
Abstract
Atherosclerotic cardiovascular disease is a leading cause of death in the western world. Increased plasma triglyceride and cholesterol levels are major risk factors for this disease. Carboxylesterase 1 (Ces1/Ces1g) has been shown to play a role in metabolic control. So far, the role of mouse Ces1/Ces1g deficiency in atherosclerosis is not elucidated. We generated Ces1/Ces1g −/− mice. Compared to wild-type mice, Ces1/Ces1g −/− mice had reduced plasma cholesterol levels. We then generated Ces1g −/− Ldlr −/− double knockout (DKO) mice, which were fed a Western diet for 16 weeks. Compared to Ldlr −/− mice, DKO mice displayed decreased plasma cholesterol and TG levels and reduced atherosclerotic lesions. Interestingly, knockdown of hepatic Ces1/Ces1g in Apoe −/− mice resulted in hyperlipidemia and exacerbated Western diet-induced atherogenesis. Mechanistically, global inactivation of Ces1/Ces1g inhibited intestinal cholesterol and fat absorption and Niemann-Pick C1 like 1 expression, and increased macrophage cholesterol efflux by inducing ATP-binding cassette subfamily A member 1 (ABCA1) and ABCG1. Ces1/Ces1g ablation also promoted M2 macrophage polarization and induced hepatic cholesterol 7α-hydroxylase and sterol 12α-hydroxylase expression. In conclusion, global loss of Ces1/Ces1g protects against the development of atherosclerosis by inhibiting intestinal cholesterol and triglyceride absorption and promoting macrophage cholesterol efflux.
Similar content being viewed by others


Introduction
Cardiovascular diseases (CVDs) are the most prevalent cause of death globally, accounting for about 31% of all global deaths1,2. Atherosclerosis is the leading cause of CVDs. Hypercholesterolemia, hypertriglyceridemia and inflammation are common risk factors for atherosclerosis. Liver and intestine play the major role in maintaining plasma lipid and lipoprotein homeostasis by regulating very low-density lipoprotein (VLDL) or chylomicron secretion and lipoprotein uptake, and therefore can regulate the development of atherosclerosis. Macrophages directly participate in the pathogenesis of atherosclerosis by regulating oxidized lipid uptake, cholesterol efflux and inflammation.
Human carboxylesterase 1 (CES1) is predominantly expressed in liver and has been shown to have triglyceride (TG) hydrolase activity3. Over-expression of human CES1 in macrophages leads to an increase in cholesteryl ester hydrolysis and free cholesterol efflux and attenuation of atherosclerosis in Ldlr −/− mice4. In addition, over-expression of human CES1 in the liver enhances reverse cholesterol transport and reduces atherosclerosis in Ldlr −/− mice5,6. These data suggest that human CES1 has cholesteryl ester hydrolase (CEH) activity. However, the CEH activity of human CES1 cannot be confirmed by other research groups7,8. Mouse Ces1 gene has multiple isoforms9. Mouse Ces1d (previously called Ces3) and Ces1g (previously called Ces1) share 78% and 74% amino acid identities with human CES1, respectively. The studies from us and Lehner’s group have previously reported that mouse Ces1g has triglyceride hydrolase (TGH) activity10,11. Bie et al. showed that hepatic deletion of Ces3/Ces1d aggravates atherosclerosis in Ldlr −/− mice12. In contrast, global loss of Ces3/Ces1d is shown to reduce atherosclerosis in Ldlr −/− mice13. So far, it is unclear whether loss of mouse Ces1/Ces1g has any impact on atherosclerosis.
In this report, we generated global Ces1/Ces1g −/− mice and Ces1/Ces1g −/− Ldlr −/− (DKO) mice. Our data show that loss of Ces1/Ces1g in Ldlr −/− mice inhibited the development of atherosclerosis by inhibiting intestinal cholesterol absorption and inducing macrophage cholesterol efflux and bile acid synthesis. Similar to loss of hepatic Ces3/Ces1d function, inactivation of hepatic Ces1/Ces1g in Apoe −/− mice aggravated the development of atherosclerosis. Our data indicate that Ces1/Ces1g plays a critical role in the pathogenesis of atherosclerosis.
