
Abstract
Deceptive pollination is key to the species richness of Orchidaceae. However, the genetic basis of species diversification is still under study. Section Trigonopedia is a monophyletic clade of genus Cypripedium distributed in the southwest of China. The species of this section are pollinated by different flies. Pollinator differentiation makes section Trigonopedia an ideal group for studying the genetic basis underlying species diversification. Here, we sequenced the transcriptomes of eight species of the genus Cypripedium, including six co-flowering species of section Trigonopedia and two species outside this section as an outgroup. We reconstructed the phylogeny of the section with the combined 1572 single-copy genes extracted from the eight species and produced a highly resolved tree of the section. Furthermore, we combined substitution rate estimation and differential expression analysis to identify candidate genes, including genes related to floral scent synthesis and environmental adaptation, involved in species differentiation. Field investigations showed that these species have adapted to different habitats. We propose that the species diversification in this section is initiated by floral scent differentiation, followed by habitat differentiation, finally leading to speciation. This study sheds novel light on the diversification of closely related orchid species in the Qinghai-Tibetan region.
Similar content being viewed by others
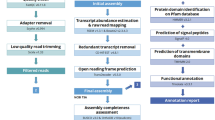

Introduction
Speciation, which is important to ecology and evolution, is a key question puzzling orchid biologists. Orchidaceae, with a large variety of species, is the largest family of monocotyledons and provides valuable material for research on evolution. Approximately one-third of orchid species are pollinated by deceit, which is important to the species richness of Orchidaceae1. Pollinators are attracted by floral colour, reward, morphology, size, and scent. Among these attractants, floral scent plays an important role in pollinator attraction2,3,4. The interactions between pollinator behaviour and floral traits lead to the prezygotic reproductive isolation of plant taxa5,6,7. Existing studies of orchid deceptive pollination mainly focus on pollination biology and the evolution of sexually deceptive orchids; although progress has been made based on the sexually deceptive orchids8,9,10, little attention has been paid to the underlying genetic basis of other deceptive pollination strategies. The inner genetic basis of deceptive pollination is still not well understood for other species. Next-generation sequencing provides an opportunity to investigate this inner genetic basis and provide clues regarding the candidate genes and pathways involved in floral scent biosynthesis11. Thus, this speciation model should be investigated in more non-model species.
The high ornamental and commercial values of orchids have promoted the transcriptome sequencing of a number of orchids8,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29. Sedeek et al.8 sequenced the transcriptomes and proteomes of three Ophrys species and identified candidate genes for pollinator attraction and reproductive isolation among sexually deceptive orchids. In addition, Orchidstra 2.0 collected 18 orchid transcriptomes, covering 12 genera in five subfamilies of Orchidaceae; these transcriptomes are important for understanding the genetic background of the biological process26. In addition, several diverse metabolic pathways are involved in floral scent synthesis, and the differential expression of candidate genes contributes to the odour difference11. Thus, additional studies should be carried out in closely related species.
The Qinghai-Tibetan Plateau is a hot spot in the study of speciation. Section Trigonopedia of Cypripedium, pollinated by different flies, is endemic to the Qinghai-Tibetan Plateau. Section Trigonopedia is a monophyly clade of genus Cypripedium30. Following the infrageneric classification of Cribb31, section Trigonopedia includes section Sinopedilum of the infrageneric classification of Perner32. The section consists of 11 species endemic to the southwest of China, including Yunnan, Sichuan, and Gansu Provinces, except for Cypripedium lichiangense, which has expanded to the north of Myanmar. Section Trigonopedia is ebracteate and has two leaves. Most species of genus Cypripedium are primarily pollinated by bees, whereas the species of this section are pollinated by flies. The floral presentation, colour, and scent also give clues to the fly pollination of this section31. Pollination studies have shown the pollinator differentiation of this section33,34,35,36. For example, C. micranthum and C. sichuanense are co-flowering sympatric species endemic to Sichuan, with the former pollinated by fruit flies and the latter pollinated by dung flies36. C. fargesii is pollinated by flat-footed flies and has a flavour of rotten leaves35. In addition, it is interesting that the predominant compound emitted by C. bardolphianum is ethyl acetate (70.4%) with a fruity wine flavour, whereas the compounds emitted by C. micranthum are dominated by pentyl ester-acetic acid (44.8%), 2-(2-methoxyethoxy)-ethanol (27.1%), and hydroxyacetic acid (15.3%)37.
Previous studies have shown that species of section Trigonopedia attract pollinators via distinct floral scents. The floral scent is the key factor promoting reproductive isolation and finally leads to species differentiation. However, the genetic basis underlying pollinator differentiation is still under study. Section Trigonopedia is distributed on the Qinghai-Tibetan Plateau, which is characterised by high mountains and deep valleys that provide diverse habitats for species differentiation. In addition, we observed habitat differentiation in the field. For instance, C. bardolphianum grows in dry, open forest, whereas C. micranthum and C. sichuanense grow in close forest. Thus, section Trigonopedia is a model system for studying the genomic background of species diversification.
