
Abstract
It is well established that brain-derived neurotrophic factor (BDNF) signaling pathway plays a key role in the pathophysiology of major depressive disorder (MDD) and in therapeutic mechanisms of antidepressants. We aim to identify genetic vairiants related to MDD susceptibility and antidepressant therapeutic response by using gene-based association analysis with genes related to the neurotrophic pathway. The present study investigated the role of genetic variants in the 10 neurotrophic-related genes (BDNF, NGFR, NTRK2, MTOR, VEGFA, S100A10, SERPINE1, ARHGAP33, GSK3B, CREB1) in MDD susceptibility through a case-control (455 MDD patients and 2,998 healthy controls) study and in antidepressant efficacy (n = 455). Measures of antidepressant therapeutic efficacy were evaluated using the 21-item Hamilton Rating Scale for Depression. Our single-marker and gene-based analyses with ten genes related to the neurotrophic pathway identified 6 polymorphisms that reached a significant level (p-value < 5.0 × 10−3) in both meta- and mega-analyses in antidepressant therapeutic response. One polymorphism was mapped to BDNF and 5 other polymorphisms were mapped to VEGFA. For case-control association study, we found that all of these reported polymorphisms and genes did not reach a suggestive level. The present study supported a role of BDNF and VEGFA variants in MDD therapeutic response.
Similar content being viewed by others

Introduction
Depression is a severe mental disorder and the leading cause of disabilities worldwide1. The reported prevalence throughout the world of depressive episodes is 1,607 per 100,000 per year for males and 2,552 per 100,000 per year for females2. Currently, the main medical treatment for depression is antidepressant medication. The selective serotonin reuptake inhibitors (SSRIs), including fluoxetine, sertraline, fluvoxamine, paroxetine and citalopram, are a popular family of antidepressants frequently prescribed at present. However, as with all antidepressant treatments, about 30–40% of major depressive disorder (MDD) patients do not respond sufficiently to SSRIs3. As evidence from earlier studies had indicated, genetic factors may play important roles in antidepressant responses4,5, and pharmacogenetic SSRI studies have attempted to identify genetic variants which predict antidepressant treatment response. Pharmacogenetics is the study of variability in drug response due to heredity, generally focusing on polymorphisms of genes related to the drug metabolizing-enzyme, drug action or disease pathophysiology6.
For candidate genes related to antidepressant therapeutic action, brain-derived neurotrophic factor (BDNF) has been a main focus of antidepressant pharmacogenetic research. BDNF, a member of the neurotrophin family, is a small dimeric protein widely expressed in adult mammalian brains with the highest levels found in the hippocampus7. BDNF plays a key role in the regulation of neuronal survival, differentiation, growth, and apoptosis by binding to two types of receptors; namely, tyrosine kinase B (trkB; encoded by the NTRK2 gene) receptor and the p75 neurotrophin receptor (p75NTR; encoded by the NGFR gene). Earlier animal studies have demonstrated that stress of immobilization can lower BDNF mRNA levels in the hippocampus and other brain regions8. The role of BDNF in depression treatments was first revealed in research conducted by Nibuya and colleagues, where long-term administration of several types of antidepressants, including SSRIs, increases in BDNF expression in the rat hippocampus9. Furthermore, centrally administered BDNF produces antidepressant-like activities in animal models of depression10. In humans, post-mortem studies demonstrated an increase in BDNF immunoreactivity in the hippocampus of MDD subjects treated with antidepressant medication at the time of death, compared with untreated controls11. Many clinical studies also found that BDNF levels were significantly lower in MDD patients than in controls, and decreased serum levels of BDNF in MDD patients recovered to normal levels associated with the recovery of depression after treatment with antidepressant medication12. The above findings suggest that BDNF may be implicated in the pathogenesis of MDD and in antidepressant actions13, and may be a good candidate gene for antidepressant pharmacogenetic study.
The human BDNF gene has been mapped to chromosome 11p13. A common single nucleotide polymorphism (SNP) consisting of a missense change (G196A), producing a non-conservative amino acid change (valine to methionine), has been identified in the coding exon of the BDNF gene at position 66 (Val66Met, rs6265). The replacement of 66Val by 66Met disrupts cellular processing, trafficking, and activity-dependent secretion of BDNF14. In our 2003 study on 110 MDD patients, we examined the association between the BDNF Val66Met polymorphism and response to 4-week antidepressant (fluoxetine) treatment15. We found a trend showing better therapeutic response for the Val/Met-heterozygote patients in comparison to those bearing the homozygote (Val/Val or Met/Met). While similar findings have been reported in some of the subsequent studies, other studies found a better response in patients carrying the Met variant16,17. With the importance of BDNF in antidepressant therapeutic mechanism, BDNF gene has been the focus of antidepressant pharmacogenetic studies. Most of the BDNF-antidepressant pharmacogenetic studies investigated only the BDNF Val66Met polymorphism which may overlook other BDNF polymorphisms. Furthermore, a single gene may play only a small part in the antidepressant therapeutic response. Genes related to the BDNF function may interact with BDNF in response to antidepressant treatment.