Results
Generation and characterization of Ces1/Ces1g −/− mice
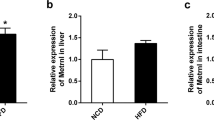
We have recently shown that hepatic Ces1/Ces1g plays a critical role in maintaining hepatic and plasma lipid homeostasis and is an important TG hydrolase in the liver11. To better understand the role of Ces1/Ces1g in peripheral tissues, we generated global Ces1/Ces1g −/− mice by replacing Ces1g exon 1 and exon 2 with an nLacZ-neomyclin cassette (Fig. 1A). The successful creation of the Ces1/Ces1g −/− mice was confirmed by Southern blotting, PCR genotyping and Western blotting (Fig. 1B–H). Other isoforms of Ces1 were not affected in the liver of Ces1/Ces1g −/− mice (Fig. 1D; for primer sequences see Supplementary Table 1). LacZ staining of tissues isolated from Ces1 +/− mice indicated that Ces1 was expressed in a number of tissues, including brain, lung, liver, stomach, gallbladder, small intestine, colon, pancreas, spleen, kidney, white adipose tissue (WAT), adrenal and brown adipose tissue (BAT), but not in heart or skeletal muscle (Fig. 1E). Since LacZ staining was not quantitative, we also measured mRNA and protein levels of Ces1g. Results from RNA profiling showed that Ces1/Ces1g was most abundantly expressed in the liver and to a less extent in other tissues (see Supplementary Fig. 1A). Western blotting results showed that Ces1/Ces1g protein was expressed in the liver, intestine and gallbladder (Fig. 1F), consistent with a previous finding by Quiroga et al.10. Interestingly, we did not observe Ces1g protein expression in the lung, kidney, WAT or BAT (Fig. 1F), which was likely due to very low expression of Ces1g in these tissues. There was a significant reduction in the expression of Ces1d protein (the faster migrating band, according to Quiroga et al.10) in the intestine and gallbladder (Fig. 1F). The reason for the reduced Ces1d expression in the intestine or gallbladder is unclear. However, we believe the reduction of Ces1d expression should be secondary to Ces1g ablation as Ces1d expression did not decrease in the liver or lung of Ces1g −/− mice (Fig. 1F). In addition, Ces1/Ces1g was also expressed in macrophages, as demonstrated by positive LacZ staining, (Fig. 1G), qRT-PCR (see Supplementary Fig. 1B) and Western blotting assays (Fig. 1H). There was no gross difference in body weight, size or food intake on a chow diet (data not shown).

Generation of global Ces1/Ces1g −/− mice and Ces1/Ces1g tissue distribution. (A) A diagram showing the Ces1/Ces1g wild-type (WT) allele and the targeting allele. Exons 1 and 2 are replaced by a nucleus LacZ (nLacZ) cassette and neomycin cassette. K, KpnI. B, BamHI. (B) Genomic DNA from WT or heterozygous ES cells was used for Southern blotting using a 5′ probe (left panel) or 3′ probe (right panel). (C) Mouse genomic DNA was genotyped by PCR. (D) Hepatic mRNA levels of different Ces1 isoforms were determined (n = 4). (E) LacZ staining of various tissues isolated from Ces1/Ces1g +/− mice. (F) Hepatic proteins were isolated from 8-weeks-old male control littermates (Ces1/Ces1g +/+ mice) or Ces1/Ces1g −/− mice. Protein levels were determined by Western blotting. The top band is Ces1/Ces1g. (G) LacZ staining of peritoneal macrophages isolated from Ces1/Ces1g +/− mice. (H) Protein levels in the liver (serve as control) or peritoneal macrophages of Ces1g +/+ or Ces1g −/− mice were determined by Western blotting. BAT, brown adipose tissue. WAT, white adipose tissue. GB, gallbladder. The Western blots are cropped from larger gels.
Ces1/Ces1g −/− mice have reduced plasma cholesterol levels
When 8-weeks-old Ces1/Ces1g −/− mice and their control littermates (Ces1/Ces1g +/+) were fed a chow diet for 6 months, plasma TG levels were not significantly different between the two genotypes (Fig. 2A), but plasma cholesterol levels were reduced by ~30% in Ces1/Ces1g −/− mice (Fig. 2B). Similar data were obtained when 8-weeks-old Ces1/Ces1g −/− mice and their control littermates were fed a Western diet for 16 weeks (Fig. 2C and D). Analysis of plasma lipoprotein profile by fast protein liquid chromatography (FPLC) indicated that Ces1/Ces1g −/− mice had reduced LDL-C and HDL-C levels (Fig. 2E). In the liver, TG or cholesterol levels were not different between the two genotypes when fed a chow or Western diet (see Supplementary Fig. 2A–D). Oil red O staining showed no difference in neutral lipid accumulation (Fig. 2F). Despite unchanged hepatic lipid levels, Ces1/Ces1g −/− mice had significantly reduced plasma AST and ALT levels (data not shown) and hepatic mRNA levels of inflammatory markers (Tnfα, Il6 and Il1β) (Fig. 2G). In addition, Western diet-fed Ces1/Ces1g −/− mice had similar body weight gain and body fat content compared to Ces1/Ces1g +/+ mice (see Supplementary Fig. 2E and F).

