whether alterations in the transcriptome potentially underlies the increased antifungal susceptibility of gcn5∆/∆ cells, we compared the effect of 15 and 45 min CSP treatment on the global transcriptomes of gcn5∆/∆ cells versus wild type cells (Fig. 3B, lanes 6 and 7). The Venn diagrams include the genes regulated at all time points from 0 min (untreated),15 min to 45 min CSP-treated in gcn5∆/∆ compared to wild type cells (Fig. 3C). We found that 15 min and 45 min CSP treatment showed approximately 70% and 50% overlap of induced (135 genes) and repressed (46 genes) genes, respectively (Fig. 3C). We also combined both 15 min and 45 min CSP-treated datasets and found that 154 genes were induced by CSP in both the gcn5∆/∆ and wild type cells. Some 81 genes were induced specifically in the absence of GCN5, while 115 genes were induced specifically upon CSP stress (Fig. 3C, upper panel). Surprisingly, given the differential sensitivities of the wild type and gcn5∆/∆ mutant to CSP, the CSP-mediated transcriptional response in wild type (Fig. 3B, lanes 1 and 2) and gcn5∆/∆ mutants (Fig. 3B, lanes 3 and 4) after 15 min and 45 min) revealed a striking overlap (Fig. 3B, lanes 1–4). The major biological processes differentially affected by the loss of Gcn5 included cell adhesion, oxidation-reduction, transmembrane transport, pathogenesis, lipid catabolism and iron homeostasis (Supplementary Fig. S3B). The inability of gcn5∆/∆ cells to fully induce the normal CSP response explains the increased antifungal susceptibility.
Lack of GCN5 affects oxidation-reduction, transport, pathogenesis and ion homeostasis
To validate key predictions of the transcriptomics datasets, we employed quantitative real-time RT-PCR on hallmark genes known to be implicated in biological processes putatively affected by loss of Gcn5 (Supplementary Fig. S3A,B). Expression of at least three genes in each biological process was validated. The superoxide dismutase genes SOD4 and SOD5, and the ferric reductase CFL2 were previously implicated in oxidative stress resistance, as well as in the yeast-to-hyphae transition. In addition, SOD5 has been linked to CSP-response62,63,64. Indeed, RT-qPCR analysis revealed approximately 4-fold, 3-fold and 29-fold increased SOD4, SOD5 and CFL2 mRNA levels, respectively, in gcn5∆/∆ cells when compared to the wild type (Fig. 4A). However, lack of GCN5, significantly elevated CFL2 levels upon short-term treatment (CSP-15′), with a reduction at CSP-45′. As expected, CSP induced SOD5 expression in wild type cells in a time-dependent manner (Fig. 4A), whereas, gcn5∆/∆ mutants were completely unable to induce SOD5 upon CSP challenge.

Genetic ablation of GCN5 affects oxidation-reduction, transport, pathogenesis, iron homeostasis and adhesion genes. Quantitative real-time RT-PCR analysis of differentially expressed genes in YPD-grown wt and gcn5Δ/Δ cells measured in the presence or absence of caspofungin after the indicated time points. Data are shown as mean of relative expression to reference gene PAT1 from three independent experiments (±SEM, *p < 0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001). Genes for validation were picked based on RNA-seq data and GO-term classification done by using online bioinformatics tool: Fungifun (https://elbe.hki-jena.de/fungifun/). (A) Genes implicated in oxido-reduction processes. (B) Genes implicated in fungal pathogenesis. (C) Genes implicated in transport and antifungal drug resistance. (D) Genes implicated in iron homeostasis.
Next, we checked the expression levels of HYR1, which encodes a GPI-anchored cell wall protein involved in hyphal regulation through the MAPK signaling genes CEK1 and CEK2. These genes are required for the pathogenesis and cell wall-mediated invasion and pheromone responsive MAPK cascade, respectively65,66. Loss of GCN5 induced approximately 4-fold, 1.5-fold and 8-fold of HYR1, CEK1 and CEK2 mRNAs, respectively. Prolonged CSP treatment slightly increased CEK1 gene expression, contrary to the expression of HYR1 and CEK2 genes that were slightly repressed. 45 min CSP treatment induced CEK1 expression by about ~3-fold (Fig. 4B). These results indicate that Gcn5 negatively regulates Cek1/Cek2 and Hyr1. Additional hyphal regulators include EFG1, NRG1, TUP1, TEC1, and hyphae-associated genes showing a predominant expression in filaments include HWP1, ECE1 and RBT529,31,67. Of note, these genes are also involved in biofilm formation and some are essential for fungal virulence31,66,67,68,69. Since, GCN5 strongly affected hyphal formation (Fig. 1A,C), we tested whether additional hyphal regulators are also regulated by GCN5. Ablation of GCN5 showed a ~5-fold and 2-fold basal upregulation of HWP1 and ECE1 transcripts, respectively. In contrast, TEC1 mRNA was reduced ~2-fold when compared to wild type cells. Furthermore, expression of the repressor genes NRG1 and TUP1 were unaffected by the lack of Gcn5 (Supplementary Fig. S4).
Next, we quantified expression of the zinc permease Zrt2 and genes encoding multidrug resistance transporters such as the Mdr1 major facilitator and the ATP-binding cassette transporter Cdr4. In wild type cells, CSP induced MDR1 expression, but significantly reduced expression of CDR4 and ZRT2. Lack of GCN5 rendered cells unable to drive CSP-responsive MDR1 expression and reduced basal levels of CDR4 and ZRT2 (Fig. 4C). In addition, we quantified expression of iron homeostasis genes. The high-affinity iron permease genes FTR1 and FTR2 were differentially regulated upon loss of GCN5 and CSP treatment, with 10-fold up and 6-fold down-regulated mRNAs in gcn5∆/∆ cells, respectively. Notably, expression of FTR1 and FTR2 encoding iron permeases was compromised upon CSP treatment in gcn5∆/∆ cells but induced in wild type in a time-dependent manner (Fig. 4D). Interestingly, gcn5∆/∆ cells showed 13-fold higher basal expression of the cell wall protein RBT5, with ~3-fold further upregulation upon CSP treatment (CSP-15′), whereas prolonged exposure significantly diminished RBT5 expression (Fig. 4D). Taken together, removal of GCN5 differentially regulates genes involved in oxido-reduction, ABC and MFS drug transporters, filamentation regulators, MAPK signaling modules, as well as iron / zinc homeostasis and heme acquisition, which requires Rbt5 and affects cell wall architecture by regulating its proteome including adhesins70,71,72,73,74.
GCN5 regulates cell adhesion through Efg1
The bHLH transcription factor Efg1 integrates signals through upstream cAMP-PKA pathways31,35,75 to control morphogenesis, CSP tolerance and adhesion76. Thus, we asked whether lack of GCN5 affects Efg1 expression. We noticed a significant reduction in EFG1 mRNA in gcn5∆/∆ cells when compared to wild type cells, which was not seen in restored strains (Fig. 5A), and confirmed the RNA-seq data (Fig. 3). Since Efg1 strongly impacted adhesion, we also quantified adherence properties by testing binding to polystyrene-coated plastic surfaces using the crystal violet readout. Indeed, cells lacking GCN5 showed drastically attenuated adherence to polystyrene surfaces, which was not seen in the restored strain (Fig. 5B).

Loss of GCN5 affects Efg1-mediated adhesion. Time course of qPCR-based quantification of mRNA levels in logarithmically growing cultures of SC5314 wild-type (wt), homozygous deletion (gcn5Δ/Δ) or restored (gcn5Δ/Δ::GCN5) Candida strains in the absence or presence of caspofungin. Data are shown as mean of relative expression to the reference gene PAT1 from three independent experiments (±SEM, *p < 0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001). (A) EFG1 expression in wt, gcn5Δ/Δ and gcn5Δ/Δ::GCN5 cells. (B) Adherence on polystyrene-coated plates was measured via crystal violet staining of wt, gcn5Δ/Δ and gcn5Δ/Δ::GCN5 cells, followed by the absorbance decay at 570 nm in the destaining solution containing 95% ethanol. Data are expressed as mean of crystal violet staining OD600 units from biological triplicates (±SEM, **p < 0.005). (C) Regulation of ALS1 expression after caspofungin treatment in the absence of GCN5 was measured as mentioned above. (D) Transcript levels of FKS1 after caspofungin treatment.
Efg1-mediated adhesion primarily engages the ALS agglutinin adhesion family76. Given that loss of GCN5 increases susceptibility to CSP and decreases Efg1-mediated cell adhesion family, we tested the effect of CSP on Als1 expression (Fig. 5C). Wild type cells dramatically elevated Als1 levels upon CSP treatment, but the upregulation of ALS1 was completely abolished in cells lacking Gcn5 (Fig. 5C). To reveal a potential role of other adhesins, we tested basal expression of all GPI-anchored ALS genes encoded in the Candida genome in wild type and gcn5∆/∆ mutants (Supplementary Fig. S5). Interestingly, like ALS1, ALS2/3/4 were downregulated in gcn5∆/∆ cells, whereas ALS5/6/7/9 mRNAs were significantly upregulated (Supplementary Fig. S5). Although the function of most ALS genes is unknown, the Gcn5-dependent dysregulation of well-established adhesins Als1 and Als3 explains the adhesion defects (Fig. 5C, Supplementary Fig. S5).