Several genes related to neurotrophic pathway have also been implicated in the mechanisms underlying depression and drug action. For example, we have demonstrated that the NTRK2 genetic variants interact with the BDNF Val66Met polymorphism contributing to the risk of geriatric depression18. One missense NGFR polymorphism (S250L) showed association with antidepressant therapeutic response19.We found that polymorphisms in the GSK3B gene (encoding glycogen synthase kinase-3 beta protein), an important component in the BDNF pathway, were associated with the antidepressant therapeutic response20. We also found that genetic variants in plasminogen activator inhibitor type 1 gene (encoded by the SERPINE1 gene), which is involved in the cleavage of pro-BDNF to mature BDNF in the brain, are related to antidepressant therapeutic response and depression susceptibility20. Vascular endothelial growth factor (VEGF) is known to play a role in the process for neuroprotection and neurogenesis, and may be involved in the pathogenesis of some neurological disorders21. VEGF is encoded by VEGFA. S100 calcium-binding protein A10 (protein encoded by the S100A10), also known as p11, has a possible role in major depression’s pathophysiology22 and may also be involved in the cleavage of proBDNF to BDNF23. ARHGAP33 is a new type of regulator for the intracellular trafficking of TrkB signaling and is essential for synapse development. Dysfunction of this mechanism may be a new molecular pathology of neuropsychiatric disorders24. CREB protein (encoded by CREB1), a cellular transcription factor, plays a crucial role in turning on the BDNF gene25. Mammalian target of rapamycin (mTOR protein encoded by MTOR), a large serine/threonine kinase, regulates the initiation of protein translation in the body. A clinical study has found that ketamine produces rapid antidepressant effects in depressive patients26. An animal study demonstrated that ketamine stimulates AMPA receptor transmission and activates BDNF/TrkB-Akt/ERK-mTOR signaling cascades, leading to a sustained increase in synaptic protein synthesis and strengthening of synaptic plasticity27.
Pharmacogenomics, which is derived from genome-wide association (GWA) studies and pharmacogenetics, are proving to be increasingly useful in personalized medicinal research. Despite the progress from single SNP studies to GWA studies in antidepressant treatment response, results were not as expected as they were often inconsistent28. GWA studies have a much lower power when the number of SNPs increases and the SNPs are correlated, especially when their effect sizes are small29. Gene-based analysis is the SNP-based method, in which each SNP is tested for association, and multiple testing corrections based on the Bonferroni procedure are applied to control the type-I error rate. The concept of a gene-based association test has been broadly applied to pharmacogenomics studies30. The most important merit of SNP/gene-based analysis for pharmacogenomic studies over GWA studies is that we can target the genotyping of SNPs/genes efficiently provided the mechanisms of drug action are known. These selected SNPs/genes are connected with treatment responses, which can help in predicting the progress of a disease and use to the selection of targeted therapies in pharmacogenetic studies31. The strategy of SNP/gene-based analysis for pharmacogenetic studies provides clinicians to better use of knowledge of a list of genetic susceptible loci to link candidate genes and clinical drug related responses in order to make a rational treatment decision32. In this study, we conducted gene-based as well as single marker analyses in 10 genes related to the BDNF signaling pathway to identify genetic variation that may affect MDD risk or antidepressant therapeutic response.
Materials and Methods
Samples
We recruited 455 patients, 268 from NHRI (The National Health Research Institutes) and 187 from TVGH (Taipei Veterans General Hospital), who met DSM-IV criteria for major depressive disorder. The diagnosis was made by board-certified psychiatrists who interviewed patients and family members, and obtained records where possible. All subjects were Taiwanese. Subjects were part of the International SSRI Pharmacogenomics Consortium (ISPC) project, which included seven member sites from five countries33. Other inclusion criteria were minimum baseline score of 14 on the 21-item Hamilton Rating Scale for Depression (HRSD), and presence of depressive symptoms for at least two weeks before entry into the study without antidepressant treatment during that period (patients were fresh cases or had quit antidepressants for more than 2 weeks). Exclusion criteria were additional current DSM-IV Axis I diagnoses (including substance abuse, generalized anxiety disorders, panic disorders, or obsessive compulsive disorders), personality disorders, pregnancy, recent suicide attempt, and major medical and/or neurological disorders.
These patients were treated with SSRIs, which include escitalopram (38.5%), paroxetine (38.5%), fluoxetine (18.3%) and citalopram (4.8%). Treatment was initiated with a recommended starting dose (for example, the escitalopram dose was 10 mg/day in the beginning), and based on the clinical response after 2-week treatment the investigator could adjust the antidepressant dosage. The final mean antidepressant doses were 12.0 mg/day for escitalopram, 16.9 mg/day for paroxetine, 20.0 mg/day for fluoxetine and 19.0 mg/day for citalopram.