Ces1/Ces1g −/− mice have reduced plasma cholesterol levels. (A and B) Male Ces1/Ces1g −/− mice and their control littermates (Ces1/Ces1g +/+) were fed a chow diet for 6 months (n = 8). Plasma TG (A) and cholesterol (B) were determined. (C-G) 8-weeks old male Ces1/Ces1g −/− mice and their control littermates (Ces1/Ces1g +/+) were fed a Western diet for 16 weeks (n = 8). Plasma TG (C) and cholesterol (D) levels were determined. FPLC was performed to determine cholesterol distribution in lipoproteins (E). Representative liver images of oil red O staining are presented (F). Hepatic mRNA levels of inflammatory marker were analyzed (G). *P < 0.05, **P < 0.01.
Global Ces1/Ces1g inactivation reduces atherosclerosis in Ldlr −/− mice
The finding that Ces1/Ces1g −/− mice have reduced plasma cholesterol levels led us to investigate whether Ces1/Ces1g inactivation affects atherogenesis. We therefore crossed Ces1/Ces1g −/− mice with Ldlr −/− mice to generate Ces1g −/− Ldlr −/− double knockout (DKO) mice, which were then fed a Western diet for 16 weeks. The control littermates were Ces1g +/+ Ldlr −/− mice. CES1 and LDLR protein levels were shown in Fig. 3A. Compared to Ces1g +/+ Ldlr −/− mice, plasma TG and cholesterol levels in DKO mice were lowered by 61% (Fig. 3B) and 44% (Fig. 3C), respectively. In contrast, hepatic TG and cholesterol levels were comparable between control and DKO mice (see Supplementary Fig. 2G and H). FPLC analysis indicated that the reduction in plasma TG and cholesterol levels were attributed to a marked decrease in VLDL-TG (Fig. 3D) and VLDL-C/LDL-C levels (Fig. 3E), respectively. Consistent with markedly improved hyperlipidemia, DKO mice had a ~ 57% reduction in atherosclerotic lesions in both the aortas (Fig. 3F and G) and aortic roots (Fig. 3H and I).

Loss of Ces1/Ces1g in Ldlr −/− mice causes hypolipidemia and protects against atherosclerosis. (A–H) 8-weeks-old male Ces1 −/− Ldlr −/− (DKO) mice and control littermates Ces1 +/+ Ldlr −/− (Ldlr −/−) mice were fed a Western diet for 16 weeks (n = 8). Hepatic Ces1/Ces1g and LDLR protein levels were determined (A). Plasma TG (B) and cholesterol (C) levels were determined. Plasma TG (D) and cholesterol (E) lipoprotein profiles were analyzed by FPLC. En face aortas were stained by oil red O and representative images are shown (F). En face aorta lesion size was quantified (G). Aortic roots were also stained with oil red O and representative images are shown (H). Aortic root lesion size was quantified (I). **P < 0.01.
Liver-specific Ces1/Ces1g deficiency exacerbates atherosclerosis in Apoe −/− mice
Ces1/Ces1g is abundantly expressed in the liver (Fig. 1). To understand how global inactivation of CES1 ameliorates atherosclerosis, we knocked down hepatic Ces1/Ces1g expression in Apoe −/− mice using an adenovirus expressing Ces1 shRNA (Ad-shCes1). Previously, we showed that Ad-shCes1 can efficiently knock down hepatic Ces1 mRNA and protein levels by ~95%11. The selection of Apoe −/− mice is because these mice can develop spontaneous atherosclerosis even on a regular chow diet. Three weeks after Ad-shCes1 infection, hepatic Ces1/Ces1g mRNA and protein levels were reduced by ~90% (Fig. 4A) whereas intestinal CES1g protein levels were unchanged (Supplementary Fig. 3). We did not observe fat malabsorption (data not shown). Similar to what we have reported previously11, knock down of hepatic Ces1/Ces1g increased plasma TG levels by 21% (Fig. 4B) and cholesterol levels by 20% (Fig. 4C), and also increased hepatic levels of TG (Fig. 4D) and cholesterol (Fig. 4E). FPLC analysis showed that hepatic Ces1/Ces1g deficiency increased VLDL-TG (Fig. 4F), VLDL-C and LDL-C (Fig. 4G). Consistent with the latter findings, hepatic Ces1/Ces1g deficiency increased atherosclerotic lesions by >50% in both the aortas (Fig. 4H and I) and aortic roots (Fig. 4J and K) of Apoe −/− mice. These data suggest that hepatic Ces1/Ces1g is protective against atherosclerosis.

Knockdown of Ces1/Ces1g in Apoe −/− mice results in hyperlipidemia and increases atherosclerosis. (A–K) 12-weeks-old male Apoe −/− mice were fed a Western diet for one week, followed by i.v. injection of Ad-shLacZ and Ad-shCes1 (n = 7). The mice continued to be fed a Western diet for another three weeks. mRNA (left panel) and protein (right panel) levels of Ces1/Ces1g were determined three weeks post adenovirus injection (A). Plasma levels of TG (B) and cholesterol (C) as well as hepatic levels of TG (D) and cholesterol (E) were determined. Plasma TG (F) and cholesterol (G) lipoprotein profiles were analyzed by FPLC. Representative en face aorta images are shown (H) and aortic lesion size was quantified (I). Representative aortic root images are also shown (J) and lesion size was quantified (K). *P < 0.05, **P < 0.01.