CSP inhibits β-1,3-D-glucan deposition by Fks1, whose overexpression confers resistance to CSP77. Wild type cells displayed significantly increased FKS1 expression after 15 and 45 min CSP treatment. Conversely, FKS1 mRNA was reduced in gcn5∆/∆ cells, and CSP treatment no longer increased FKS1 mRNA levels (Fig. 5D), whereas both FKS2 and FKS3 were significantly upregulated (Fig. 2D). These data explain the CSP hyper-susceptibility and suggest a pivotal role of Gcn5 in regulating glucan deposition and cell wall function, including a coordinated action in concert with EFG1-mediated adhesion.
Gcn5 is required for intracellular survival in BMDMs and essential for fungal virulence
Several phenotypes adopted by gcn5∆/∆ cells implied that Gcn5 may be involved in fungal virulence, as suggested by a previous report56. Thus, we investigated the role of GCN5 in virulence in vivo using mouse models, and quantified the susceptibility of fungal cells to killing by innate immune cells. We used a murine Candia infection model and injected 1 × 105 Candida cells through the lateral tail vein78. Mice infected with gcn5∆/∆ cells or mice receiving only PBS showed no weight loss. By contrast, mice receiving wild type or restored Candida strains lost approximately 20% of their weight over seven days (Fig. 6A). Remarkably, wild type and GCN5 reconstituted cells efficiently killed animals, whereas gcn5∆/∆ cells were avirulent with all mice being able to clear the systemic infections (Fig. 6B).

Lack of GCN5 impairs survival inside BMDMs and leads to avirulence in mice. (A,B) Equal number of cells of logarithmically growing SC5314 wild-type (wt), homozygous deletion (gcn5Δ/Δ) and restored (gcn5Δ/Δ::GCN5) Candida strains were tail vein-injected (1 × 105 cells/21 g mouse body weight). Mice were continuously monitored and weight loss was recorded as described in materials and methods. For survival experiments, mice were monitored for 15 days. A group five mice were used for each strain and log-rank test and the Kaplan Meier survival method was applied (*p < 0.05). (C) Logarithmically growing cells of SC5314 wild-type (wt), homozygous deletion (gcn5Δ/Δ) and restored (gcn5Δ/Δ::GCN5) Candida strains were used for infecting primary bone marrow-derived macrophages (BMDMs) at a multiplicity of infection (MOI) of 10:1 (fungi to macrophages). Fungal cells were harvested 2 h and 4 h post infection and viability quantified by cfu-counting on YPD plates as described in materials & methods to calculate the percentage of survival after 48 h incubation. Data represent the mean of three independent experiments (±SEM, *p < 0.05). (D) Confocal microscopy images of Alexafluor 488-labeled SC5314 wild-type (wt), homozygous deletion (gcn5Δ/Δ) or restored (gcn5Δ/Δ::GCN5) Candida strains infecting primary bone-marrow derived macrophages (BMDMs) at an MOI of 5:1 (fungi to macrophages). At 45 minutes post infection, cells were stained with Lyso Tracker Red DND-99 to visualize the phagolysosome, then fixed and further stained with DAPI to visualize the nucleus. Images shown are representative overlays of all four channels red, blue, grey and green to indicate staining of lysosomes, nucleus, Differential Interference Contrast (DIC) and Candida albicans, respectively. Scale bar = 5 µM. (E) Primary BMDMs were infected with Alexafluor 488-labeled wt, gcn5Δ/Δ and gcn5Δ/Δ::GCN5 cells at an MOI of 2:1 (fungi to macrophages). At 45 minutes post infection, fluorescence of extracellular or adherent Candida was quenched by 0.4% trypan blue. After washing, intracellular phagocytosed Candida cells were quantified by flow cytometry. Data represent the mean of percent of phagocytosis (±SEM, **p < 0.005) from three independent assays. Representative overlay histogram to demonstrate the shift in fluorescence of gcn5Δ/Δ-infected BMDMs (red colour histogram). The mean fluorescence intensity of macrophage- internalized Candida were normalized to Candida alone or outside and represented as percent phagocytosis in a bar diagram shown next to the histogram.
To uncover a possible cellular mechanism, we challenged primary BMDMs with fungal strains in host-pathogen interaction experiments to quantify the killing capacity of macrophages. Macrophages are key components of the innate immune system as they engage in protecting the host by effective oxidative killing of C. albicans. To determine whether GCN5 is required for intracellular survival of fungal cells after phagocytosis, we infected BMDMs with wild type, gcn5∆/∆ and restored strains using an MOI of 10:1 (Candida to BMDMs). Surviving fungal cells were quantified by CFU-counting after 2 h and 4 h of infection. Strikingly, gcn5∆/∆ were much more sensitive to killing by BMDMs when compared to wild type cells or GCN5 reconstituted strains (Fig. 6C). Thus, loss of GCN5 significantly reduced fungal viability inside BMDMs.
The impaired survival in macrophages prompted us to inspect the rate and degree of fungal phagocytosis using both fluorescence microscope and FACS-based detection of macrophage-internalized Alexafluor-488-labelled Candida cells (Fig. 6D,E). BMDMs showed enhanced engulfment and phagocytosis of fungal cells lacking GCN5 when compared to wild type cells (Fig. 6D). These data are also consistent with the result showing abolished induction of SOD5 in cells lacking Gcn5 (Fig. 4A), explaining the increased oxidative killing of fungal cells by BMDMs (Fig. 6E). Taken together, this works establishes that the fungal Gcn5 histone acetyltransferase is essential for fungal virulence and that pathogenicity is regulated through several independent mechanisms, including adhesion, antifungal susceptibility and cell wall architecture and a strikingly increased susceptibility to killing by host-derived oxidative stress. The data establish Gcn5 as a new potential target gene for antifungal drug discovery.
Discussion
The prevalence and mortality of invasive fungal infections has been concerning. Importantly, the dramatic increases for fungal pathogens such as Candida glabrata in the past years, and the staggering increase in multidrug-resistant Candida auris infections, make fungal infections a global healthcare threat1,79. Therapeutic treatment options are limited to a small armory of antifungal drugs, some of which can lead to antifungal resistance or show restricted species-specificity. Hence, there is need to develop new and improved antifungal drugs, including an expansion of available chemical entities that could target new and understudied genes80. Compelling evidence indicates that enzymes that control chromatin modification could constitute a novel family of antifungal target genes, since they participate in numerous pathophysiological processes that regulate virulence traits40. Our work and data reported earlier56, are entirely consistent, and demonstrate that C. albicans Gcn5 is required for the hyphal formation and virulence in murine model of candidiasis. Here, we show that C. albicans Gcn5 controls morphogenesis and susceptibility to killing by phagocytes such as macrophages. Moreover, loss of Gcn5 is accompanied by Efg1-controled susceptibility to fungicidal caspofungin, as well as differentially altered MAPK signaling, which controls cell wall remodeling, adhesion and filamentation.
The Gcn5 histone lysyl acetyltransferaseis an intrinsic component of evolutionary conserved transcriptional co-activator complexes, such as SAGA, ADA and SLIK46, which regulate several fundamental processes81,82,83 in yeast84,85,86 and humans45,46. The knowledge about fungal Gcn5 primarily comes from studies in baker’s yeast50,81,87 and other fungal species88,89. Here, we use global transcriptomics and phenotypic profiling to identify regulatory networks and signaling pathways whose control engages Gcn5 in the human pathogen C. albicans. Although several phenotypes emerging upon deletion of GCN5 in C. albicans phenocopy those seen in yeast84,85,86, our data suggest that Candida spp Gcn5 may regulate additional networks required for virulence.
Gcn5-mediated regulation affects signaling pathways that are essential for controling morphogenetic changes such as filamentation as well as virulence. Hypersensitivities to caspofungin and other stress agents most likely arise as a consequence of altered cell wall composition and dysregulated glucan synthesis enzymes such as Fks1 and Fks2, Fks3. Interestingly, while we observed a strong repressive impact on FKS1 in gcn5∆/∆ cells, compensatory adaptive mechanisms may drive FKS2 and FKS3 upregulation. However, we do not know whether this regulation is a direct or indirect mechanism activated upon loss of Gcn5. While C. glabrata FKS1 and FKS2 are functionally redundant, a recent report suggests that C. albicans FKS2 and FKS3 negatively regulate FKS1, which modulates echinocandin susceptibility90,91. These data suggest severe perturbations of cell wall homeostasis derived from loss of Gcn5 control cell wall architecture and function through dysregulated MAPK signaling92. The Hog1 and Cek1 pathways engage in molecular cross-talk24 and lack of Gcn5 indeed affects both pathways C. albicans.
Moreover, Gcn5 affects the synthesis of cell wall glucan and chitin through the Mkc1 cell integrity pathway27. Hence, our data suggest that Gcn5 differentially regulates MAPK signaling events, thereby altering cell surface composition. Indeed, the cell wall superoxide dismutase Sod5 is dramatically downregulated upon Gcn5 loss. Moreover, Gcn5 loss has a severe impact on adhesion as indicated by the dysregulation of the major Als1 adhesin, and the inability to induce Fks1 upon caspofungin stress. This dual regulation perhaps creates synthetic lethality under stress conditions, which could explain the attenuated virulence and dramatically impaired fitness in the host.