The species of the section are closely related, co-flowering, morphologically similar, and pollinated by different species, providing an ideal system with which to investigate speciation on the Qinghai-Tibetan Plateau. In this study, we performed transcriptome sequencing to obtain massive protein-coding genes of the section and unravel the fundamental characteristics of these species. We first constructed the phylogeny of the section based on single-copy genes at the genomic level. Then, substitution rates were estimated in the six species. This approach allowed us to ascertain the positively selected genes possibly related to species diversification. Finally, we compared the differentially expressed genes (DEGs) between the flowers and leaves of each species to identify the candidate genes involved in floral scent differentiation. We aim to bridge the gap between pollination studies and species differentiation and unravel the molecular basis of the species diversification of this section. The study of diversification in this section will provide new insights into the speciation, ecological habits and evolution of Orchidaceae.
Results
Transcriptome Assembly
In total, we obtained 15 transcriptome sequences representing eight species of genus Cypripedium, and each species had leaf and flower transcriptome sequences except C. fargesii, for which only the flower transcriptome was obtained (Table 1). After filtering adapter sequences and low-quality sequences, we obtained 33,748,574 (C. sichuanense, flower) to 69,005,058 (C. micranthum, leaf) clean reads (Table 1). After Trinity assembly, 68,870–133,597 unique transcripts were identified with a GC content of 42.77–44.76% and mean length of 583.89 bp–767.29 bp (Fig. 1, Table 1). Then, approximately 50% of the transcripts were filtered during transcription by TransDecoder. Finally, 32,949–45,037 proteins were translated from the assembly.

Sequence length distribution of unigenes.
Phylogenetic Analyses of Section Trigonopedia
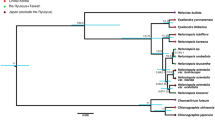
A total of 1572 single-copy genes were identified from the eight sequenced species. The phylogenetic tree showed that the section was classified into two reciprocally monophyletic groups: one formed by C. bardolphianum and C. micranthum and the other formed by the other four species (C. lentiginosum, (C. margaritaceum, (C. fargesii, C. sichuanense)) (Fig. 2).

Phylogenetic tree of section Trigonopedia derived from 1572 concatenated single-copy genes.
Substitution Rate Estimation
In the site model analysis, we detected footprints of positive selection in 67 genes, but only 8 of these genes could be KEGG annotated. Some of these genes may be associated with the diversification of this section (Table S1). One gene related to floral scent (accB), which takes part in several biosynthesis pathways, initiates the fatty acid biosynthesis. The other three genes were related to stress tolerance (SOD2, rpoB, and MKS1). In the branch model analysis, we detected 61 (C. sichuanense) to 216 (C. micranthum) positively selected genes in each lineage; C. micranthum (216) and C. fargesii (204) have more genes under positive selection than the other four species (61–131) (Fig. 3). The frequency distribution of dN/dS ratios showed that C. lentiginosum (485 genes) has more genes with elevated dN/dS ratios (3 > dN/dS > 0.5) than the other five species (354–389 genes) (Fig. 3). The boxplot results showed that C. lentiginosum had the lowest dN/dS median value (0.2649), while C. sichuanense had the highest median value (0.4207). The median values of the other four species were similar (Fig. 3). For each branch, most dN/dS ratios were in the range 0.5–1.0, except C. lentiginosum (Fig. 3). We also conducted KEGG functional classification for these PSGs in each branch. The distribution of KEGG classifications of PSGs included “Genetic Information Processing”, “Cellular Processes”, “Environmental Information Processing”, “Energy Metabolism”, “Lipid Metabolism”, “Metabolism of Cofactors and Vitamins”, “Carbohydrate Metabolism”, etc. “Genetic Information Processing” processes received the most hits in all the tested species. Each species has species-specific positive selected genes, especially genes with roles in floral scent (accB, nadC, EPT1, GLYR, URH1, and FAB2) and environmental adaptation. These genes may be closely related to the species differentiation in this section (Table S1); for example, nadC is involved in nicotinate and nicotinamide metabolism, and FAB2 (K03921) is involved in fatty acid biosynthesis (Table S1). Most interestingly, we detected five genes (PPIE, SNRPD3, HNRNPA1_3, SF3B2, and BCAS2) involved in the spliceosome in C. micranthum (Table S1). In addition, both the site model and branch model indicated that SOD2 was involved in the FOXO signalling pathway, which is related to stress tolerance.

Number of orthologues with the given dN/dS ratios for the six species. (A) Frequency distribution of the dN/dS ratios. (B) The box plot shows the ratio of non-synonymous to synonymous mutations (dN/dS) for terminal branches estimated for each orthologue.