All participants were evaluated using the 21-item HRSD to measure depression severity. Patients were assessed repeatedly at baseline and week 2, 4, and 8. All raters for the HRSD received the same investigators’ training module. For the evaluation of the NHRI subjects, the inter-rater reliability coefficient was consistent, with a value of 0.9. The TVGH patients were evaluated by a senior psychiatrist (YWY).
Healthy controls were drawn from the Taiwan Biobank with a Taiwanese Han ancestry34, consisted of 2,998 participants in the current study (nearly equal proportions of males and females), whom were interviewed by trained interviewers with standard questionnaires to collect information of demographic variables, life-style and physical health conditions as well as clinical data. All controls were found to have no definite diagnosis of any major medical or mental illnesses, they underwent genotyping at the genome-wide level and were thus treated as controls in the current study.
Experiments were conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of Taipei Veterans General Hospital (VGHIRB No.: 2014-06-001B). Written informed consents were obtained from all participants ensuring adequate understanding of the study.
Measurement
We used four repeated phenotypes to measure different treatment responses, including remitted, response, and stem-depressed. First, we calculated the sum score of 21-item HRSD at week 8, and recoded as 1 if the sum score greater than 7 and 2 (i.e., remitted) otherwise. Second, we calculated the percentage change of HRSD (i.e., %ΔHRSD), and recoded the results as 1 if the percentage change greater than −50% and 2 (i.e., response) otherwise. Third, we also used the percentage change of HRSD as a treatment-response index. Last, we used the score of ‘depressed mood’ item to measure depression, and recoded the results as 1 if the score greater than or equal to 3 and 2 (i.e., stem-depressed) otherwise. For detailed definitions of treatment-response phenotypes, please refer to Lin et al.35.
Genotyping data and quality controls
All participants were genotyped using Illumina HumanOmniExpressExome BeadChips in the International SSRI Pharmacogenomics Consortium. A total of 455 subjects were genotyped with 951,123 SNPs. We selected 10 SSRIs treatment-responses or depression related candidate genes (BDNF, NGFR, NTRK2, MTOR, VEGFA, S100A10, SERPINE1, ARHGAP33, GSK3B, CREB1), which are related to the neurotrophic pathway. A total of 1,251 and 2,656 SNPs were mapped into the 10 selected candidate genes in mega- and meta-analysis, respectively.
Quality control procedures were done firstly with each individual, including sample quality, kinship and population stratification. We first check plate-wise genotyping biases. Samples with plate pass rate greater than 97% were retained in the analysis. A total of 18 samples (11 from NHRI and 7 from TVGH) were removed during this step. Second, we checked the inbreeding coefficient and identity by state (IBS), so that samples with strong kinship were eliminated. In total, 9 individuals (4 from NHRI and 5 from TVGH) were removed from the samples due to having similar measures which are far away from the clustering. Third, we used a multidimensional scaling analysis on the genome-wide IBS pairwise distance to eliminate samples with outliers. Our results showed that none was away from the clustering on the scatter plot. Finally, 7 patients treated with sertraline (SSRI) and venlafaxine (SNRI) were excluded from the analysis. As a result, 421 (mean age of 43.7 years and 71.3% of females) MDD patients were retained.
We also performed quality control procedures for markers. We removed markers which failed the Hardy-Weinberg tests with a p-value less than 0.0001, genotype missing rate greater than 5%, minor allele frequency (MAF) smaller than 0.05, or ones with bad calling in clustering. As a result, a total of 647,030 SNPs in the samples were retained for imputation. The genotyping call rate was 99.9% for all subjects.
Imputation and gene mapping
Imputation was carried out using IMPUTE2 v336, with haplotype reference panels released in March/April 2012 from the 1000 Genomes Project on the basis of HapMap build 37 (https://mathgen.stats.ox.ac.uk/impute/data_download_1000G_phase1_integrated_SHAPEIT2.html). Only imputed SNPs with high genotype information content (i.e. IMPUTE info score > 0.5) were used in association analyses. In total, 30,040,257 SNPs were imputed with high confidence for each individual in the samples. Following the same quality control procedures for markers, a total of 4,241,701 SNPs were retained for analyses. Gene-mapping was conducted using 50 kb upstream and downstream of the gene boundaries.
Multiple imputation method for missing phenotypic data
To deal with the issue of missing data, we used multiple imputation approach to account uncertainty of the imputed data. We first created multiple-imputed datasets, by fitting regression models based on observed complete data from other variables to predict missing data multiple times (number of iterations was set to 30 by default) to account for uncertainty. Second, we performed standard statistical analyses on each of imputed datasets. Third, we pooled the results of each of these analyses to produce one set of results. The method of multiple imputation overcame the limitations of single imputation (e.g. LOCF) to provide more realistically estimated precision and less biased results. This approach is beneficial with several advantages, including unbiased parameter estimates, robust departures from normality assumptions, adequate results when sample size is small or high rates of missing data, computationally simple, and tractable solution to missing data problems.