Ces1/Ces1g inactivation reduces intestinal cholesterol and triglyceride absorption
The data of Fig. 4 suggest that hepatic Ces1/Ces1g is unlikely to contribute to the atheroprotective phenotype in the DKO mice. Since Ces1/Ces1g is expressed in intestine (Fig. 1), we investigated whether CES1 inactivation affected cholesterol and/or fat (triglyceride) absorption. The data of Fig. 5A show that Ces1/Ces1g −/− mice had a ~33% reduction in cholesterol absorption. In DKO mice, cholesterol absorption was reduced by 48% (Fig. 5B) and fat (triglyceride) absorption was also significantly reduced (Fig. 5C). Consistent with decreased intestinal cholesterol absorption, the mRNA levels of Nieman-Pick C1-like 1 (Npc1l1), a protein that facilities cholesterol transport, were repressed (Fig. 5D). However, other genes involved in cholesterol metabolism (Abca1, Abcg5, Abcg8, Cd36) were not significantly changed in Ces1/Ces1g −/− mice vs. control mice (Fig. 5D).

Loss of Ces1/Ces1g inhibits intestinal cholesterol absorption. (A–C) 8-weeks-old male WT and Ces1/Ces1g −/− mice (A) or 8-weeks-old male Ldlr −/− and Ces1 −/− Ldlr −/− (DKO) mice (B) were fed a Western diet for 8 weeks. Cholesterol absorption was then performed (n = 8). Fat absorption was determined in male Ldlr −/− vs DKO mice (n = 8) (C). (D) mRNA levels in the intestine of 6-months-old male WT mice and Ces1 −/− mice were quantified (n = 8). *P < 0.05, **P < 0.01.
Ces1/Ces1g inactivation induces hepatic CYP7A1 and CYP8B1 expression and bile acid synthesis
In the liver, cholesterol is converted to bile acids via cholesterol 7α-hydroxylase (CYP7A1) and sterol 12α-hydroxylase (CYP8B1) or secreted directly to the bile. Transgenic expression of CYP7A1 in the liver reduces both HDL-C and non-HDL-C levels14 and protects against atherosclerosis15. In the liver of Ces1/Ces1g −/− mice, both Cyp7a1 and Cyp8b1 mRNA levels were induced by ~1.8 fold (Fig. 6A), which was accompanied by a 99% reduction in fibroblast growth factor 15 (Fgf15) in the intestine (Fig. 6A). We did not observe much change in mRNA levels of small heterodimer partner (Shp) (Fig. 6A), scavenger receptor class B type 1 (SR-BI), ATP-binding cassette subfamily G member 5 (Abcg5), Abcg8, HMG-CoA reductase (Hmgcr), Apoe or Ldlr (see Supplementary Fig. 4).

Ces1/Ces1g −/− mice have increased Cyp7a1 and Cyp8b1 expression. (A–F) 6-months-old male chow-fed mice were sacrificed (n = 6). Hepatic and intestinal mRNA levels were quantified by qRT-PCR (A). Bile acids (BA) in gallbladder (B), intestine (C) or liver (D) were quantified. Total BA levels were calculated by addition of BA levels in gallbladder, intestine and liver (E). Cholesterol concentration in bile was analyzed (F). (G–I) 8-weeks-old male Ces1 −/− Ldlr −/− (DKO) mice and Ldlr −/− control littermates were fed a Western diet for 16 weeks (n = 8). Hepatic and intestinal mRNA levels were quantified by qRT-PCR (G). Bile acids in liver, gallbladder, intestine as well as total bile acid levels were determined (H). Biliary cholesterol levels were also determined (I). *P < 0.05, **P < 0.01.
Gain- and loss-of-function studies have demonstrated that intestinal FGF15 is an important repressor of hepatic CYP7A1 and CYP8B1 expression16,17. Consistent with these data, bile acid (BA) levels in gallbladder (Fig. 6B) and intestine (Fig. 6C) were increased whereas BA levels in the liver were unchanged (Fig. 6D). As a result, total BA pool size was increased (Fig. 6E). In addition, biliary cholesterol levels were also increased (Fig. 6F). Similarly, mRNAs levels of hepatic Cyp7a1 and Cyp8b1 were elevated and intestinal Fgf15 level was repressed in DKO mice compared with control littermates (Fig. 6G). In addition, BA levels in gallbladder or intestine were increased in DKO mice, resulting in increased total BA pool size (Fig. 6H). These data suggest that the induction of Cyp7a1 and Cyp8b1 may contribute to hypocholesterolemia in Ces1/Ces1g −/− mice.