The prevailing notion that KATs in general, and Gcn5 specifically, functions as classical co-activators must be viewed with caution, since our RNA-seq data demonstrate that Gcn5, and possibly other Candida KATs, might also act as co-repressors, given the large number of about 235 genes upregulated after loss of Gcn5. For example, the elevated glucan and chitin contents imply a possible direct or indirect repressor function of Gcn5 by directly or indirectly modulating a positive transcriptional regulator required for cell wall homeostasis or adhesion. Further, the activation of both Cek1 and Hog1 MAPK pathways in the absence of stress may arise from constitutive cell wall alterations triggered by ectopic signaling. We speculate that Gcn5 alters chromatin remodelling in regulatory networks through its ability to acetylate histone H3 lysines (K14, K9, K18, K23, K27 and K36), as well as histone H4 and H2B93 when cells encounter stress or immune defense. However, it is tempting to further speculate that Gcn5 could also acetylate non-histone proteins, including signaling components of MAPK pathways or dedicated transcription factors to regulate their activity. Alternatively, Gcn5 could control the chromatin landscapes in promoters in virulence genes in cells that encounter active host immune defense63,94. Indeed, our initial characterization of Gcn5-associated proteins by native co-immunoprecipitation, and the identification of the Gcn5-dependent acetylome, suggests that Gcn5 interacts and acetylates several hundred target proteins, including numerous components of the ADA, SLIK, SAGA transcriptional regulatory complexes (Shivarathri et al., unpublished data). The RNA-seq data also reveal that Gcn5 modulates additional regulatory networks governing ergosterol lipid biosynthesis, oxido-reduction and cell adhesion, ABC and MFS transporter expression and antifungal resistance95, as well as iron homeostasis as in C. glabrata96. Interestingly, iron homeostasis is tightly linked to cell wall architecture, and Gcn5 regulates expression of Rbt5 that affects cell wall architecture by regulating adhesins and the entire surface proteome70,71,72,73,74. In addition, Gcn5 selectively controls hyphal regulator genes such as EFG1, TEC1, but also hyphal genes, including HWP1 and ECE1. Of note, expression of NRG1 and TUP1 encoding hyphal repressors was not affected, implying that Gcn5 regulates hyphal elongation and filamentation primarily through Efg1-mediated control. In addition, deletion of GCN5 considerably diminishes expression of ergosterol biosynthesis genes, including ERG3, ERG250, ERG11, ERG13 and the binuclear Zn2-Cys6 transcription factor UPC297,98 involved in the sterol uptake in yeast and Candida97. Hence, Upc2 may sense altered membrane lipid permeability, which is also linked to the overexpression of membrane transporters99,100,101,102. Indeed, gcn5 mutants show changes in non-protein-mediated membrane lipid permeability and upregulation of the Cdr1 efflux pump103. This may explain, at least in part, UPC2-mediated alterations in antifungal drug susceptibilities emerging upon GCN5 deletion97.
Moreover, ablation of Gcn5, strongly attenuates adhesion104, which requires Efg1 and determines immune cell interactions16,105. This is in line with previous reports showing that Efg1 modulates CSP tolerance through activating the major GPI-anchored cell surface adhesion protein Als176. The Gcn5-mediated integration of filamentation signals also engages downstream transcription factors such as Efg1 and Tec129,106,107,108 as both EFG1 and TEC1 expression is impaired in gcn5 cells, thereby dysregulating Als1-mediated adhesion in gcn5∆/∆ cells facing CSP challenge. Of note, both ALS1 and ALS3 are also critically involved in fungal biofilm formation109, but a role for Gcn5 in biofilm control has not been tested.
Importantly, lack of Gcn5 significantly increases the susceptibility to killing by macrophages, because gcn5∆/∆ cells suffer from reduced fitness inside macrophages. The increased phagocytosis and or killing enables macrophages increased clearance of gcn5∆/∆ cells. This can explain for the most part the striking avirulence phenotype observed in a mouse model of systemic candidiasis, but additional unexplored mechanisms may also affect fitness of gcn5∆/∆ mutants. Of note, killing of cells lacking Gcn5 by dendritic cells is not affected, which may relate to their function as key antigen-presenting cells initiating adaptive immune responses110. The enhanced phagocytosis for gcn5∆/∆ may be due to increased exposure of cell surface 1,3-β-glucan, which triggers elevated ROS production by macrophages111. Similarly, yeast Gcn5 also modulates ROS-mediated cell death and oxidative stress48, although our results indicate that Gcn5-mediated ROS regulation in Candida excludes ROS produced upon caspofungin challenge76. Taken together, the impaired ROS scavenging by Sod5, the altered cell wall composition and adherence contribute to abrogating fungal virulence. However, the filamentation defects of gcn5∆/∆ cells might also cause reduced Sod5 induction, leading to increased susceptibility to killing. In fact, the reduced fitness may actually be a combinatorial effect, arising from defective Gcn5-mediated SOD5 regulation (the major ROS-detoxifying SOD in Candida), impaired filamentation, along with the inability to escape from the phagosome. We speculate that Gcn5 may act in concert with Efg1 that integrates environmental and morphogenetic signals from upstream MAPK cascades, as well as protein kinase A (PKA) pathways31,75,112.
The C. albicans genome contains at least ten catalytic subunits40,113 with KAT activity, some of which may share redundant functions with Gcn5. For instance, other Candida KATs implicated in growth, morphogenesis and virulence include Ngg1, Esa1, as well as Hat1, Sas2 and Rtt10936,38,39,114,115,116. Of note, GCN5, NGG1 and ESA1 are required for filamentation and yeast-to-hyphal growth, whereas genetic ablation of HAT1 and SAS2 induces hyperfilamentation37,115 suggesting both positive and negative regulatory input by Candida KATs. Of note, the Gcn5-related acetyltransferase Ngs1 is required for sensing of the key filament inducer N-acetylglucosamine GlcNac. Ngs1 binds GlcNAc to activate its C-terminal acetyl transferase domain, driving promoter histone acetylation and transcription, including its promoter recruitment by the transcriptional regulator Rep1117.
KATs always act in concert with histone deacetylase in the regulation of both histone and non-histone targets, and like KATs, several C albicans KDACs such as Set3C, Rpd3, Rpd31, Hda1, Hos2, and Hst3 play essential roles in fungal virulence34,40,118. Therefore, KATs/KDACs are emerging drug targets18,39,40,43,119 amenable to antifungal drug discovery, as the current antifungal arsenal may become insufficient owing to intrinsic and pronounced drug resistance in Candida glabrata, and broad-spectrum antifungal resistance14,120 in emerging pathogens such as C. auris12,121,122. For example, the fungal KDAC inhibitor MGCD290, proved active in combination with fluconazole and echinocandins against drug-resistant Candida, as well as filamentous fungi123,124. Importantly, several Candida KATs target specific lysine residues on histones tails which are either absent or not modified by mammalian KAT orthologues. This suggests that fungal KAT inhibitors are unlikely to adversely affect mammalian KATs, making them especially suitable for drug discovery, and possibly useful for combination therapies40,125.
Taken together, our data suggest that Gcn5 holds new promises for therapeutic options to treat of invasive fungal infections, as Gcn5 controls several independent cellular and molecular pathways, all of which are critical for fungal fitness under immune surveillance. Interestingly, emerging evidence indicates that non-histone targets of KATs and perhaps KDACs could also play pivotal roles in fungal virulence and drug resistance40,119, opening yet another new window of opportunity for exploiting KATs/KDACS and their protein targets for antifungal drug discovery.
Materials and Methods
Ethics statement
All animal experiments were evaluated by the ethics committee of the Medical University of Vienna and approved by the Federal Ministry for Science and Research, Vienna, Austria (GZ: BMWF- 68.20n5/231-II/3b/2011) adhering to European legislation for animal experimentation.
Strains and growth conditions
Candida albicans wild type and mutant strains were grown either in rich medium (YPD; 1% yeast extract, 2% peptone, and 2% dextrose) or in synthetically defined yeast nitrogen base (SC; 0.67% yeast nitrogen base and 2% dextrose) medium at 30 °C with shaking at 200 rpm. Logarithmic phase cells were obtained by growing overnight cultures in fresh yeast YPD/nitrogen base medium for 4 h at 30 °C. 200 µg/ml Nourseothricin was used as a selection marker for C. albicans. Bacterial strains were grown at 37 °C in LB medium containing either 120 µg g/ml ampicillin or 40 µg g/ml chloramphenicol. 2% agar was added to the plates. For bacterial transformations, SOC (add 20 mM glucose, 10 mM MgSO4, 10 mM KCl, 2.5 mM KH2PO4 to LB medium) media was used. The strains, plasmids, and primers used are listed in Supplementary Tables S1–S3.
Fungalgene deletions and plasmid construction
Deletion of GCN5 was performed by using the modified recyclable NAT1 flipper method36,57. Briefly, upstream and downstream flanking regions of the GCN5 gene were amplified using appropriate primers (Supplementary Table S1) covering approximately 50 bp of the homologous region to NAT1 marker cassette (to replace the whole coding region of GCN5) and YEP352 plasmid which is containing ampicillin resistance marker and E. coli origin of replication. The FRT-FLP-NAT1-FRT cassette was amplified by using the plasmid pSFS3b. PCR amplified upstream, downstream fragments, NAT1 marker cassette and YEP352 plasmids were cloned in vivo recombination either in E. coli EL350 cells or DH5α resulting in gcn5∆ plasmid. Purified restriction digested gnc5∆ plasmid was used to transform into C. albicans wild type cells. Two rounds of integration and excision generate homozygous gcn5∆/∆ mutant. Transformation of C. albicans was done via electroporation exactly as described previously57.