Genetic association analyses
We performed single-marker association and gene-based association tests for treatment-response phenotypes. Both linear and logistic regression analysis were conducted with additive genetic model. All models were adjusted for gender and age to correct for different distributions in gender and age across treatment-response phenotypes in MDD patients.
Gene-based association analyses were conducted to obtain gene-level empirically significant estimations. Information from a set of SNPs (association p-value < 0.1 by default) within a gene was aggregated. To account for the linkage disequilibrium (LD) among markers, only SNPs having r2 < 0.5 with each other were retained for each gene. We also fitted both linear and logistic regression models to perform gene-based association tests. Empirical p-values were calculated with 50,000 permutations.
To combine individual association results (i.e. p-values) from the two samples (i.e., NHRI and TVGH), meta-analysis was applied using the inverse Gamma model with a shape parameter (α) of 1, that is, the Fisher’s method37 to summarize association information across the two samples. Alternatively, we conducted association analyses to an integrated database of NHRI and TVGH. We adopted a method of LD adjusted multiple testing correction developed by Duggal et al.38 to account for the interdependence among SNPs to balance between false-negative and false-positive findings. Only SNPs and genes with p-values less than 5.0 × 10−4 (or 5.0 × 10−3) and 5.0 × 10−2 (or 1.0 × 10−2), either in mega-analysis or in meta-analysis, were considered to be significant (or suggestive), and were reported in single-marker association analysis and gene-based association analysis, respectively. Additionally, we conducted a case-control (455 MDD patients and 2,998 healthy controls) study using SNP-based and gene-based association analyses to investigate whether the treatment-response associated loci were also MDD susceptible loci.
All aforementioned analyses were conducted with R version 3.0.2, PLINK version 1. 90b3.37 64-bit, and haploview version 4.1. Additionally, to explore the potential roles of these six SNPs as expression quantitative trait locus (eQTL), we used HaploReg (http://compbio.mit.edu/HaploReg) to search gene regulation databases.
Results
Among 428 subjects, 394 (92.06%) cases were observed at all four time points; 30 cases (7.02%) were missing at just one time-point of follow-up; and the remaining 4 cases (0.92%) were missing at two or more time-points of follow-up. Table 1 exhibits detailed patterns of missing data in phenotypes across different time points, indicating the issue of incomplete data. Here we conducted multiple imputation method to impute these missing data using available observed data, such that our data are complete. Furthermore, we excluded 7 subjects (4 treated with sertraline and 3 with venlafaxine) from the analysis. As a result, 421 MDD patients were retained for the following analyses. Table 2 represents summary statistics for treatment-response phenotypes. All three treatment-response phenotypes (remitted, response, and stem-depressed) showed significant difference (p-value < 2.2 × 10−16) between treatment responders and treatment non-responders. We found that 373 (88.6%) treatment-responders had moderate or less depressed mood compared to 48 (11.4%) treatment non-responders who had moderately severe or severe depressed mood. No gender difference was observed among all treatment-response phenotypes.
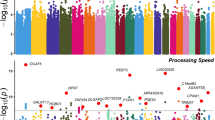
In single-marker association analyses, 64 SNPs with odds ratios (ORs) ranged from 0.18 to 2.88 in NHRI samples and ranged from 0.19 to 4.22 in TVGH samples (0.92–1.99 in combined samples) were reported to have suggestive signals with treatment-response phenotypes, which reached p-value < 5.0 × 10−3 either in mega-analysis or in meta-analysis (Table 3). Among them, only six SNPs reached significant levels (p-value < 5.0 × 10−3) in both meta-analysis and mega-analysis. One SNP, chr11:27682769:D (mapped to BDNF) was significantly associated with remitted (Score) (please see Figs 1 and 2 for their Manhattan plot of meta-analysis and mega-analysis), and 5 SNPs (rs6920449, rs1535507, rs833051, rs833052, and rs2148252), which were mapped to VEGFA, were significantly associated with response (%ΔHRSD, continuous) (please see Figs 3 and 4 for their Manhattan plot of meta-analysis and mega-analysis). Other 58 SNPs were significantly associated with treatment-response phenotypes in either meta-analysis or mega-analysis. Results of gene-based association analyses are listed in Table 4. Only BDNF gene in remitted (Score) and VEGFA gene in response (%ΔHRSD, binary and continuous) showed suggestive signals (p-values < 5.0 × 10−2) in mega-analysis and/or meta-analysis. These indicated that the possible mechanisms of association may be due to the neurotrophic pathway and serotonin system pathway. Furthermore, the most studied polymorphism Val66Met (rs6265) in BDNF is included in particular and the results of single-marker association and gene-based association were non-suggestive.

Manhattan plot of meta-analysis of selected candidate genes for Remitted (Score).

Manhattan plot of mega-analysis of selected candidate genes for Remitted (Score).

Manhattan plot of meta-analysis of selected candidate genes for Response (%ΔHRSD, continuous).

Manhattan plot of mega-analysis of selected candidate genes for Response (%ΔHRSD, continuous).