Cholesterol efflux is increased in Ces1/Ces1g deficient macrophages
Macrophage cholesterol efflux is the first step in reverse cholesterol transport (RCT). Next, we determined whether loss of Ces1/Ces1g in macrophages affected cholesterol efflux. Peritoneal macrophages were isolated from Ces1/Ces1g +/+ mice and Ces1/Ces1g −/− mice and then treated with or without acetylated LDL (Ac-LDL). Ces1/Ces1g inactivation resulted in a 1.7-fold increase in cholesterol efflux to ApoA-I and this increase was further enhanced in the presence of Ac-LDL (Fig. 7A). In addition, Ces1 inactivation caused a 2.1-fold increase in cholesterol efflux to HDL (Fig. 7B).

Loss of Ces1/Ces1g increases cholesterol efflux from macrophages. (A and B) Cholesterol efflux to ApoA-I (A) or HDL (B) was performed in the presence or absence of 25 μg/L Ac-LDL (n = 6). (C) mRNA levels were quantified in peritoneal macrophages in the presence of PBS for 24 h (n = 6). (D and E) Peritoneal macrophages were isolated and treated with 10 μM T-0901317 for 24 h (n = 6). mRNA levels of Abca1 (D) and Abcg1 (E) were quantified. (F,G) Peritoneal macrophages were treated in the presence or absence of 25 μg/L Ac-LDL for 24 h (n = 5). Intracellular free cholesterol (F) and total cholesterol (G) were quantified. *P < 0.05, **P < 0.01.
In addition to human CES1, mouse hormone sensitive lipase (Hsl)7,18 and neutral cholesterol ester hydrolase 1 (NCEH1)8 are reported to promote hydrolysis of cholesterol esters in macrophages. ATP-binding cassette subfamily A member 1 (ABCA1) and ABCG1 are the major transporters that efflux free cholesterol to ApoA-I or HDL, respectively19. Ces1/Ces1g inactivation significantly increased Hsl but not Nceh1 mRNA levels (Fig. 7C). Ces1/Ces1g inactivation in macrophages also caused 2- and 3-fold increase in Abca1 and Abcg1 mRNA levels, respectively (Fig. 7C). In contrast, Ces1/Ces1g inactivation had no much effect on scavenger receptor class A (Sra), fatty acid translocase (Cd36) or acetyl-Coenzyme A acetyltransferase 2 (Acat2) (Fig. 7C and see Supplementary Fig. 5A–C). T-0901317, a liver X receptor agonist, drastically induced Abca1 (Fig. 7D) and Abcg1 (Fig. 7E) mRNA levels in wild-type macrophages, and this induction was further increased in Ces1/Ces1g −/− macrophages (Fig. 7D and E), suggesting that Ces1/Ces1g inactivation may lead to increased production of endogenous LXR agonists (oxysterols). In the absence of Ac-LDL treatment, the intracellular free cholesterol and total cholesterol contents were decreased in Ces1/Ces1g −/− macrophages compared with wild-type macrophages, but this decrease became less pronounced when macrophages were treated with Ac-LDL (Fig. 7F and G). Taken together, these data suggest that Ces1/Ces1g inactivation increases macrophage cholesterol efflux by inducing both ABCA1 and ABCG1.
Ces1/Ces1g inactivation drives M2 macrophage polarization
Recent evidence links inflammatory M1 macrophages to atherosclerotic progression and anti-inflammatory M2 macrophages to atherosclerotic regression20. M0 macrophages were treated with interferon γ (IFNγ) plus lipopolysaccharide (LPS) or interleukin 4 (IL-4) to induce M1 or M2 macrophages, respectively. Both tumor necrosis factor α (Tnfα) (Fig. 8A) and Il1β (Fig. 8B) were markedly induced in both Ces1/Ces1g +/+ and Ces1/Ces1g −/− M1 macrophages (grey bars). In contrast, both Tnfα and Il1β were significantly reduced in Ces1/Ces1g −/− M2 macrophages (black bars) (Fig. 8A and B).

Loss of Ces1/Ces1g promotes M2 macrophage polarization. (A–E) Peritoneal macrophages were isolated from wild-type or Ces1/Ces1g −/− mice and subjected to induction to M1 or M2 macrophages (n = 4). mRNA levels were determined by qRT-PCR. (F) Mechanism for Ces1/Ces1g inactivation to repress atherosclerosis. a,b,c, or d P < 0.05.