Differential Expression Analysis
Differential expression analysis between flowers and leaves indicated that C. bardolphianum has up to 2831 DEGs, followed by C. lentiginosum (2636), C. micranthum (2602), C. sichuanense (2207), and C. margaritaceum (1947), whereas 2294 DEGs (C. flavum) and 1860 DEGs (C. singchii) were found in the outgroup species. Species in section Trigonopedia have more up-regulated genes in leaves than in flowers (Fig. 4). The GO annotation of the differential expression genes was classified into three categories: biological process, molecular function, and cellular component (Fig. S1). The DEG enrichment analysis and Venn diagram both indicated that these species have section-shared DEGs and species-specific DEGs (Figs 4, S1). Five species of this section share DEGs related to fatty acid biosynthesis (ACSL); fatty acid degradation (ACSL and MFP2); ether lipid metabolism (plc); nicotinate and nicotinamide metabolism (URH1); diterpenoid biosynthesis (GA3 and E1.14.11.13); and biosynthesis of terpenoids, steroids (crtB), etc., related to floral scent synthesis. Meanwhile, we identified ispF as being related to terpenoid backbone biosynthesis in C. lentiginosum and ispD as being related to terpenoid backbone biosynthesis in C. bardolphianum.

Comparison of the expression patterns of differential unigenes identified between flowers and leaves of section Trigonopedia. (A) Number of DEG unigenes between flowers and leaves. (B) Venn diagram showing unique and shared DEG unigenes.
Discussion
The Phylogeny of Section Trigonopedia
Considering the tree of a single gene or few cpDNA/mtDNA cannot reveal the true relationships among species. Increasing studies use transcriptome data for phylogenetic construction28,38,39,40,41. Zhang et al.28 indicated that single copy genes were powerful tools for orchid phylogeny construction. Here, we used transcriptome methods to screen the genome-wide differentiation of closely related species. The phylogeny of the section, based on 1572 single-copy genes, shows that the section forms two reciprocally monophyletic groups with high support: the first clade formed by C. bardolphianum and C. micranthum and the other clade formed by the other four species (C. lentiginosum, (C. margaritaceum, (C. fargesii, C. sichuanense))) (Fig. 2). The species of the former clade have tiny flowers and no spots on the leaves, while the species of the other clade have relatively larger flowers and blackish purple spots on the leaves (Fig. 2). Our results are congruent with the cpDNA tree of previous studies and incongruent with the nrITS tree and concatenated gene tree (nrITS + cpDNA)30. The non-monophyly of the nonconcerted evolution of orthology and paralogy of nrITS may mislead the phylogenetic inference42, which may explain the inconsistency between this study and the nrITS gene tree. Moreover, the single-copy nuclear genes developed in this study could be beneficial for the phylogenetic construction of genus Cypripedium.
The classification of the two clades indicates the early diversification of these species. Species characterised by flowers without a bract below the ovary were gathered in section Trigonopedia31. Then, the species with tiny flowers and leaves were placed in section Sinopedilum, while the other species were placed in section Trigonopedia32,43. Our study supports the delimitation of two sections, which was also supported by the morphological characters. However, studies combining these species would further elucidate the evolution of the genus.
Species on the Qinghai-Tibetan Plateau are crucial for understanding species diversification in this region. The common ancestor of the section is dated to the Pliocene, and many species diversified in the Pleistocene44. Our results showed that neighbouring species are more closely related, e.g., C. bardolphianum and C. micranthum, with sister relationships grow in a sympatric distribution in Sichuan; C. lentiginosum is distributed in Yunnan at the base of the other clade, followed by C. margaritaceum; while C. fargesii and C. sichuanense, with overlapping distributions, form the sister clade, which agrees with the south to north niche shift of the genus45.
The field investigations indicated that the species of the genus have adapted to different habitats. For example, C. bardolphianum and C. micranthum are sister species but occupy different habitats; C. bardolphianum grows in the dry, open forest; and C. micranthum grows in the dense forest. The populations of C. bardolphianum and C. sichuanense are no more than 50 m apart in Danyunxia. C. fargesii may be moisture tolerant, and its population in Jiuzhaizhou is near a calcareous pool, which fills with water during the rainy period. However, the habitat of C. margaritaceum is dry in Zhongdian and Lijiang. Moreover, the altitude varies (Table 1). For example, C. bardolphianum and C. margaritaceum are found at 3000 m; C. micranthum, C. sichuanense and C. fargesii are found at 2200 m; and C. lentiginosum is found at 1800 m. Interestingly, species with a more restricted distribution also show more distinctive morphology, especially C. lentiginosum, which has larger flowers and longer leaves but is restricted to Malipo of Yunan, whereas C. micranthum is found only in Danyunxia of Huanglong. Species with peripheral distributions are strongly differentiated from other species. The frequency distribution of dN/dS ratios showed that C. micranthum has more genes under positive selection than the other five species (Fig. 2). The elevated dN/dS ratio of C. lentiginosum (485 genes) was higher than that of its relatives (354–389 genes), and more species-specific DEGs were found in C. lentiginosum than those in other species in this section (Fig. 4); these findings may explain the endemic distribution and morphology of C. lentiginosum. This habitat diversification may accelerate the inner genetic diversification in this section.