Table 5 exhibits summary of eQTL prediction. We found that 4 SNPs (rs1535507, rs833051, rs833052, rs2148252) exhibited direct eQTL effects (in total 9 hits). The rs1535507 SNP is involved in regulating expressions of RSPH9 and MAD2L1BP |VEGF in dendritic cells and in blood tissues. The rs833051 SNP is involved in regulating expressions of RP1-261G23.7 in cells transformed fibroblasts. The rs833052 SNP is involved in regulating expressions of MAD2L1BP and MRPS18A in blood tissue and in dendritic cells. The rs2148252 SNP is involved in regulating expressions of VEGFA in dendritic cells. The other two SNPs (rs6920449 and chr11:27682769:D) do not act as eQTLs, but SNPs in high LD (r2 ≥ 0.8) with rs6920449 are predicted to be eQTLs, which regulated several gene expressions, including MAD2L1BP (blood tissue), VEGFA (blood tissue), and RSPH9 (dendritic cells).
We also conducted a case-control association study using samples from NHRI and TVGH (421 MDD patients) and Taiwan Biobank (2,998 healthy controls). We found that all reported SNPs and genes did not reach the suggestive association level.
Discussion
The major finding is that 64 SNPs were reported to have suggestive signals with treatment-response phenotypes either in mega-analysis or in meta-analysis. Among them, 6 SNPs reached a significant level (p-value < 5.0 × 10−3) in both meta-analysis and mega-analysis. The genetic association directions of the 6 SNPs are the same in both study groups. The other 58 SNPs were significantly associated with treatment-response phenotypes in either meta-analysis or mega-analysis. Further, among the 10 genes tested, BDNF gene (in remitted) and VEGFA gene (in response) were suggestive markers for treatment-response phenotypes using samples from NHRI, TVGH, and the combined.
BDNF is essential for the mechanism of pharmacological therapies currently used to treat MDD39,40. Both BDNF mRNA and protein expressions are increased after long-term medication with antidepressants, including SSRIs, in the hippocampus in animal models9,41. Moreover, infusion of BDNF into the dentate gyrus of hippocampus produced antidepressant-like behaviors42. In support of these findings, a recent study demonstrated that knockdown of TrkB, but not Bdnf, in the dorsal raphe nucleus of mice results in loss of antidepressant efficacy43. Recent meta-analyses showed strong evidence that peripheral BDNF levels were lower in MDD subjects than healthy control subjects, and BDNF levels significantly increased after antidepressant treatment12,41. From the above findings, BDNF has been tested in many antidepressant pharmacogenetic studies, especially the functional BDNF Val66Met polymorphism16,44. However, most studies showed no association between this polymorphism and antidepressant response16. Meta-analysis of seven studies and the STAR*D data including a total of 3,128 subjects showed that the BDNF Val66Met polymorphism was not significantly associated with antidepressant therapeutic response45. In this study we found no association between this BDNF Val66Met polymorphism and SSRI therapeutic response. However, we found one SNP, chr11:27682769:D (mapped to BDNF), to be significantly associated with the remitted (Score). This SNP had ORs that were in the same direction in NHRI samples and TVGH samples, and our results of meta-analysis and mega-analysis suggested a statistically significant association between this SNP and the remitted. There was no eQTL effect for this SNP. It is still not clear how the mechanism of the chr11:27682769:D SNP is related to the SSRI therapeutic mechanism. Further studies are required to explore the functions of this BDNF SNP.
The other 5 SNPs (rs6920449, rs1535507, rs833051, rs833052, and rs2148252), which were significantly associated with SSRI therapeutic response, were mapped to VEGFA. VEGF is a member of growth factor family and has been implicated in neurotrophy and neurogenesis, which plays a pivotal role in brain development46. An animal study found that antidepressant drugs were shown to induce hippocampal expression of VEGF and that VEGF signaling through the Flk-1 receptor is required for antidepressant-induced cell proliferation47. In addition, the same research group demonstrated that infusions of inhibitors of VEGF-Flk-1 receptor signaling blocks the antidepressant-like activity of fluoxetine47. Study involving 30 MDD patients found that plasma VEGF levels significantly increased its association with clinical response with antidepressant48. With the significant role of VEGF in antidepressant action, we have tested 7 VEGFA polymorphisms and SSRI antidepressant response in 351 MDD patients49. No significant association with SSRI antidepressant therapeutic effect was shown in the alleles and genotypes of single loci, or haplotypes constructed from these polymorphisms. In this study, we also found no significant association between the 5 VEGFA SNPs and SSRI antidepressant response in both SNP-based and haplotype-based (data not shown) analyses using binary outcome (i.e., dichotomized %ΔHRSD), which is similar to the 7 VEGFA polymorphisms reported in our previous study. However, the 5 VEGFA SNPs found to be associated with SSRI antidepressant response using continuous outcome (i.e., %ΔHRSD) were different from the 7 VEGFA polymorphisms reported in our previous study, suggesting its simplicity of dichotomizing continuous variables gained at some cost (i.e., some of the information lost) in pharmacogenetic research. One benefit of using high-density imputed SNP mapping of antidepressant candidate genes is that some potentially significant signals for antidepressant treatment response could not be missed in pharmacogenetic studies.