As expected, Ces1/Ces1g +/+ M2 macrophages had increased levels of arginase 1 (Arg1) (Fig. 8C), resistin like alpha (Retnla/Fizz1) (Fig. 8D) and chitinase-like 3 (Chil3/Ym1) (Fig. 8E), all of which are markers for M2 macrophages. Ces1/Ces1g inactivation further increased Arg1, Fizz1 and Ym1 expression (Fig. 8C–E). In addition, Inactivation of Ces1/Ces1g increased the basal levels of Arg1 (Fig. 8C). Consistent with the ex vivo data, in the lesions of Ces1g −/− Ldlr −/− mice vs. Ces1g +/+ Ldlr −/− mice, Abca1, Arg1 and Mrc1 mRNA levels were significantly increased (see Supplementary Fig. 6). Together, these data suggest that Ces1/Ces1g inactivation promotes M2 macrophage polarization.
Discussion
The studies on the role of the carboxylesterase family in regulating lipid metabolism are challenged by the structural and functional similarities shared by their family members9. Mouse Ces3/Ces1d and Ces1/Ces1g share high amino acid identities with human CES1, and both Ces1d and Ces1g have been reported to play roles in lipid metabolism11,21. Ces3/Ces1d and Ces1/Ces1g share 76% amino acid identity and have the similar protein size, but display different tissue expression profiles. Global or liver-specific loss of Ces3/Ces1d/TGH is shown to improve blood lipids by attenuating VLDL secretion22,23. Ces3/Ces1d/TGH knockout mice are reported to have increased energy metabolism and glucose tolerance23. In contrast, loss of Ces1/Ces1g/Es-x is reported to induce obesity and hyperlipidemia10. In addition, loss of Ces3/Ces1d/TGH attenuates atherosclerosis in Ldlr −/− mice, but liver specific loss of Ces3/Ces1d/TGH promotes atherosclerosis in Ldlr −/− mice12,13. Similar results are found in this study when we inactive Ces1/Ces1g in Ldlr −/− mice or Apoe −/− mice. In this report, we provide the first evidence demonstrating that global Ces1/Ces1g inactivation markedly attenuates the development of atherosclerosis by reducing intestinal cholesterol absorption, promoting macrophage cholesterol efflux and M2 macrophage polarization, and inducing bile acid synthesis (Fig. 8F) whereas loss of hepatic Ces1/Ces1g aggravates the development of atherosclerosis.
Previously, Lehner group show that female, 5–6 months old Ces1/Ces1g/Es-x −/− mice are obese10. We do not see any difference in body weight or adiposity between male Ces1 +/+ mice and male Ces1g −/− mice that are fed either a chow diet or high fat diet. We realize that we may have used mice with different sex (male vs female) and background. Lehner group also reports that Ces1/Ces1g/Es-x −/− mice have increased secretion of chylomicron and decreased chylomicron clearance24. We find that loss of Ces1/Ces1g −/− leads to decreased TG and cholesterol absorption in the intestine. Interestingly, in our Ces1g −/− mice intestinal Ces1d expression is also reduced, which may contribute to the reduced fat absorption and overall lipid homeostasis.
In our global Ces1/Ces1g −/− mice, we do not see any change in the levels of hepatic neutral lipids. Previously, we show that liver-specific knockdown of Ces1/Ces1g causes hepatic triglyceride accumulation and increased VLDL secretion and plasma lipids11. In global Ces1/Ces1g −/− mice, the reduction in intestinal fat and cholesterol absorption may be sufficient to counteract the increased lipogenic activity in the liver, thus resulting in reduced plasma cholesterol levels and unchanged hepatic lipids.
Reverse cholesterol transport begins with macrophage cholesterol efflux to the acceptors ApoA-I or HDL by ABCA1 or ABCG1. Prior to cholesterol efflux, cholesterol esters are hydrolyzed by cholesterol ester hydrolase. Loss-of-function studies have established both HSL and NCEH1 as important cholesterol ester hydrolases in macrophages7,8. Our data show that Ces1/Ces1g is present in peritoneal macrophages. So far, it is unclear whether mouse Ces1/Ces1g has cholesterol ester hydrolase activity. Interestingly, loss of Ces1/Ces1g in macrophages induces Hsl, Abca1 and Abcg1 expression. HSL over-expression in macrophages is reported to increase cholesterol ester hydrolysis18. The induction of Abca1 and Abcg1 suggests that intracellular oxysterols may be increased in Ces1/Ces1g −/− macrophages. Indeed, LXR activation is further enhanced in Ces1/Ces1g −/− macrophages. In the future, we will determine whether oxysterols and other lipid metabolites are changed in Ces1/Ces1g −/− macrophages.
In addition to regulating cholesterol efflux, our data also show that Ces1/Ces1g inactivation promotes M2 macrophage polarization. M2 macrophages are anti-inflammatory and play an important role in regression of atherosclerosis20. Although the mechanism underlying the regulation of macrophage polarization remains to be determined, our data indicate that loss of Ces1/Ces1g in macrophages has two beneficial effects − promoting cholesterol efflux and inhibiting inflammation.