Growth and phenotypic characterization
For monitoring growth curves, overnight-grown Candida cultures were inoculated into YPD with or without caspofungin or into SC medium containing various carbon sources as indicated (initial OD600 of 0.1). Absorbance was recorded after various intervals and OD600 values were plotted versus time. Phenotypic characterization of Candida albicans mutants was done via serial-dilution spotting analysis on agar plates. Equal volumes (3 µl) of 10-fold serial dilutions of logarithmically growing C. albicans strains were spotted onto YPD plates containing different stress agents such as temperature stress (37 °C), serum (10%), caffeine (10 mM), SDS (0.05%), Congo Red (150 µg/ml), caspofungin (CSP, 100 and 150 ng/ml), hydrogen peroxide (H2O2, 4, 5 and 6 mM), Itraconazole (ITZ, 0.02 µg/ml), fluconazole (FLC, 2, 4 and 8 µg/ml), ketoconazole (KTZ, 0.05 µg/ml) and voriconazole (VCZ, 0.02 µg/ml) along with vitamin C (VitC, 25 mM) when indicated. Colony growth was scored after 48 h and compared to the YPD control plate. SC media plates containing different carbon sources such as glucose (2%), ethanol (2%), citric acid (2%), sodium acetate (2%), and glycerol (2%) were used to assess the utilization of alternative carbon sources.
Filamentation assay
Logarithmically growing cells of SC5314 wild type (wt), homozygous deletion (gcn5∆/∆) and restored (gcn5∆/∆::GCN5) were plated on YPD plate supplemented with 10% fetal calf serum (FCS). Colony morphology was analysed after incubating plates for 3 days at 37 °C. Photographs were taken using a Discovery V12 Stereoscope equipped with an Axiocam MR5 camera (Zeiss). Scale bar corresponds to 1 mm.
Calcoflour white staining and microscopy
To stain cell wall chitin with Calcoflour White (CFW; Fluorescent Brightener 28, Sigma), 1 ml aliquots of logarithmically growing cells were fixed in 4% p-formaldehyde for 1–2 hours, stained with CFW 1 mg/ml for 5 min. Differential Interference Contrast (DIC) and UV light images (UV) of the same cells are shown at 60x magnification. Scale bar = 5 µM. To inspect hyphal morphologies, 1 ml of logarithmically growing cells at 30 °C and 37 °C in YPD supplemented with 10% FCS were washed twice with PBS and fixed in 4% p-formaldehyde for 2 hours. Fixed cells were washed and images were taken with an LSM 700 Zeiss Confocal microscope at 60x magnification. Scale bar = 5 µM.
ROS assay
Intracellular reactive oxygen species (ROS) was measured as described previously37 using the following modifications. Overnight grown Candida cultures were inoculated in SC medium containing 10% glucose at an initial OD600 of 0.3 and cultured at 30 °C for 1 h. The cultures contained 10% instead of 2% glucose to avoid CSP-induced flocculation76. Further, cultures were incubated with 20 mM dihydroethidium (DHE, Invitrogen) for 1 h, followed by caspofungin treatment (CSP, 150 ng/ml) for 150 min. Then, cells were washed once with water and samples were analysed by flow cytometry using FACS Calibur (BD Biosciences) at FL3-H channel. A minimum of 10,000 events were recorded for each sample. The data were analysed using Flowjo software (Flowjo LLC) and expressed mean relative fluorescence units from three independent different experiments.
Flourescein diacetate (FDA) uptake assay
Candida strains were grown in the presence or absence of caspofungin (CSP, 50 ng/ml) for 4 h. About 5 × 106 cells were resuspended and washed twice in 1 ml of FDA buffer before supplementing with 50 nm FDA. A 200 µl volume of cell mixture with or without FDA was added to an optical-bottom 96-well plate. The kinetics of FDA uptake was recorded every 5 min for 30 reads or until saturation was reached with simultaneous shaking of samples on the H1 Synergy plate reader with an excitation and emission wavelengths 485 and 535 nm, respectively. Data represent the mean fluorescence intensity over time. The slope rate was calculated using GraphPad Prism software.
Western blot analysis
Logarithmically growing Candida cultures were washed once with ice-cold water and whole-cell extracts were prepared by trichloroacetic acid (TCA) method as described previously126. Extracts corresponding to 1 OD600 (1 × 107 cells) were fractionated by 12% SDS-Page and blotted for proteins as indicated. Signals from the same whole cell extracts were detected using antibodies for total and active phosphorylated MAP kinases. The commercial antibodies recognized Mkc1 and Cek1 (p44/42 MAPK Erk1/2, Cell Signaling), and Hog1 (y-215, Santa Cruz), and phosphorylated Mkc1-P and Cek1-P (Phospho-p44/42 MAPK (Erk1/2), Cell Signaling) and Hog1-P (Phospho-p38, Cell Signaling). Reprobing with PSTAIR antibody (Sigma) recognizing Cdc28 served as a loading control. Protein bands on the nitrocellulose membrane were visualized using an Odysee® CLx scanner (Li-Cor®). Quantification of the protein band intensity was performed by using image studio software (LI-Cor®).
Quantification of cell wall components by flow cytometry
Quantification of cell wall components by flow cytometry was performed as described previously60. Briefly, logarithmically growing cells of SC5314 wild type (wt), homozygous deletion (gcn5∆/∆) and restored (gcn5∆/∆::GCN5) strains were washed and stained with Concanavalin A-conjugated Texas Red, Dectin-1/Fc + 488 and CFW to decorate mannans, glucan and chitin, respectively. These triple-stained cells were measured in a BD Fortessa flow cytometer (BD biosciences) to quantify the amount of chitin, glucan and mannan using the BV421 (violet 405 nm, 50 mW power), FITC (blue 488 nm wavelength, 50 mW power) and Texas Red (red 640 nm wavelength, 40 mW power) lasers, respectively. A minimum of 10,000 events were recorded for each sample and the data were analysed using Flowjo software (Flowjo LLC). Unstained and single-stained samples served as controls and the data expressed as the mean fluorescence intensity from three independent experiments.
Quantitative Real-time PCR (qPCR) analysis, RNA-seq analysis and bioinformatics
Total RNA isolation, cDNA synthesis and qPCR analysis was done as described previously37. The efficiency-corrected ΔΔCt method was used to quantify mRNA expression levels of a target gene transcript in comparison to a reference gene transcript127. The mRNA of the gene associated with Topoisomerase II (PAT1) was used as a reference gene128. GraphPad Prism software was used to perform statistical analyses of independent biological replicates as indicated.
For RNA-seq analysis, 5 μg DNase-treated total RNA was subjected to mRNA purification using Dynabeads mRNA purification kit (Invitrogen). Remaining rRNA contamination was checked in the bioanalyzer using Agilent RNA 6000 Pico kit (Agilent). Purified mRNA was subjected to fragmentation using the NEBNext® Magnesium RNA Fragmentation Module (New England Biolabs) and purified with RNeasy Plus Mini kit (Qiagen). Then, first-strand cDNA was synthesized with 50 ng/μl random hexamer primers using SuperScript ® III First-Strand Synthesis System for RT-PCR (Invitrogen). dNTPs were eliminated by purifying with MiniQuickSpin DNA Columns129 and subjected to second strand cDNA synthesis followed by clean-up with Minielute Reaction cleanup kit (Qiagen). cDNA concentration was measured using Quant-iT Picogreen dsDNA Reagents (Invitrogen) in a NanoDrop Fluorospectrophotometer ND-3300 (Thermo Fisher). cDNA samples were further processed for library preparation and sequenced on a HiSeq. 2500 (Illumina) at the Next Generation Sequencing Core Facility, Vienna Biocenter Core Facilities (VBCF, https://www.vbcf.ac.at/facilities/next-generation-sequencing). Three biological replicates for each time-points of wild type (wt) treated with caspofungin (CSP, 0, 15 and 45 min), as well as homozygous deletion (gcn5∆/∆) and two biological replicates for the remaining two time-points of gcn5∆/∆ (CSP, 15 min and 45 min) were sequenced. The RNA-seq data has been deposited in Gene Expression Omnibus (GEO) under the accession number GSE123412.
The bioinformatics analysis pipeline relied on the C. albicans genome Assembly 22 (http://www.candidagenome.org) to map sequence reads using TopHat, allowing for uniquely mapped reads130. HTSeq131 with union mode was used to assess the read counts using a reference annotation (C_albicans_SC5314_version_A22-s07-m01-r70; http://www.candidagenome.org). To identify the differentially expressed genes, read counts utilized DESeq2 R package132 with an adjusted P-value cut- off ≤ 0.05.
Heat map of hierarchical clustered and differentially expressed genes was generated using data mining tool Orange3133. Hierarchical clustering used the Euclidian distance and average cluster linking. The resulting dendrograms show the degree of similarity in gene expression. Venn diagrams were generated using Venny 2.1 (http://bioinfogp.cnb.csic.es/tools/venny)134. Gene ontology (GO) annotations were used the GO slim mapper tool (http://www.candidagenome.org/cgi-bin/GO/goTermMapper), and the online bioinformatics tool Fungifun2 (https://elbe.hki-jena.de/fungifun/)135.
Adherence assays
Adherence on polystyrene-coated plates was measured by crystal violet staining as described previously136 with minor modifications. Samples containing 2 × 107 Candida cells in YPD were loaded into flat-bottomed 96-well microtiter plate (Corning) and incubated for 4 hours at 30 °C. The culture medium was aspirated and cells washed once with PBS to remove non-adherent Candida. Plate wells were allowed to dry at room temperature after 15 min methanol fixation and cells were stained for 5 min with 200 µl of 1% crystal violet (v/v). Then, cells were washed gently once with water, followed by the addition of 200 µl of 33% acetic acid for destaining, before absorbance was measured at 570 nm. Data are expressed as mean of absorbance of Candida strains from three independent technical as well as biological replicates.