We used HaploReg to predict a possible functional role of these 5 VEGFA SNPs. We found that 4 SNPs (rs1535507, rs833051, rs833052, and rs2148252) exhibited direct eQTL effects. Of note, functional annotation by HaploReg indicated that transcriptional regulation activity exists at the rs1535507, rs833052, and rs2148252 loci for the VEGFA gene in blood tissue and/or dendritic cells. Their functional impacts in antidepressant mechanism still need to be elucidated by further experimental studies.
Neurotrophic factors comprise major regulatory mechanisms controlling normal brain plasticity and neurogenesis. Dysfunctions of these systems form the basis for development of MDD50. For case-control association study using samples from NHRI and TVGH (421 MDD patients) and HCCGB (2,998 healthy controls), all these reported SNPs and genes did not reach significant level. This suggested that these tested SNPs and genes related to the neurotrophic pathway were not associated with MDD susceptibility. MDD is a complex mental disorder with a partly genetic etiology and each risk genetic locus confers relatively small increments in risk51. The negative association finding could be due to our sample size which lacks the statistical power to detect common variants of MDD susceptibility.
The major limitation of this study is the lack of a placebo lead-in design. It is well known that the placebo response plays an important role in antidepressant therapeutic response52. The second limitation of this study is that some potential confounding factors, such as side effects, incompliance or drop-out, were not under consideration, which might disturb the analysis in detecting a difference between the two treatment response groups. The third limitation is that our inclusion criterion for HRSD minimum score is 14, which is lower than other studies. Finally, plasma levels of antidepressants were not analyzed. Poor antidepressant response may result from sub-therapeutic plasma drug concentrations53.
In conclusion, our gene-based association identifies loci in BDNF and VEGFA as potential genetic predictors for SSRI therapeutic response in MDD patients. Future studies are needed to delineate the precise mechanism by which these genetic variants may influence antidepressant therapeutic response.
References
Whiteford, H. A. et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet 382, 1575–1586, https://doi.org/10.1016/S0140-6736(13)61611-6 (2013).
Ustun, T. B., Ayuso-Mateos, J. L., Chatterji, S., Mathers, C. & Murray, C. J. Global burden of depressive disorders in the year 2000. Br J Psychiatry 184, 386–392 (2004).
Hirschfeld, R. M. et al. Partial response and nonresponse to antidepressant therapy: current approaches and treatment options. J Clin Psychiatry 63, 826–837 (2002).
Pare, C. M. & Mack, J. W. Differentiation of two genetically specific types of depression by the response to antidepressant drugs. J Med Genet 8, 306–309 (1971).
O’Reilly, R. L., Bogue, L. & Singh, S. M. Pharmacogenetic response to antidepressants in a multicase family with affective disorder. Biol Psychiatry 36, 467–471 (1994).
Schosser, A. & Kasper, S. The role of pharmacogenetics in the treatment of depression and anxiety disorders. Int Clin Psychopharmacol 24, 277–288, https://doi.org/10.1097/YIC.0b013e3283306a2f (2009).
Hofer, M., Pagliusi, S. R., Hohn, A., Leibrock, J. & Barde, Y. A. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J 9, 2459–2464 (1990).
Ueyama, T. et al. Immobilization stress reduced the expression of neurotrophins and their receptors in the rat brain. Neurosci Res 28, 103–110 (1997).
Nibuya, M., Morinobu, S. & Duman, R. S. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 15, 7539–7547 (1995).
Siuciak, J. A., Lewis, D. R., Wiegand, S. J. & Lindsay, R. M. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol Biochem Behav 56, 131–137, https://doi.org/10.1016/S0091-3057(96)00169-4 (1997).
Chen, B., Dowlatshahi, D., MacQueen, G. M., Wang, J. F. & Young, L. T. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry 50, 260–265 (2001).
Zhou, C. et al. Meta-analyses of comparative efficacy of antidepressant medications on peripheral BDNF concentration in patients with depression. PLoS One 12, e0172270, https://doi.org/10.1371/journal.pone.0172270 (2017).
Duman, R. S. & Li, N. A neurotrophic hypothesis of depression: role of synaptogenesis in the actions of NMDA receptor antagonists. Philos Trans R Soc Lond B Biol Sci 367, 2475–2484, https://doi.org/10.1098/rstb.2011.0357 (2012).
Egan, M. F. et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112, 257–269 (2003).
Tsai, S. J., Cheng, C. Y., Yu, Y. W., Chen, T. J. & Hong, C. J. Association study of a brain-derived neurotrophic-factor genetic polymorphism and major depressive disorders, symptomatology, and antidepressant response. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics 123b, 19–22, https://doi.org/10.1002/ajmg.b.20026 (2003).