Genes Related to Species Diversification of Section Trigonopedia
The positive selection and DEGs provide clues to the species differentiation of this section. Floral scents function as olfactory cues for pollinator attraction and play a vital role in reproductive isolation. Studies of sexually deceptive orchids have indicated that the interaction between floral scent and pollinators promotes speciation, e.g., Chiloglottis46 and Ophrys3,47,48. This phenomenon has also been discovered in other non-deceptive pollination groups, e.g., Silene49, Petunia50, and Mimulus51. Waelti et al.49 found that a difference in a single compound can affect the pollination efficiency. Previous studies have indicated that the metabolisation of floral scent compounds is related to multiple biosynthesis pathways, including the biosynthesis of terpenoids, phenylpropanoids/benzenoids, volatile fatty acid derivatives, and amino acid-derived volatiles52. Section Trigonopedia is pollinated by different species and exhibits a differentiation of floral scent composition33,34,35,36, which indicates that the floral scent-related genes are important for the species differentiation in this section.
We identified 12 candidate genes related to the biosynthesis of floral scent compounds: five were identified from selection analysis, and seven were identified from differential expression analysis. These genes cover nine gene pathways, e.g., fatty acid biosynthesis, fatty acid degradation, ether lipid metabolism, nicotinate and nicotinamide metabolism, diterpenoid biosynthesis, and biosynthesis of terpenoids and steroids (Tables S1, S2). These genes provide clues to the molecular basis of floral scent differentiation. The site model indicated that accB (dN/dS = 1.58077) has an elevated evolutionary rate. accB (the biotin carboxyl carrier) is the subunit of acetyl-CoA carboxylase, which initiates the synthesis of fatty acids. In addition, the branch model indicated that FAB2 in C. bardolphianum (dN/dS = 1.0167) and C. micranthum (dN/dS = 1.1665) have an elevated evolutionary rate. The FAB2 gene encoding acyl-[acyl-carrier-protein] desaturase is a homolog of SAD, which has an important role in the reproductive isolation in sexually deceptive orchids53,54. Sedeek et al.54 even proposed that SAD5 is a speciation gene in Ophrys.
Our DEG findings indicate that species-specific and section-shared genes both contribute to the floral scent differentiation. The interplay between section-shared DEGs and species-specific DEGs may leads to the differentiation of floral scent. We identified seven section-shared candidate genes involved in the biosynthesis of floral scent compounds; for example, ACSL was a section-shared DEG involved in both fatty acid biosynthesis and fatty acid degradation, MFP2 is related to fatty acid degradation, GA3 and E1.14.11.13 are related to diterpenoid biosynthesis, and crtB is related to the biosynthesis of terpenoids and steroids (Table S2). In addition, we identified species-specific DEGs; for example, we identified ispF as being related to terpenoid backbone biosynthesis in C. lentiginosum and ispD as being related to terpenoid backbone biosynthesis in C. bardolphianum. Due to the limitations of our study, there may be more species-specific DEGs involved in floral scent differentiation.
The other group of candidate genes involved in speciation is related to environmental adaptation. E.g. crassulacean acid metabolism might resulted from differential gene expression28. Our findings indicate that the “Environmental Information Processing” and “Energy Metabolism” genes in this section would be important for habitat differentiation. For example, rpoB encodes the RNA polymerase beta subunit, and its mutation causes extensive changes in gene expression in Escherichia coli55. We identified the positive selection of genes of the arachidonic acid metabolism pathway in C. bardolphianum (LTA4H) and C. fargesii (gpx); reportedly, this pathway is involved in desert adaptation in animals (Table 1). Interestingly, we detected seven positively selected genes involved in the spliceosome, five in C. micranthum (PPIE, SNRPD3, HNRNPA1_3, SF3B2, and BCAS2), four in C. fargesii (SNRPD3, SF3B2, SF3A2, and TXNL4A), and one in C. lentiginosum (BCAS2) (Table S1). Alternative splicing is closely associated with many abiotic stresses (salt stress, drought, flooding, temperature, etc.)56 and contributes to the evolution of phenotypic novelty57. We also detected genes related to DNA repair and recombination, including nucleotide excision repair and homologous recombination. Qiao et al.58 detected these genes in Crucihimalaya himalaica, with positive selection related to environmental adaptation on the Qinghai-Tibetan Plateau.