Phagocytosis assay
Flow cytometry-based phagocytosis assays were performed as described previously36. Wild type (wt), homozygous deletion (gcn5∆/∆) and restored (gcn5∆/∆::GCN5) cells were grown overnight to an OD600 of 1 and washed twice with PBS. Cells were stained with 10 mg/ml Alexa Fluor 488 (Life Technologies) in 100 mM HEPES buffer (pH 7.5) for 60 min at 30 °C with shaking in the dark. Stained Candida cells were washed thrice and resuspended in HEPES buffer, before infecting primary bone marrow-derived macrophages (BMDMs) at a multiplicity of infection (MOI) of 2:1. Phagocytosis was allowed and for 45 min at 37 °C and 5% CO2, before stopped by chilling samples on ice. Cells were fixed in 1% p-formaldehyde and stained with 0.4% trypan blue to quench the fluorescence of extracellular or adherent Candida. Control samples were kept on ice throughout the experiment. Intracellular phagocytosed Candida cells were quantified by flow cytometry analysis with FL1-H on a FACSCalibur (BD Biosciences). The data were analysed using Flowjo software (Flowjo LLC) and expressed as mean percentage of phagocytosis from three biological replicates.
Fluorescence microscopy-based phagocytosis assays were performed as described previously137, using minor modifications. BMDMs were infected with Alexafluor 488-labeled Candida strains at an MOI of 1:5 for 45 min, washed with PBS and further stained with 100 nM LysoTracker Red (DND-99) for 30 min prior to fixation. After 20 min in 3.7% formaldehyde fixation medium, cell were permeabilized for 15 min with 0.7% Triton X-100. DNA in permeabilized cells was stained with DAPI (49,6-diamidino-2-phenylindole) for 30 min. Microscopic slides were examined with a laser scanning microscope (LSM 700; Carl ZEISS). Images shown are representative overlays of all four channels red, blue, grey and green to indicate staining of lysosomes, nucleus, Differential Interference Contrast (DIC) and Candida albicans, respectively. Scale bar = 5 µM.
Immune cells and mouse strains
Age-matched C57BL/6 wild type mice of 8–10 weeks were used for all experiments. Primary cultures of bone marrow-derived macrophages (BMDMs) and myeloid dendritic cells (mDCs) were isolated, and cultivated exactly as described before138. Survival of C. albicans in BMDMs and mDCs was quantified as described previously63,139 using an MOI of 10:1 (fungi to macrophages). Fungal cells were harvested 2 h and 4 h post infection and viability quantified by cfu-counting of samples on YPD. Survival was calculated as percentage of viable cfus after 48 h infection by comparing with uninfected Candida strains.
For virulence experiments, mouse infections were carried out as described previously36,78. Briefly, C. albicans strains were grown from frozen stocks overnight to an OD600 of around 1, washed twice and finally resuspended in PBS. For infection, 1 × 105 Candida cells per 21 g body weight were injected into mice via the lateral tail vein. For survival experiments, mice were monitored for 15 days, including the recording of weight loss. A group of five mice were used for each strain. Statistical analysis was carried out using the GraphPad Prism software (Graphpad Software Inc.). Mouse survival curves used the log-rank (Mantle-Cox) test. For statistical analysis unpaired two-tailed Student’s t-test with 95% confidence intervals were used. P-values such as *p-value < 0.05; **p-value < 0.01; ***p-value < 0.005 were considered significant.
Data Availability
The raw datasets and annotated gene expression analysis files have been deposited in the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) database under the accession number GSE123412.
References
Brown, G. D. et al. Hidden killers: human fungal infections. Sci Transl Med 4, 165rv113, https://doi.org/10.1126/scitranslmed.3004404 (2012).
Pfaller, M. A. & Diekema, D. J. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20, 133–163, https://doi.org/10.1128/CMR.00029-06 (2007).
Perlroth, J., Choi, B. & Spellberg, B. Nosocomial fungal infections: epidemiology, diagnosis, and treatment. Med Mycol 45, 321–346, https://doi.org/10.1080/13693780701218689 (2007).
Kuhbacher, A., Burger-Kentischer, A. & Rupp, S. Interaction of Candida Species with the Skin. Microorganisms 5, https://doi.org/10.3390/microorganisms5020032 (2017).
Iliev, I. D. & Leonardi, I. Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat Rev Immunol 17, 635–646, https://doi.org/10.1038/nri.2017.55 (2017).
Pappas, P. G., Lionakis, M. S., Arendrup, M. C., Ostrosky-Zeichner, L. & Kullberg, B. J. Invasive candidiasis. Nat Rev Dis Primers 4, 18026, https://doi.org/10.1038/nrdp.2018.26 (2018).
Netelenbos, T. et al. The burden of invasive infections in neutropenic patients: incidence, outcomes, and use of granulocyte transfusions. Transfusion, https://doi.org/10.1111/trf.14994 (2018).
Spampinato, C. & Leonardi, D. Candida infections, causes, targets, and resistance mechanisms: traditional and alternative antifungal agents. Biomed Res Int 2013, 204237, https://doi.org/10.1155/2013/204237 (2013).
Bassetti, M., Mikulska, M. & Viscoli, C. Bench-to-bedside review: therapeutic management of invasive candidiasis in the intensive care unit. Crit Care 14, 244, https://doi.org/10.1186/cc9239 (2010).
Yapar, N. Epidemiology and risk factors for invasive candidiasis. Ther Clin Risk Manag 10, 95–105, https://doi.org/10.2147/TCRM.S40160 (2014).
Jeffery-Smith, A. et al. Candida auris: a Review of the Literature. Clin Microbiol Rev 31, https://doi.org/10.1128/CMR.00029-17 (2018).
Wiederhold, N. P. Antifungal resistance: current trends and future strategies to combat. Infect Drug Resist 10, 249–259, https://doi.org/10.2147/IDR.S124918 (2017).
Cortegiani, A. et al. Epidemiology, clinical characteristics, resistance, and treatment of infections by Candida auris. J Intensive Care 6, 69, https://doi.org/10.1186/s40560-018-0342-4 (2018).
Perlin, D. S., Rautemaa-Richardson, R. & Alastruey-Izquierdo, A. The global problem of antifungal resistance: prevalence, mechanisms, and management. Lancet Infect Dis 17, e383–e392, https://doi.org/10.1016/S1473-3099(17)30316-X (2017).
Arendrup, M. C. & Patterson, T. F. Multidrug-Resistant Candida: Epidemiology, Molecular Mechanisms, and Treatment. J Infect Dis 216, S445–S451, https://doi.org/10.1093/infdis/jix131 (2017).
Hofs, S., Mogavero, S. & Hube, B. Interaction of Candida albicans with host cells: virulence factors, host defense, escape strategies, and the microbiota. J Microbiol 54, 149–169, https://doi.org/10.1007/s12275-016-5514-0 (2016).
Gow, N. A., van de Veerdonk, F. L., Brown, A. J. & Netea, M. G. Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nat Rev Microbiol 10, 112–122, https://doi.org/10.1038/nrmicro2711 (2011).
Hnisz, D., Tscherner, M. & Kuchler, K. Targeting chromatin in fungal pathogens as a novel therapeutic strategy: histone modification gets infectious. Epigenomics 3, 129–132, https://doi.org/10.2217/epi.11.7 (2011).
McCarthy, M. W., Kontoyiannis, D. P., Cornely, O. A., Perfect, J. R. & Walsh, T. J. Novel Agents and Drug Targets to Meet the Challenges of Resistant Fungi. J Infect Dis 216, S474–S483, https://doi.org/10.1093/infdis/jix130 (2017).
Mazu, T. K., Bricker, B. A., Flores-Rozas, H. & Ablordeppey, S. Y. The Mechanistic Targets of Antifungal Agents: An Overview. Mini Rev Med Chem 16, 555–578 (2016).
Gow, N. A. R., Latge, J. P. & Munro, C. A. The Fungal Cell Wall: Structure, Biosynthesis, and Function. Microbiol Spectr 5, https://doi.org/10.1128/microbiolspec.FUNK-0035-2016 (2017).
Ene, I. V. et al. Cell Wall Remodeling Enzymes Modulate Fungal Cell Wall Elasticity and Osmotic Stress Resistance. MBio 6, e00986, https://doi.org/10.1128/mBio.00986-15 (2015).
Hopke, A., Brown, A. J. P., Hall, R. A. & Wheeler, R. T. Dynamic Fungal Cell Wall Architecture in Stress Adaptation and Immune Evasion. Trends Microbiol 26, 284–295, https://doi.org/10.1016/j.tim.2018.01.007 (2018).
Cullen, P. J. & Edgerton, M. Unmasking fungal pathogens by studying MAPK-dependent cell wall regulation in Candida albicans. Virulence 7, 502–505, https://doi.org/10.1080/21505594.2016.1177695 (2016).
de Dios, C. H., Roman, E., Monge, R. A. & Pla, J. The role of MAPK signal transduction pathways in the response to oxidative stress in the fungal pathogen Candida albicans: implications in virulence. Curr Protein Pept Sci 11, 693–703 (2010).
Monge, A. R., Román, E., Nombela, C. & Pla, J. The MAP kinase signal transduction network in Candida albicans. Microbiology 152, 905–912, https://doi.org/10.1099/mic.0.28616-0 (2006).