Liver is one of the most important organs regulating the pathogenesis of atherosclerosis. Our data show that Ces1/Ces1g knockdown in the liver promotes the development of atherosclerosis in Apoe −/− mice whereas global loss of Ces1/Ces1g protects against atherosclerosis in Ldlr −/− mice. Over-expression of hepatic Ces1g produces less atherogentic lipoproteins11,25, which may account for the atheroprotective effect of hepatic Ces1g. Since we used mice with different genetic background (Apoe −/− mice versus Ldlr −/− mice), we are not able to reach a firm conclusion at this point that hepatic Ces1/Ces1g deficiency is not part of the mechanism underlying the atheroprotective effect found in DKO mice. However, Apoe −/− mice and Ldlr −/− mice are both widely used for atherosclerosis study and the results achieved using these mouse lines are often very similar. The reduced intestinal TG and cholesterol absorption plays an important role in atheroprotection in DKO mice. In addition, hepatic Cyp7a1 and Cyp8b1 are induced in Ces1/Ces1g −/− mice likely as a result of inhibition of intestinal Fgf15. Since Cyp7a1 and Cyp8b1 are key enzymes in cholesterol catabolism in the liver, the induction of Cyp7a1 and Cyp8b1 may also contribute to the atheroprotective effect of Ces1/Ces1g inactivation.
In summary, we demonstrate that global inactivation of Ces1/Ces1g ameliorates the development of atherosclerosis by inhibiting intestinal cholesterol absorption, promoting macrophage cholesterol efflux and M2 macrophage polarization, and inducing bile acid production. In contrast, inactivation of hepatic Ces1/Ces1g aggravates the development of atherosclerosis likely by producing atherogenic lipoproteins11.
Methods
Mice and diet
To generate Ces1/Ces1g −/− mice, the Ces1 (Ces1g) targeting vector (see Fig. 1) was electroporated into G4 ES cells (129/B6 background; Samuel Lunenfeld Research Institute at Mount Sinai Hospital, Toronto, Canada). After screening by PCR and Southern blotting, positive ES clones were injected into blastocysts from C57BL/6 mice, which were then implanted into pseudopregnant foster mothers in the Transgenic Mouse Facility in University of California, Irvine. Genotyping was performed using primers 5′-CTCAGAGGTTAGCCAGCTGGGAGGA-3′ (forward), 5′-GGCCTTCAGACAGGGAAAAGCTTTG-3′ (reverse, WT, 720 bp) and 5′-TAGCAGCCAGTGGGGTTCTCAGTG-3′ (reverse, mutant, 450 bp). Ces1/Ces1g +/− mice were back-crossed with C57BL/6 J mice for 6 generations to get 99.3% C57BL/6 background before self-crossing to generate Ces1/Ces1g −/− mice and Ces1/Ces1g +/+ mice (control), or crossing with Ldlr −/− mice (Jackson Laboratory, Maine, USA) to generate Ces1/Ces1g −/− Ldlr −/− mice and Ces1/Ces1g +/+ Ldlr −/− mice (control). Apoe −/− mice were purchased from the Jackson Laboratory (Maine, USA). All the mice were male and were fed a chow diet or a Western diet containing 42% kcal fat and 0.2% cholesterol (TD.88139, Envigo, Wisconsin, USA) for up to 16 weeks, starting from 8 weeks old. All the mice were fasted for 5–6 hours prior to euthanization for samples (tissue, blood, etc) collection. All the animal studies have been approved by the Institutional Animal Care and Use Committee at Northeast Ohio Medical University. All the experiments were performed in accordance with the relevant guidelines and regulations.
RNA isolation and quantitative real-time PCR
Total RNA was isolated using TRIzol Reagent (Life Technologies, NY). mRNA levels were determined by quantitative reverse-transcription polymerase chain reaction (qRT-PCR) on a 7500 real-time PCR machine from Applied Biosystems (Foster City, CA). Relative mRNA levels were calculated using the comparative cycle threshold (Ct) method and were normalized to the values of 36B4 mRNA levels.
Southern and Western blotting
Southern blotting was performed as described26. Western blotting was performed using a CES1 antibody (Cat # ab45957) or Tubulin antibody (Cat # ab4074) from Abcam (Cambridage, MA) or LDLR antibody from Novus Biologicals (Cat # NBP1-06709, Littleton, CO).
Lipid analysis
Approximately 100 mg liver was homogenized in methanol and lipids were extracted in chloroform/methanol (2:1 v/v). Hepatic and plasma triglyceride and cholesterol levels were quantified using Infinity reagents from Thermo Scientific (Waltham, MA). For analyzing plasma lipoprotein levels, FPLC was performed as described11. In brief, 100 μL plasma was injected into the BioLogic DuoFlow QuadTec 10 system (Bio-Rad, Hercules, CA). Lipoproteins were run at 0.5 mL/min in a buffer containing 0.15 M NaCl, 0.01 M Na2HPO4, 0.1 mM EDTA, pH7.5, and separated on a Superose 6 10/300 GL column (GE Healthcare). A 500 μL sample per fraction was collected for analyzing lipid levels using Infinity reagents.