These genes involved in floral scent and environmental adaptation may be related to the species diversification of this section. This finding is compatible with field observations. The complex topography of the Qinghai-Tibetan Plateau provides diverse habitats, and some species of this section also inhabit differential habitats, which accelerates the speciation process of this genus. Floral scent pathway genes induced species differentiation, and then habitat adaptation accelerated the speciation of the section. Our findings provide insight into the role of floral scent in pollinator attraction, followed by adaptation to different habitats, and finally leading to speciation.
Conclusions
This study provides valuable genomic resources for studying the molecular mechanism of closely related deceptively pollinated species with large and complex genomes. Our findings indicate that multiple genetic changes (positive selection and gene expression changes) may have contributed to the speciation process of this section and give clues to the molecular basis of floral scent differentiation. The Qinghai-Tibetan Plateau is the diversity centre of many species, and the differentiation of the species here is important for understanding species diversity in this region. Though a series of studies have been carried out, the genetic basis of species diversification is still under study. In this study, we investigated the diversification of section Trigonopedia through high-throughput sequencing and found candidate genes related to species diversification in this section through substitution rate analysis and differential expression analysis. Species diversification in this section involves the interplay between pollinator differentiation and habitat adaptation. Furthermore, the genetic basis of species differentiation provides new insights into orchid diversity and sheds light on the species diversification of orchids on the Qinghai-Tibetan Plateau.
Materials and Methods
RNA Extraction and Transcriptome Sequencing
We collected all the samples from fields in Sichuan and Yunnan, including six species (C. bardolphianum, C. fargesii, C. lentiginosum, C. margaritaceum, C. micranthum, and C. sichuanense) of section Trigonopedia and two species (C. flavum and C. singchii) outside this section that served as an outgroup. The detailed sample information is shown in Table 1. The RNA materials collected were stored in RNAlater solution (Ambion) and preserved at −80 °C before RNA isolation.
Total RNA was isolated from leaves and flowers using a Tiangen® RNAprep Pure Plant Kit (Tiangen, Beijing). The transcriptome library construction and Illumina sequencing were performed by Novogene (Beijing, China).
Sequence Assembly, Functional Annotation, and Orthologue Identification
The raw reads were filtered to obtain high-quality clean reads by removing the adapter sequences or low-quality reads. The clean reads were deposited in the NCBI Short Read Archive (SRA) under the accession number SRP151832.
The clean reads were de novo assembled using Trinity (released 20140717)59, and contigs less than 300 bp were excluded. Cd-Hit-Est60 was used for a clustering analysis to reduce sequence redundancy. Then, the filtered sequences were translated with TransDecoder61. The translated amino acid sequences were annotated through blast to the PFAM database by InterProScan 562. The annotated sequences were aligned with all-to-all blast by OrthoMCL63 with an e-value cut-off of 10−5 for gene family clustering. We constructed two databases: the first database comprised all eight species, and the other database was composed of the six ingroup species. The CDS sequences of each single-copy gene were aligned with MUSCLE64. Then, the alignments were manually refined in BioEdit 7.2.565. Finally, 1572 single-copy genes remained for phylogenetic analyses and 2592 single-copy genes remained for substitution rate estimation.
Phylogenetic Analyses
We screened single-copy genes for the phylogeny construction. We aligned each fragment with MUSCLE64 and concatenated the fragments for phylogeny construction. Then, we used RAxML-HPC BlackBox 8.2.1066 for the construction of the ML tree using the GTR + GAMMA model; this analysis was performed on the CIPRES Science Gateway V.3.3 platform67. We used the ML tree for the downstream analyses for positive selection.
Substitution Rate Estimation
The CodeML programme implemented in PAML 4.968 was used to estimate the non-synonymous substitution rate (dN) and synonymous substitution rate (dS) for each gene using the F3X4 codon model. The species tree ((C. lentiginosum, (C. margaritaceum, (C. fargesii, C. sichuanense))), (C. bardolphianum, C. micranthum)) was used as the guide tree. In addition, we estimated the dN/dS ratio of each branch using the free-ratio model (Model = 1) for each orthogroup. We conducted the boxplot analysis using the dN/dS values derived from the free-ratio model results and filtered for dN/dS > 3 or dN/dS < 0.0001. We also established frequency distribution plots of all the dN/dS ratios of the six species. To elucidate the biological functions of positive selected genes, we performed KEGG pathway analysis.
Differential Expression Analysis, Annotation of DEGs, and Cluster Analysis
Gene expression levels were estimated with RSEM by assembling the clean reads of each sample to Trinity assemblies69. We then performed differential expression analysis using the edgeR package in R70. The identified DEGs were used for GO analysis. GO functional annotations were obtained for all DEGs using InterProScan 562. GO enrichment analysis of the DEGs was performed using the WEGO online tool (http://wego.genomics.org.cn/)71. We then performed KEGG analysis for the DEGs.