Navarro-Garcia, F., Eisman, B., Fiuza, S. M., Nombela, C. & Pla, J. The MAP kinase Mkc1p is activated under different stress conditions in Candida albicans. Microbiology 151, 2737–2749, https://doi.org/10.1099/mic.0.28038-0 (2005).
Herrero-de-Dios, C., Alonso-Monge, R. & Pla, J. The lack of upstream elements of the Cek1 and Hog1 mediated pathways leads to a synthetic lethal phenotype upon osmotic stress in Candida albicans. Fungal Genet Biol 69, 31–42, https://doi.org/10.1016/j.fgb.2014.05.010 (2014).
Sudbery, P. E. Growth of Candida albicans hyphae. Nat Rev Microbiol 9, 737–748, https://doi.org/10.1038/nrmicro2636 (2011).
Shapiro, R. S., Robbins, N. & Cowen, L. E. Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiology and molecular biology reviews: MMBR 75, 213–267, https://doi.org/10.1128/mmbr.00045-10 (2011).
Biswas, S., Van Dijck, P. & Datta, A. Environmental sensing and signal transduction pathways regulating morphopathogenic determinants of Candida albicans. Microbiol Mol Biol Rev 71, 348–376, https://doi.org/10.1128/MMBR.00009-06 (2007).
Noble, S. M., Gianetti, B. A. & Witchley, J. N. Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat Rev Microbiol 15, 96–108, https://doi.org/10.1038/nrmicro.2016.157 (2017).
Xie, J., Jenull, S., Tscherner, M. & Kuchler, K. The Paralogous Histone Deacetylases Rpd3 and Rpd31 Play Opposing Roles in Regulating the White-Opaque Switch in the Fungal Pathogen Candida albicans. MBio 7, https://doi.org/10.1128/mBio.01807-16 (2016).
Hnisz, D. et al. A histone deacetylase adjusts transcription kinetics at coding sequences during Candida albicans morphogenesis. PLoS Genet 8, e1003118, https://doi.org/10.1371/journal.pgen.1003118 (2012).
Hnisz, D., Majer, O., Frohner, I. E., Komnenovic, V. & Kuchler, K. The Set3/Hos2 histone deacetylase complex attenuates cAMP/PKA signaling to regulate morphogenesis and virulence of Candida albicans. PLoS pathogens 6, https://doi.org/10.1371/journal.ppat.1000889 (2010).
Tscherner, M. et al. The Candida albicans Histone Acetyltransferase Hat1 Regulates Stress Resistance and Virulence via Distinct Chromatin Assembly Pathways. PLoS Pathog 11, https://doi.org/10.1371/journal.ppat.1005218 (2015).
Tscherner, M., Stappler, E., Hnisz, D. & Kuchler, K. The histone acetyltransferase Hat1 facilitates DNA damage repair and morphogenesis in Candida albicans. Molecular microbiology 86, 1197–1214, https://doi.org/10.1111/mmi.12051 (2012).
Lopes da Rosa, J., Boyartchuk, V. L., Zhu, L. J. & Kaufman, P. D. Histone acetyltransferase Rtt109 is required for Candida albicans pathogenesis. Proc Natl Acad Sci USA 107, 1594–1599, https://doi.org/10.1073/pnas.0912427107 (2010).
Wurtele, H. et al. Modulation of histone H3 lysine 56 acetylation as an antifungal therapeutic strategy. Nat Med 16, 774–780, https://doi.org/10.1038/nm.2175 (2010).
Kuchler, K., Jenull, S., Shivarathri, R. & Chauhan, N. Fungal KATs/KDACs: A New Highway to Better Antifungal Drugs? PLoS Pathog 12, e1005938, https://doi.org/10.1371/journal.ppat.1005938 (2016).
Lopes da Rosa, J. & Kaufman, P. D. Chromatin-mediated Candida albicans virulence. Biochim Biophys Acta 1819, 349–355, https://doi.org/10.1016/j.bbagrm.2011.08.007 (2012).
Bauer, I. et al. A Class 1 Histone Deacetylase with Potential as an Antifungal Target. MBio 7, https://doi.org/10.1128/mBio.00831-16 (2016).
Garnaud, C., Champleboux, M., Maubon, D. & Cornet, M. & Govin, J. Histone Deacetylases and Their Inhibition in Candida Species. Front Microbiol 7, 1238, https://doi.org/10.3389/fmicb.2016.01238 (2016).
Allis, C. D. & Jenuwein, T. The molecular hallmarks of epigenetic control. Nature reviews. Genetics, https://doi.org/10.1038/nrg.2016.59 (2016).
Helmlinger, D. & Tora, L. Sharing the SAGA. Trends Biochem Sci 42, 850–861, https://doi.org/10.1016/j.tibs.2017.09.001 (2017).
Lee, K. K. & Workman, J. L. Histone acetyltransferase complexes: one size doesn’t fit all. Nature Reviews Molecular Cell Biology 8, 284–295, https://doi.org/10.1038/nrm2145 (2007).
Pray-Grant, M. G. et al. The Novel SLIK Histone Acetyltransferase Complex Functions in the Yeast Retrograde Response Pathway. Molecular and Cellular Biology 22, 8774–8786, https://doi.org/10.1128/mcb.22.24.8774-8786.2002 (2002).
Gaupel, A. C., Begley, T. J. & Tenniswood, M. Gcn5 Modulates the Cellular Response to Oxidative Stress and Histone Deacetylase Inhibition. J Cell Biochem 116, 1982–1992, https://doi.org/10.1002/jcb.25153 (2015).
Xue-Franzen, Y., Henriksson, J., Burglin, T. R. & Wright, A. P. Distinct roles of the Gcn5 histone acetyltransferase revealed during transient stress-induced reprogramming of the genome. BMC Genomics 14, 479, https://doi.org/10.1186/1471-2164-14-479 (2013).
Xue-Franzen, Y. et al. Genome-wide characterisation of the Gcn5 histone acetyltransferase in budding yeast during stress adaptation reveals evolutionarily conserved and diverged roles. BMC Genomics 11, 200, https://doi.org/10.1186/1471-2164-11-200 (2010).
Martinez-Soto, D., Gonzalez-Prieto, J. M. & Ruiz-Herrera, J. Transcriptomic analysis of the GCN5 gene reveals mechanisms of the epigenetic regulation of virulence and morphogenesis in Ustilago maydis. FEMS Yeast Res 15, https://doi.org/10.1093/femsyr/fov055 (2015).
Gonzalez-Prieto, J. M., Rosas-Quijano, R., Dominguez, A. & Ruiz-Herrera, J. The UmGcn5 gene encoding histone acetyltransferase from Ustilago maydis is involved in dimorphism and virulence. Fungal Genet Biol 71, 86–95, https://doi.org/10.1016/j.fgb.2014.09.002 (2014).
Canovas, D. et al. The histone acetyltransferase GcnE (GCN5) plays a central role in the regulation of Aspergillus asexual development. Genetics 197, 1175–1189, https://doi.org/10.1534/genetics.114.165688 (2014).
O’Meara, T. R., Hay, C., Price, M. S., Giles, S. & Alspaugh, J. A. Cryptococcus neoformans histone acetyltransferase Gcn5 regulates fungal adaptation to the host. Eukaryot Cell 9, 1193–1202, https://doi.org/10.1128/EC.00098-10 (2010).
Cai, Q., Wang, J. J., Fu, B., Ying, S. H. & Feng, M. G. Gcn5-dependent histone H3 acetylation and gene activity is required for the asexual development and virulence of Beauveria bassiana. Environ Microbiol 20, 1484–1497, https://doi.org/10.1111/1462-2920.14066 (2018).
Chang, P., Fan, X. & Chen, J. Function and subcellular localization of Gcn5, a histone acetyltransferase in Candida albicans. Fungal Genet Biol 81, 132–141, https://doi.org/10.1016/j.fgb.2015.01.011 (2015).
Reuss, O., Vik, A., Kolter, R. & Morschhauser, J. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341, 119–127, https://doi.org/10.1016/j.gene.2004.06.021 (2004).
Martinez-Soto, D. & Ruiz-Herrera, J. Functional analysis of the MAPK pathways in fungi. Rev Iberoam Micol 34, 192–202, https://doi.org/10.1016/j.riam.2017.02.006 (2017).
Alonso-Monge, R., Roman, E., Arana, D. M., Pla, J. & Nombela, C. Fungi sensing environmental stress. Clin Microbiol Infect 15(Suppl 1), 17–19, https://doi.org/10.1111/j.1469-0691.2008.02690.x (2009).
Nogueira, M. et al. Quantitative Analysis of Candida Cell Wall Components by Flow Cytometry with Triple-Fluorescence Staining. Journal of Microbiology and Modern Techniques 2, 1–9 (2017).
Breeuwer, P. et al. Characterization of uptake and hydrolysis of fluorescein diacetate and carboxyfluorescein diacetate by intracellular esterases in Saccharomyces cerevisiae, which result in accumulation of fluorescent product. Appl Environ Microbiol 61, 1614–1619 (1995).
Blankenship, J. R., Fanning, S., Hamaker, J. J. & Mitchell, A. P. An extensive circuitry for cell wall regulation in Candida albicans. PLoS Pathog 6, e1000752, https://doi.org/10.1371/journal.ppat.1000752 (2010).
Frohner, I. E., Bourgeois, C., Yatsyk, K., Majer, O. & Kuchler, K. Candida albicans cell surface superoxide dismutases degrade host-derived reactive oxygen species to escape innate immune surveillance. Molecular microbiology 71, 240–252, https://doi.org/10.1111/j.1365-2958.2008.06528.x (2009).