Tsai, S. J., Hong, C. J. & Liou, Y. J. Effects of BDNF polymorphisms on antidepressant action. Psychiatry investigation 7, 236–242, https://doi.org/10.4306/pi.2010.7.4.236 (2010).
Hong, C. J., Liou, Y. J. & Tsai, S. J. Effects of BDNF polymorphisms on brain function and behavior in health and disease. Brain Res Bull 86, 287–297, https://doi.org/10.1016/j.brainresbull.2011.08.019 (2011).
Lin, E. et al. Gene-gene interactions of the brain-derived neurotrophic-factor and neurotrophic tyrosine kinase receptor 2 genes in geriatric depression. Rejuvenation Res 12, 387–393, https://doi.org/10.1089/rej.2009.0871 (2009).
Gau, Y. T. et al. Evidence for association between genetic variants of p75 neurotrophin receptor (p75NTR) gene and antidepressant treatment response in Chinese major depressive disorder. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics 147B, 594–599, https://doi.org/10.1002/ajmg.b.30646 (2008).
Tsai, S. J., Liou, Y. J., Hong, C. J., Yu, Y. W. & Chen, T. J. Glycogen synthase kinase-3beta gene is associated with antidepressant treatment response in Chinese major depressive disorder. Pharmacogenomics J 8, 384–390, https://doi.org/10.1038/sj.tpj.6500486 (2008).
Storkebaum, E., Lambrechts, D. & Carmeliet, P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bioessays 26, 943–954, https://doi.org/10.1002/bies.20092 (2004).
Svenningsson, P. et al. Alterations in 5-HT1B receptor function by p11 in depression-like states. Science 311, 77–80, https://doi.org/10.1126/science.1117571 (2006).
Tsai, S. J. The P11, tPA/plasminogen system and brain-derived neurotrophic factor: Implications for the pathogenesis of major depression and the therapeutic mechanism of antidepressants. Med Hypotheses 68, 180–183, https://doi.org/10.1016/j.mehy.2006.06.005 (2007).
Nakazawa, T. et al. Emerging roles of ARHGAP33 in intracellular trafficking of TrkB and pathophysiology of neuropsychiatric disorders. Nat Commun 7, 10594, https://doi.org/10.1038/ncomms10594 (2016).
Lonze, B. E. & Ginty, D. D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 35, 605–623 (2002).
Krystal, J. H., Sanacora, G. & Duman, R. S. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry 73, 1133–1141, https://doi.org/10.1016/j.biopsych.2013.03.026 (2013).
Li, N. et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964, https://doi.org/10.1126/science.1190287 (2010).
Fabbri, C. & Serretti, A. Pharmacogenetics of major depressive disorder: top genes and pathways toward clinical applications. Curr Psychiatry Rep 17, 50, https://doi.org/10.1007/s11920-015-0594-9 (2015).
Kang, G., Jiang, B. & Cui, Y. Gene-based Genomewide Association Analysis: A Comparison Study. Curr Genomics 14, 250–255, https://doi.org/10.2174/13892029113149990001 (2013).
Baye, T. M. & Wilke, R. A. Mapping genes that predict treatment outcome in admixed populations. Pharmacogenomics J 10, 465–477, https://doi.org/10.1038/tpj.2010.71 (2010).
Ritchie, M. D. The success of pharmacogenomics in moving genetic association studies from bench to bedside: study design and implementation of precision medicine in the post-GWAS era. Hum Genet 131, 1615–1626, https://doi.org/10.1007/s00439-012-1221-z (2012).
Reynolds, G. P. The impact of pharmacogenetics on the development and use of antipsychotic drugs. Drug Discov Today 12, 953–959, https://doi.org/10.1016/j.drudis.2007.07.018 (2007).
Biernacka, J. M. et al. The International SSRI Pharmacogenomics Consortium (ISPC): a genome-wide association study of antidepressant treatment response. Transl Psychiatry 5, e553, https://doi.org/10.1038/tp.2015.47 (2015).
Fan, C. T., Lin, J. C. & Lee, C. H. Taiwan Biobank: a project aiming to aid Taiwan’s transition into a biomedical island. Pharmacogenomics 9, 235–246, https://doi.org/10.2217/14622416.9.2.235 (2008).
Lin, K. M. et al. CYP1A2 genetic polymorphisms are associated with treatment response to the antidepressant paroxetine. Pharmacogenomics 11, 1535–1543, https://doi.org/10.2217/pgs.10.128 (2010).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5, e1000529 (2009).
Zaykin, D. V., Zhivotovsky, L. A., Czika, W., Shao, S. & Wolfinger, R. D. Combining p-values in large scale genomics experiments. Pharm Stat 6, 217–226 (2007).
Duggal, P., Gillanders, E. M., Holmes, T. N. & Bailey-Wilson, J. E. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genomics 9, 516, https://doi.org/10.1186/1471-2164-9-516 (2008).
Autry, A. E. & Monteggia, L. M. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev 64, 238–258, https://doi.org/10.1124/pr.111.005108 (2012).