References
Cozzolino, S. & Widmer, A. Orchid diversity: an evolutionary consequence of deception? Trends Ecol. Evol. 20, 487–494 (2005).
Raguso, R. A. Start making scents: the challenge of integrating chemistry into pollination ecology. Entomol. Exp. Appl. 128, 196–207 (2008).
Gervasi, D. D. et al. Floral scent and species divergence in a pair of sexually deceptive orchids. Ecol. Evol. 7, 6023–6034 (2017).
Wong, D. C. J. & Matus, J. T. Constructing Integrated Networks for Identifying New Secondary Metabolic Pathway Regulators in Grapevine: Recent Applications and Future Opportunities. Front. Plant Sci. 8, 505, https://doi.org/10.3389/fpls.2017.00505 (2017).
Grant, V. Pollination Systems as Isolating Mechanisms in Angiosperms. Evolution 3, 82–97 (1949).
Johnson, S. D. Pollinator-driven speciation in plants. In The ecology and evolution of flowers, (Harder L. D., Barrett S. C. H. eds). Oxford University Press, Oxford, pp. 295–310 (2006).
Schiestl, F. & Schluter, P. Floral Isolation, Specialized Pollination, and Pollinator Behavior in Orchids. Annu. Rev. Entomol. 54, 425–446 (2009).
Sedeek, K. E. M. et al. Transcriptome and proteome data reveal candidate genes for pollinator attraction in sexually deceptive orchids. PLoS ONE 8, e64621, https://doi.org/10.1371/journal.pone.0064621 (2013).
Wong, D. C. J. et al. Tissue-Specific Floral Transcriptome Analysis of the Sexually Deceptive Orchid Chiloglottis trapeziformis Provides Insights into the Biosynthesis and Regulation of Its Unique UV-B Dependent Floral Volatile, Chiloglottone 1. Front. Plant Sci. 8, 1260, https://doi.org/10.3389/fpls.2017.01260 (2017).
Xu, H. et al. Complex Sexual Deception in an Orchid Is Achieved by Co-opting Two Independent Biosynthetic Pathways for Pollinator Attraction. Curr. Biol. 27, 1867–1877 (2017).
Wong, D. C. J., Pichersky, E. & Peakall, R. The Biosynthesis of Unusual Floral Volatiles and Blends Involved in Orchid Pollination by Deception: Current Progress and Future Prospects. Front. Plant Sci. 8, 1955, https://doi.org/10.3389/fpls.2017.01955 (2017).
Chang, Y.-Y. et al. Characterization of Oncidium ‘Gower Ramsey’transcriptomes using 454 GS-FLX pyrosequencing and their application to the identification of genes associated with flowering time. Plant Cell Physiol. 52, 1532–1545 (2011).
Fu, C.-H. et al. OrchidBase: a collection of sequences of the transcriptome derived from orchids. Plant Cell Physiol. 52, 238–243 (2011).
Su, C. et al. De novo assembly of expressed transcripts and global analysis of the phalaenopsis aphrodite transcriptome. Plant Cell Physiol. 52, 1501–1514 (2011).
Liang, S. et al. Transcriptional regulations on the low-temperature-induced floral transition in an Orchidaceae species, Dendrobium nobile: an expressed sequence tags analysis. Comp. Funct. Genomics 2012 https://doi.org/10.1155/2012/757801 (2012).
Monteiro, F. et al. Labellum transcriptome reveals alkene biosynthetic genes involved in orchid sexual deception and pollination-induced senescence. Funct. & Integr. Genomics 12, 693–703 (2012).
Chou, M.-L. et al. Global transcriptome analysis and identification of a CONSTANS-like gene family in the orchid Erycina pusilla. Planta 1–17 (2013).
Li, X. et al. Deep sequencing-based analysis of the Cymbidium ensifolium floral transcriptome. PLoS ONE 8, e85480, https://doi.org/10.1371/journal.pone.0085480 (2013).
Tsai, W.-C. et al. OrchidBase 2.0: Comprehensive collection of Orchidaceae floral transcriptomes. Plant Cell Physiol. 54, e7 (2013).
Zhang, J. et al. Transcriptome analysis of Cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genom. 14, 279, https://doi.org/10.1186/1471-2164-14-279 (2013).
Zhao, M.-M., Zhang, G., Zhang, D.-W., Hsiao, Y.-Y. & Guo, S.-X. ESTs analysis reveals putative genes involved in symbiotic seed germination in Dendrobium officinale. PLoS ONE 8, e72705, https://doi.org/10.1371/journal.pone.0072705 (2013).
De Paolo, S. et al. De novo transcriptome assembly from inflorescence of Orchis italica: analysis of coding and non-coding transcripts. PLoS ONE 9, e102155, https://doi.org/10.1371/journal.pone.0102155 (2014).