Liu, T. T. et al. Genome-wide expression profiling of the response to azole, polyene, echinocandin, and pyrimidine antifungal agents in Candida albicans. Antimicrobial agents and chemotherapy 49, 2226–2236, https://doi.org/10.1128/AAC.49.6.2226-2236.2005 (2005).
Csank, C. et al. Roles of the Candida albicans mitogen-activated protein kinase homolog, Cek1p, in hyphal development and systemic candidiasis. Infect Immun 66, 2713–2721 (1998).
Dwivedi, P. et al. Role of Bcr1-activated genes Hwp1 and Hyr1 in Candida albicans oral mucosal biofilms and neutrophil evasion. PLoS One 6, e16218, https://doi.org/10.1371/journal.pone.0016218 (2011).
Nobile, C. J. & Mitchell, A. P. Regulation of cell-surface genes and biofilm formation by the C. albicans transcription factor Bcr1p. Curr Biol 15, 1150–1155, https://doi.org/10.1016/j.cub.2005.05.047 (2005).
Bailey, D. A., Feldmann, P. J., Bovey, M., Gow, N. A. & Brown, A. J. The Candida albicans HYR1 gene, which is activated in response to hyphal development, belongs to a gene family encoding yeast cell wall proteins. J Bacteriol 178, 5353–5360 (1996).
Braun, B. R. & Johnson, A. D. TUP1, CPH1 and EFG1 make independent contributions to filamentation in Candida albicans. Genetics 155, 57–67 (2000).
Sorgo, A. G., Brul, S., de Koster, C. G., de Koning, L. J. & Klis, F. M. Iron restriction-induced adaptations in the wall proteome of Candida albicans. Microbiology 159, 1673–1682, https://doi.org/10.1099/mic.0.065599-0 (2013).
Moran, G. P. Transcript profiling reveals rewiring of iron assimilation gene expression in Candida albicans and C. dubliniensis. FEMS Yeast Res 12, 918–923, https://doi.org/10.1111/j.1567-1364.2012.00841.x (2012).
Perez, A. et al. Some biological features of Candida albicans mutants for genes coding fungal proteins containing the CFEM domain. FEMS Yeast Res 11, 273–284, https://doi.org/10.1111/j.1567-1364.2010.00714.x (2011).
Kuznets, G. et al. A relay network of extracellular heme-binding proteins drives C. albicans iron acquisition from hemoglobin. PLoS Pathog 10, e1004407, https://doi.org/10.1371/journal.ppat.1004407 (2014).
Chakravarti, A., Camp, K., McNabb, D. S. & Pinto, I. The Iron-Dependent Regulation of the Candida albicans Oxidative Stress Response by the CCAAT-Binding Factor. PLoS One 12, e0170649, https://doi.org/10.1371/journal.pone.0170649 (2017).
Bockmuhl, D. P. & Ernst, J. F. A potential phosphorylation site for an A-type kinase in the Efg1 regulator protein contributes to hyphal morphogenesis of Candida albicans. Genetics 157, 1523–1530 (2001).
Gregori, C. et al. Efg1 Controls caspofungin-induced cell aggregation of Candida albicans through the adhesin Als1. Eukaryot Cell 10, 1694–1704, https://doi.org/10.1128/EC.05187-11 (2011).
Perlin, D. S. Mechanisms of echinocandin antifungal drug resistance. Ann N Y Acad Sci 1354, 1–11, https://doi.org/10.1111/nyas.12831 (2015).
Wirnsberger, G. et al. Jagunal homolog 1 is a critical regulator of neutrophil function in fungal host defense. Nat Genet 46, 1028–1033, https://doi.org/10.1038/ng.3070 (2014).
Bongomin, F., Gago, S., Oladele, R. O. & Denning, D. W. Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J Fungi (Basel) 3, https://doi.org/10.3390/jof3040057 (2017).
Wiederhold, N. P. The antifungal arsenal: alternative drugs and future targets. Int J Antimicrob Agents 51, 333–339, https://doi.org/10.1016/j.ijantimicag.2017.09.002 (2018).
Grant, P. A., Duggan, L., Côté, J. & Roberts, S. M. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes & …. https://doi.org/10.1101/gad.11.13.1640 (1997).
Burgess, R. J., Zhou, H., Han, J. & Zhang, Z. A role for Gcn5 in replication-coupled nucleosome assembly. Mol Cell 37, 469–480, https://doi.org/10.1016/j.molcel.2010.01.020 (2010).
Daniel, J. A. & Grant, P. A. Multi-tasking on chromatin with the SAGA coactivator complexes. Mutat Res 618, 135–148, https://doi.org/10.1016/j.mrfmmm.2006.09.008 (2007).
Georgakopoulos, T. & Thireos, G. Two distinct yeast transcriptional activators require the function of the GCN5 protein to promote normal levels of transcription. EMBO J 11, 4145–4152 (1992).
Timmers, H. T. & Tora, L. SAGA unveiled. Trends Biochem Sci 30, 7–10, https://doi.org/10.1016/j.tibs.2004.11.007 (2005).
Wang, L. et al. Histone acetyltransferase activity is conserved between yeast and human GCN5 and is required for complementation of growth and transcriptional activation. Mol Cell Biol 17, 519–527 (1997).
Teng, Y., Yu, Y. & Waters, R. The Saccharomyces cerevisiae histone acetyltransferase Gcn5 has a role in the photoreactivation and nucleotide excision repair of UV-induced cyclobutane pyrimidine dimers in the MFA2 gene. J Mol Biol 316, 489–499, https://doi.org/10.1006/jmbi.2001.5383 (2002).
Gao, T., Zheng, Z., Hou, Y. & Zhou, M. Transcription factors spt3 and spt8 are associated with conidiation, mycelium growth, and pathogenicity in Fusarium graminearum. FEMS Microbiol Lett 351, 42–50, https://doi.org/10.1111/1574-6968.12350 (2014).
Brownell, J. E. et al. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84, 843–851, https://doi.org/10.1016/s0092-8674(00)81063-6 (1996).
Suwunnakorn, S., Wakabayashi, H., Kordalewska, M., Perlin, D. S. & Rustchenko, E. FKS2 and FKS3 Genes of Opportunistic Human Pathogen Candida albicans Influence Echinocandin Susceptibility. Antimicrob Agents Chemother 62, https://doi.org/10.1128/AAC.02299-17 (2018).
Katiyar, S., Pfaller, M. & Edlind, T. Candida albicans and Candida glabrata clinical isolates exhibiting reduced echinocandin susceptibility. Antimicrob Agents Chemother 50, 2892–2894, https://doi.org/10.1128/AAC.00349-06 (2006).
de Nobel, H. et al. Cell wall perturbation in yeast results in dual phosphorylation of the Slt2/Mpk1 MAP kinase and in an Slt2-mediated increase in FKS2-lacZ expression, glucanase resistance and thermotolerance. Microbiology 146((Pt 9)), 2121–2132, https://doi.org/10.1099/00221287-146-9-2121 (2000).
Cieniewicz, A. M. et al. The bromodomain of Gcn5 regulates site specificity of lysine acetylation on histone H3. Mol Cell Proteomics 13, 2896–2910, https://doi.org/10.1074/mcp.M114.038174 (2014).
Duhring, S. et al. Host-pathogen interactions between the human innate immune system and Candida albicans-understanding and modeling defense and evasion strategies. Front Microbiol 6, 625, https://doi.org/10.3389/fmicb.2015.00625 (2015).
Ramirez-Zavala, B. et al. SAGA/ADA complex subunit Ada2 is required for Cap1- but not Mrr1-mediated upregulation of the Candida albicans multidrug efflux pump MDR1. Antimicrob Agents Chemother 58, 5102–5110, https://doi.org/10.1128/AAC.03065-14 (2014).
Koch, K. A., Allard, S., Santoro, N., Cote, J. & Thiele, D. J. The Candida glabrata Amt1 copper-sensing transcription factor requires Swi/Snf and Gcn5 at a critical step in copper detoxification. Mol Microbiol 40, 1165–1174 (2001).
Silver, P. M., Oliver, B. G. & White, T. C. Role of Candida albicans transcription factor Upc2p in drug resistance and sterol metabolism. Eukaryot Cell 3, 1391–1397, https://doi.org/10.1128/EC.3.6.1391-1397.2004 (2004).
Dunkel, N. et al. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot Cell 7, 1180–1190, https://doi.org/10.1128/EC.00103-08 (2008).
Berkow, E. L. et al. Multidrug Transporters and Alterations in Sterol Biosynthesis Contribute to Azole Antifungal Resistance in Candida parapsilosis. Antimicrob Agents Chemother 59, 5942–5950, https://doi.org/10.1128/AAC.01358-15 (2015).
Lohberger, A., Coste, A. T. & Sanglard, D. Distinct roles of Candida albicans drug resistance transcription factors TAC1, MRR1, and UPC2 in virulence. Eukaryot Cell 13, 127–142, https://doi.org/10.1128/EC.00245-13 (2014).
Vasicek, E. M., Berkow, E. L., Flowers, S. A., Barker, K. S. & Rogers, P. D. UPC2 is universally essential for azole antifungal resistance in Candida albicans. Eukaryot Cell 13, 933–946, https://doi.org/10.1128/EC.00221-13 (2014).
Whaley, S. G. et al. UPC2A is required for high-level azole antifungal resistance in Candida glabrata. Antimicrob Agents Chemother 58, 4543–4554, https://doi.org/10.1128/AAC.02217-13 (2014).