Castren, E. & Rantamaki, T. Neurotrophins in depression and antidepressant effects. Novartis Found Symp 289, 43–52 (2008). discussion 53-49, 87-93.
Balu, D. T. et al. Differential regulation of central BDNF protein levels by antidepressant and non-antidepressant drug treatments. Brain Res 1211, 37–43, https://doi.org/10.1016/j.brainres.2008.03.023 (2008).
Shirayama, Y., Chen, A. C., Nakagawa, S., Russell, D. S. & Duman, R. S. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci 22, 3251–3261, 20026292 (2002).
Adachi, M., Autry, A. E., Mahgoub, M., Suzuki, K. & Monteggia, L. M. TrkB Signaling in Dorsal Raphe Nucleus is Essential for Antidepressant Efficacy and Normal Aggression Behavior. Neuropsychopharmacology 42, 886–894, https://doi.org/10.1038/npp.2016.201 (2017).
Tsai, S. J., Hong, C. J. & Liou, Y. J. Brain-derived neurotrophic factor and antidepressant action: another piece of evidence from pharmacogenetics. Pharmacogenomics 9, 1353–1358, https://doi.org/10.2217/14622416.9.9.1353 (2008).
Niitsu, T., Fabbri, C., Bentini, F. & Serretti, A. Pharmacogenetics in major depression: a comprehensive meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry 45, 183–194, https://doi.org/10.1016/j.pnpbp.2013.05.011 (2013).
Nowacka, M. M. & Obuchowicz, E. Vascular endothelial growth factor (VEGF) and its role in the central nervous system: a new element in the neurotrophic hypothesis of antidepressant drug action. Neuropeptides 46, 1–10, https://doi.org/10.1016/j.npep.2011.05.005 (2012).
Warner-Schmidt, J. L. & Duman, R. S. VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci USA 104, 4647–4652, https://doi.org/10.1073/pnas.0610282104 (2007).
Fornaro, M. et al. VEGF plasma level variations in duloxetine-treated patients with major depression. J Affect Disord 151, 590–595, https://doi.org/10.1016/j.jad.2013.06.055 (2013).
Tsai, S. J. et al. Haplotype analysis of single nucleotide polymorphisms in the vascular endothelial growth factor (VEGFA) gene and antidepressant treatment response in major depressive disorder. Psychiatry Res 169, 113–117, https://doi.org/10.1016/j.psychres.2008.06.028 (2009).
Calabrese, F., Molteni, R., Racagni, G. & Riva, M. A. Neuronal plasticity: a link between stress and mood disorders. Psychoneuroendocrinology 34(Suppl 1), S208–216, https://doi.org/10.1016/j.psyneuen.2009.05.014 (2009).
Wray, N. R. et al. Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry 17, 36–48, https://doi.org/10.1038/mp.2010.109 (2012).
Khan, A. & Brown, W. A. Antidepressants versus placebo in major depression: an overview. World Psychiatry 14, 294–300, https://doi.org/10.1002/wps.20241 (2015).
Florio, V., Porcelli, S., Saria, A., Serretti, A. & Conca, A. Escitalopram plasma levels and antidepressant response. Eur Neuropsychopharmacol 27, 940–944, https://doi.org/10.1016/j.euroneuro.2017.06.009 (2017).
Acknowledgements
We would like to thank Translational Resource Center for Genomic Medicine of National Research Program for Biopharmaceuticals for their data service. Genotyping of MDD subjects was funded by the RIKEN Center for Integrative Medical Science, Yokohama, Japan. We also thank Taiwan Han Chinese Cell and Genome Bank of Academic Sinica that provided controls data and computational support. This work was supported by grant MOST 103-2314-B-075 −067-MY3 from Taiwan Ministry of Science and Technology, and grant VGHUST103-G1-4-1 from the Taipei Veterans General Hospital. We thank Emily Ting for English editing.
Author information
Authors and Affiliations
Contributions
Study conception and design: E.L., A.C.Y., Y.W.Y. and S.-J.T. Acquisition of data: P.-H.K. and C.-F.K. Analysis and interpretation of data: C.-F.K., P.-H.K. and S.-J.T. Draft and revise manuscript: C.-F.K., P.-H.K. and S.-J.T. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kao, CF., Liu, YL., Yu, Y.WY. et al. Gene-based analysis of genes related to neurotrophic pathway suggests association of BDNF and VEGFA with antidepressant treatment-response in depressed patients. Sci Rep 8, 6983 (2018). https://doi.org/10.1038/s41598-018-25529-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-25529-y
This article is cited by
-
Exploring the mechanism of Icariin in the treatment of depression through BDNF-TrkB pathway based on network pharmacology
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
Correlation between variants of the CREB1 and GRM7 genes and risk of depression
BMC Psychiatry (2023)
-
Evaluation of animal model congruence to human depression based on large-scale gene expression patterns of the CNS
Scientific Reports (2022)
-
Molecular mechanism of Epicedium treatment for depression based on network pharmacology and molecular docking technology
BMC Complementary Medicine and Therapies (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