Huang, J.-Z. et al. A De Novo Floral Transcriptome Reveals Clues into Phalaenopsis Orchid Flower Development. PLoS ONE 10, e0123474, https://doi.org/10.1371/journal.pone.0123474 (2015).
Xu, C., Zeng, B., Huang, J., Huang, W. & Liu, Y. Genome-Wide Transcriptome and Expression Profile Analysis of Phalaenopsis during Explant Browning. PLoS ONE 10, e0123356, https://doi.org/10.1371/journal.pone.0123356 (2015).
Zhang, J. et al. Transcriptome analysis of Dendrobium officinale and its application to the identification of genes associated with polysaccharide synthesis. Front. Plant Sci. 7, 5, https://doi.org/10.3389/fpls.2016.00005 (2016).
Chao, Y.-T. et al. Orchidstra 2.0-A Transcriptomics Resource for the Orchid Family. Plant cell Physiol. 58, e9, https://doi.org/10.1093/pcp/pcw220 (2017).
Yang, F. et al. Integrated mRNA and microRNA transcriptome variations in the multi-tepal mutant provide insights into the floral patterning of the orchid Cymbidium goeringii. BMC Genom. 18, 367, https://doi.org/10.1186/s12864-017-3756-9 (2017).
Zhang, L. et al. Origin and mechanism of crassulacean acid metabolism in orchids as implied by comparative transcriptomics and genomics of the carbon fixation pathway. Plant J. 86, 175–185 (2016).
Deng, H. et al. Evolutionary history of PEPC genes in green plants: Implications for the evolution of CAM in orchids. Mol. Phylogenet. Evol. 94, 559–564 (2015).
Li, J.-H. et al. Molecular phylogeny of Cypripedium (Orchidaceae: Cypripedioideae) inferred from multiple nuclear and chloroplast regions. Mol. Phylogenet. Evol. 61, 308–320 (2011).
Cribb, P. J. The Genus Cypripedium. Timber Press, Portland, Oregon (1997).
Perner, H. Sinopedilum — a new section of the genus. Cypripedium. Die Orchidee 59, 35–51 (2008).
Liu, Z.-J., Chen, L.-J., Rao, W.-H., Li, L.-Q. & Zhang, Y.-T. Correlation between numeric dynamics and reproductive behaviour in Cypripedium lentiginosum. Acta Ecol. Sin. 28, 111–121 (2008).
Zheng, G.-L. et al. Flowering and fruit set dynamics in Cypripedium. Acta Ecol. Sin. 30, 3182–3187 (2010).
Ren, Z. X., Li, D.-Z., Bernhardt, P. & Wang, H. Flowers of Cypripedium fargesii (Orchidaceae) fool flat-footed flies (Platypezidae) by faking fungus-infected foliage. Proc. Natl. Acad. Sci. USA 108, 7478–7480 (2011).
Li, P., Pemberton, R., Zheng, G. & Luo, Y. Fly pollination in Cypripedium: a case study of sympatric C. sichuanense and C. micranthum. Bot. J. Linn. Soc. 170, 50–58 (2012).
Li, P. Pollination Biology of Cypripedium (Orchidaceae) in Huanglong, Sichuan. Chinese Academy of Sciences, Beijing (2006).
Wen, J. et al. Transcriptome sequences resolve deep relationships of the grape family. PLoS ONE 8, e74394, https://doi.org/10.1371/journal.pone.0074394 (2013).
Ai, B. et al. Comparative transcriptome resources of eleven Primulina species, a group of “stone plants” from a biodiversity hotspot. Mol. Ecol. Resour. 15, 619–632 (2015).
Wang, H.-J., Li, W.-T., Liu, Y.-N., Yang, F.-S. & Wang, X.-Q. Resolving interspecific relationships within evolutionarily young lineages using RNA-seq data: An example from Pedicularis section Cyathophora (Orobanchaceae). Mol. Phylogenet. Evol. 107, 345–355 (2017).
Zeng, L. et al. Resolution of deep eudicot phylogeny and their temporal diversification using nuclear genes from transcriptomic and genomic datasets. New Phytol. 214, 1338–1354 (2017).
Álvarez, I. & Wendel, J. F. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 29, 417–434 (2003).
Chen, S.-C. et al. The genus Cypripedium in China. Science Press. (2013).
Li, J.-H. Molecular Systematics and Biogeography of Cypripedium L. (Orchidaceae). Chinese Academy of Sciences, Beijing (2010).
Guo, Y.-Y., Luo, Y.-B., Liu, Z.-J. & Wang, X.-Q. Evolution and biogeography of the slipper orchids: Eocene vicariance of the conduplicate genera in the Old and New World tropics. PLoS ONE 7, e38788, https://doi.org/10.1371/journal.pone.0038788 (2012).
Peakall, R. et al. Pollinator specificity, floral odour chemistry and the phylogeny of Australian sexually deceptive Chiloglottis orchids: implications for pollinator-driven speciation. New Phytol. 188, 437–450 (2010).