Sanglard, D. et al. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob Agents Chemother 39, 2378–2386 (1995).
de Groot, P. W., Bader, O., de Boer, A. D., Weig, M. & Chauhan, N. Adhesins in human fungal pathogens: glue with plenty of stick. Eukaryot Cell 12, 470–481, https://doi.org/10.1128/EC.00364-12 (2013).
Bourgeois, C. & Kuchler, K. Fungal pathogens-a sweet and sour treat for toll-like receptors. Front Cell Infect Microbiol 2, 142, https://doi.org/10.3389/fcimb.2012.00142 (2012).
Brown, A. J. et al. Stress adaptation in a pathogenic fungus. J Exp Biol 217, 144–155, https://doi.org/10.1242/jeb.088930 (2014).
Leng, P., Lee, P. R., Wu, H. & Brown, A. J. Efg1, a morphogenetic regulator in Candida albicans, is a sequence-specific DNA binding protein. J Bacteriol 183, 4090–4093, https://doi.org/10.1128/JB.183.13.4090-4093.2001 (2001).
Lu, Y. et al. Efg1-mediated recruitment of NuA4 to promoters is required for hypha-specific Swi/Snf binding and activation in Candida albicans. Mol Biol Cell 19, 4260–4272, https://doi.org/10.1091/mbc.E08-02-0173 (2008).
Lohse, M. B., Gulati, M., Johnson, A. D. & Nobile, C. J. Development and regulation of single- and multi-species Candida albicans biofilms. Nat Rev Microbiol 16, 19–31, https://doi.org/10.1038/nrmicro.2017.107 (2018).
Richardson, J. P. & Moyes, D. L. Adaptive immune responses to Candida albicans infection. Virulence 6, 327–337, https://doi.org/10.1080/21505594.2015.1004977 (2015).
Wellington, M., Dolan, K. & Krysan, D. J. Live Candida albicans suppresses production of reactive oxygen species in phagocytes. Infect Immun 77, 405–413, https://doi.org/10.1128/IAI.00860-08 (2009).
Hogan, D. A. & Sundstrom, P. The Ras/cAMP/PKA signaling pathway and virulence in Candida albicans. Future Microbiol 4, 1263–1270, https://doi.org/10.2217/fmb.09.106 (2009).
Kim, J., Park, S. & Lee, J. S. Epigenetic Control of Oxidative Stresses by Histone Acetyltransferases in Candida albicans. J Microbiol Biotechnol 28, 181–189, https://doi.org/10.4014/jmb.1707.07029 (2018).
Li, D. D. et al. Histone acetyltransferase encoded by NGG1 is required for morphological conversion and virulence of Candida albicans. Future Microbiol 12, 1497–1510, https://doi.org/10.2217/fmb-2017-0084 (2017).
Wang, X., Chang, P., Ding, J. & Chen, J. Distinct and redundant roles of the two MYST histone acetyltransferases Esa1 and Sas2 in cell growth and morphogenesis of Candida albicans. Eukaryot Cell 12, 438–449, https://doi.org/10.1128/EC.00275-12 (2013).
Stevenson, J. S. & Liu, H. Regulation of white and opaque cell-type formation in Candida albicans by Rtt109 and Hst3. Mol Microbiol 81, 1078–1091, https://doi.org/10.1111/j.1365-2958.2011.07754.x (2011).
Su, C., Lu, Y. & Liu, H. N-acetylglucosamine sensing by a GCN5-related N-acetyltransferase induces transcription via chromatin histone acetylation in fungi. Nat Commun 7, 12916, https://doi.org/10.1038/ncomms12916 (2016).
Li, X., Robbins, N., O’Meara, T. R. & Cowen, L. E. Extensive functional redundancy in the regulation of Candida albicans drug resistance and morphogenesis by lysine deacetylases Hos2, Hda1, Rpd3 and Rpd31. Mol Microbiol 103, 635–656, https://doi.org/10.1111/mmi.13578 (2017).
Lamoth, F., Juvvadi, P. R. & Steinbach, W. J. Histone deacetylase inhibition as an alternative strategy against invasive aspergillosis. Front Microbiol 6, 96, https://doi.org/10.3389/fmicb.2015.00096 (2015).
Robbins, N., Caplan, T. & Cowen, L. E. Molecular Evolution of Antifungal Drug Resistance. Annu Rev Microbiol 71, 753–775, https://doi.org/10.1146/annurev-micro-030117-020345 (2017).
Chowdhary, A., Sharma, C. & Meis, J. F. Candida auris: A rapidly emerging cause of hospital-acquired multidrug-resistant fungal infections globally. PLoS Pathog 13, e1006290, https://doi.org/10.1371/journal.ppat.1006290 (2017).
Spivak, E. S. & Hanson, K. E. Candida auris: an Emerging Fungal Pathogen. J Clin Microbiol 56, https://doi.org/10.1128/JCM.01588-17 (2018).
Pfaller, M. A. et al. Activity of MGCD290, a Hos2 histone deacetylase inhibitor, in combination with azole antifungals against opportunistic fungal pathogens. J Clin Microbiol 47, 3797–3804, https://doi.org/10.1128/JCM.00618-09 (2009).
Pfaller, M. A., Rhomberg, P. R., Messer, S. A. & Castanheira, M. In vitro activity of a Hos2 deacetylase inhibitor, MGCD290, in combination with echinocandins against echinocandin-resistant Candida species. Diagn Microbiol Infect Dis 81, 259–263, https://doi.org/10.1016/j.diagmicrobio.2014.11.008 (2015).
Hill, J. A. & Cowen, L. E. Using combination therapy to thwart drug resistance. Future Microbiol 10, 1719–1726, https://doi.org/10.2217/fmb.15.68 (2015).
Mamnun, Y. M., Schuller, C. & Kuchler, K. Expression regulation of the yeast PDR5 ATP-binding cassette (ABC) transporter suggests a role in cellular detoxification during the exponential growth phase. FEBS Lett 559, 111–117, https://doi.org/10.1016/S0014-5793(04)00046-8 (2004).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45 (2001).
Zordan, R. E., Galgoczy, D. J. & Johnson, A. D. Epigenetic properties of white-opaque switching in Candida albicans are based on a self-sustaining transcriptional feedback loop. Proc Natl Acad Sci USA 103, 12807–12812, https://doi.org/10.1073/pnas.0605138103 (2006).
Coffey, K. et al. Characterisation of a Tip60 specific inhibitor, NU9056, in prostate cancer. PloS one 7, https://doi.org/10.1371/journal.pone.0045539 (2012).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111, https://doi.org/10.1093/bioinformatics/btp120 (2009).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, https://doi.org/10.1093/bioinformatics/btu638 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Demsar Janez, C. T. et al: Data Mining Toolbox in Python. Journal of Machine Learning Research 14, 2349-2353 (2013).
Oliveros, J. C. Venny. An interactive tool for comparing lists with Venn’s diagrams.
Priebe, S., Kreisel, C., Horn, F., Guthke, R. & Linde, J. FungiFun2: a comprehensive online resource for systematic analysis of gene lists from fungal species. Bioinformatics 31, 445–446, https://doi.org/10.1093/bioinformatics/btu627 (2015).
Negri, M. et al. Crystal violet staining to quantify Candida adhesion to epithelial cells. Br J Biomed Sci 67, 120–125 (2010).
Rai, M. N., Balusu, S., Gorityala, N., Dandu, L. & Kaur, R. Functional genomic analysis of Candida glabrata-macrophage interaction: role of chromatin remodeling in virulence. PLoS Pathog 8, e1002863, https://doi.org/10.1371/journal.ppat.1002863 (2012).
Zwolanek, F. et al. The non-receptor tyrosine kinase Tec controls assembly and activity of the noncanonical caspase-8 inflammasome. PLoS Pathog 10, e1004525, https://doi.org/10.1371/journal.ppat.1004525 (2014).
Majer, O. et al. Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog 8, e1002811, https://doi.org/10.1371/journal.ppat.1002811 (2012).
Acknowledgements
This work was supported by a grant from the National Institute of Health to NC and KK (R01AI124499), and in part by grants from the Austrian Science Fund (FWF-P25333-B22) and the FP7 project (FUNGITECT) to KK. The FP7 Initial Training Network ImResFun (MC-ITN-2013-606786) supported KK and RS, and NKS was supported through a Marie-Curie ESR early stage researcher fellowship and an experienced researcher ER grant, respectively. MT was supported by an Erwin Schroedinger Fellowship (J3835) of the Austrian Science Fund. We thank all NC and KK laboratory members for helpful discussions. We highly appreciate and acknowledge the gifts of fungal strains and antibodies from Dominique Sanglard and Joachim Morschhäuser.
Author information
Authors and Affiliations
Contributions
R.S., N.C. and K.K. conceived and designed the study; R.S., F.Z. and M.T. performed experiments, R.S., F.Z., N.K.S., K.K. and N.C. analysed the data, R.S. and K.K. prepared figures. R.S., N.C. and K.K. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shivarathri, R., Tscherner, M., Zwolanek, F. et al. The Fungal Histone Acetyl Transferase Gcn5 Controls Virulence of the Human Pathogen Candida albicans through Multiple Pathways. Sci Rep 9, 9445 (2019). https://doi.org/10.1038/s41598-019-45817-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-45817-5
This article is cited by
-
Molecular mechanisms governing antifungal drug resistance
npj Antimicrobials and Resistance (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