Vereecken, N. J., Cozzolino, S. & Schiestl, F. P. Hybrid floral scent novelty drives pollinator shift in sexually deceptive orchids. BMC Evol. Biol. 10, 103, https://doi.org/10.1186/1471-2148-10-103 (2010).
Cuervo, M., Rakosy, D., Martel, C., Schulz, S. & Ayasse, M. Sexual Deception in the Eucera-Pollinated Ophrys leochroma: A Chemical Intermediate between Wasp- and Andrena-Pollinated Species. J. Chem. Ecol. 43, 469–479 (2017).
Waelti, M. O., Muhlemann, J. K., Widmer, A. & Schiestl, F. P. Floral odour and reproductive isolation in two species of Silene. J. Evol. Biol. 21, 111–121 (2008).
Klahre, U. et al. Pollinator choice in Petunia depends on two major genetic loci for floral scent production. Curr. Biol. 21, 730–739 (2011).
Byers, K. J. R. P. et al. Floral volatile alleles can contribute to pollinator-mediated reproductive isolation in monkeyflowers (Mimulus). Plant J. 80, 1031–1042 (2014).
Dudareva, N., Klempien, A., Muhlemann, J. K. & Kaplan, I. Biosynthesis, function and metabolic engineering of plant volatile organic compounds. New Phytol. 198, 16–32 (2013).
Schlüter, P. M. et al. Stearoyl-acyl carrier protein desaturases are associated with floral isolation in sexually deceptive orchids. Proc. Natl. Acad. Sci. USA 108, 5696–5701 (2011).
Sedeek, K. E. M. et al. Amino Acid Change in an Orchid Desaturase Enables Mimicry of the Pollinator’s Sex Pheromone. Curr. Biol. 26, 1505–1511 (2016).
Rodríguez-Verdugo, A., Tenaillon, O. & Gaut, B. S. First-Step Mutations during Adaptation Restore the Expression of Hundreds of Genes. Mol. Biol. Evol. 33, 25–39 (2015).
Mei, W., Boatwright, L., Feng, G., Schnable, J. C. & Barbazuk, W. B. Evolutionarily Conserved Alternative Splicing Across Monocots. Genetics 207, 465–480 (2017).
Bush, S. J., Chen, L., Tovar-Corona, J. M. & Urrutia, A. O. Alternative splicing and the evolution of phenotypic novelty. Philos. Trans. Royal Soc. B 372, 20150474, https://doi.org/10.1098/rstb.2015.0474 (2017).
Qiao, Q. et al. Transcriptome sequencing of Crucihimalaya himalaica (Brassicaceae) reveals how Arabidopsis close relative adapt to the Qinghai-Tibet Plateau. Sci. Rep. 6, 21729, https://doi.org/10.1038/srep21729 (2016).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98 (1999).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), 1–8; https://doi.org/10.1109/GCE.2010.5676129. (2010).
Yang, Z. PAML4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Li, B. & Dewey, C. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 12, 323, https://doi.org/10.1186/1471-2105-12-323 (2011).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Ye, J. et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297 (2006).
Acknowledgements
The authors acknowledge support from Funds for forestry reform and development of National Forestry and Grassland Administration (No. 2017, 126) and Funds for Forestry development and protection of Guangdong Province (No. 2017, 94). The authors thank also Dr. Yi-Bo Luo and Hai-Qin Sun from the Institute of Botany, Chinese Academy of Sciences, and Xu-Hui Chen from The National Orchid Conservation Center of China for their kind help in sample collection. The authors also thank Mr. Chun-Ping Liang and Yong Zheng (Wanglang Nature Reserve), Mr. Chang-Bao Tian (Huanglong Nature Reserve), Mr. Zhong-Fu Zhu (Jiuzhaigou Nature Reserve), and Mr. Xi-Bin Guo (Malipo, Yunnan) for their assistance in field collection; Hong-Juan Wang, Yan-Yan Liu, and Jie-Yu Wang for data analysis; and Shan-Ce Niu, Mei-Na Wang, and Xin-Yi Wu for RNA isolation.
Author information
Authors and Affiliations
Contributions
Y.Y.G. and Z.J.L. designed the study. Y.Y.G. performed the laboratory work. Z.J.L., L.Q.H. and G.Q.Z. contributed reagents, materials and analysis tools. Y.Y.G. and Y.Q.Z. analysed the data. Y.Y.G. wrote the article.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Guo, YY., Zhang, YQ., Zhang, GQ. et al. Comparative transcriptomics provides insight into the molecular basis of species diversification of section Trigonopedia (Cypripedium) on the Qinghai-Tibetan Plateau. Sci Rep 8, 11640 (2018). https://doi.org/10.1038/s41598-018-30147-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-30147-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