β-galactosidase (LacZ) staining
Peritoneal macrophages or tissues were washed with PBS, fixed with a fixative solution containing 0.5% glutaraldehyde, washed with PBS and stained with a solution containing 1 mg/ml X-gal at 37 °C.
Peritoneal macrophage isolation
Wild-type and Ces1/Ces1g −/− mice were injected with 1 mL of 3% (w/v) brewer thioglycollate medium into their peritoneal cavity. Four days later, mice were euthanized. The outer skin of the peritoneum was cut with gentle and pulled back to expose the inner skin lining the peritoneal cavity. Dulbecco’s Modified Eagle Medium (DMEM) medium with 10% fetal bovine serum (FBS) was injected into the peritoneal cavity using a 21.5 G needle, followed by gently massage of the peritoneum to dislodge any attached cells into the medium. The cells were collected into a 5-ml syringe. DMEM medium was injected 1–2 more time to retrieve more cells. Cell suspension was spun at 1500 rpm, resuspended and cultured in 6-well plates.
Intestinal fat absorption
After overnight fasting, mice were injected intravenously with 500 mg/kg of Triton WR1339. Immediately after the Triton injection, mice were given an intragastric 200 μL olive oil containing 7 μCi of [3H]triolein (Perkin Elmer). Blood samples were drawn via retro-orbital bleeding at indicated time points after administration of [3H]triolein. The amount of 3H in plasma was determined using a liquid scintillation counter.
Intestinal cholesterol absorption
Intestinal cholesterol absorption was determined by a dual-isotope plasma ratio method27. Briefly, mice were intravenously injected with 2.5 μCi 3H-cholesterol in Intralipid (Sigma, St Louis, MO), immediately followed by oral gavage of 1 μCi 14C-cholesterol in median-chain triglycerides (MCT oil, Mead Johnson, Evansville, IN). Mice were returned to cage with free access to food and water. After 72 h, blood samples were collected and the radioactivity of 14C and 3H were determined by scintillation counting. Intestinal cholesterol absorption was determined as the ratio of 14C/3H in 1 ml of plasma.
Cholesterol efflux
Cholesterol efflux was performed as described28,29. Peritoneal macrophages from wild-type and Ces1 −/− mice were isolated and incubated in DMEM containing 0.2% bovine serum albumin (BSA). Cells were then incubated with acetylated LDL (50 μg/mL) and [3H] cholesterol (1 μCi/mL) for 24 hours. After 24 h, cells were washed twice with PBS, and further incubated in DMEM containing 0.2% BSA for 2–4 hours. Cholesterol efflux was performed in the presence of fresh DMEM containing 0.2% BSA, 50 μg/mL of HDL, ApoA-I or Ac-LDL. After 4-h incubation, the radioactivity in the medium and the cell lysate was measured. The percent efflux was calculated as (medium dpm)/(cell dpm + medium dpm) × 100%.
Bile acid analysis
Bile acids in gallbladder, liver and intestine were extracted with 95% ethanol overnight, followed by 80% ethanol for 2 hours and methanol/chloroform (2:1) for 2 hours at 50 °C. Total bile acids were determined using a bile acid assay kit (Diazyme, San Diego, CA).
Body composition analysis
Body fat mass and lean mass were measured by Echo-MRI (EchoMRI, LLC, Houston, TX).
Atherosclerosis study
The aorta, including the ascending arch, thoracic and abdominal segments were dissected, gently cleaned of the adventitia and stained with Oil Red O as described previously30,31. In addition, the aortic roots were collected from the base of the heart including the atria and embedded in optimal cutting temperature (OCT) compound. Sections (5 μm) were obtained every 50 μm from the base of the aortic leaflets to 400 μm above. After staining with Oil Red O, images were captured with a microscope, and the lesion area for each aortic ring was analyzed using Image Pro (Media Cybernetics).
Statistical Analysis
The data were analyzed using unpaired Student t test and ANOVA (GraphPad Prisim, CA). All values were expressed as mean ± SEM. Differences were considered statistically significant at P < 0.05.
Data availability
All the data are available upon request.
References
Lloyd-Jones, D. et al. Executive summary: heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation 121, 948–954, https://doi.org/10.1161/CIRCULATIONAHA.109.192666 (2010).
Lloyd-Jones, D. et al. Heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation 121, e46–e215, https://doi.org/10.1161/CIRCULATIONAHA.109.192667 (2010).
Alam, M., Ho, S., Vance, D. E. & Lehner, R. Heterologous expression, purification, and characterization of human triacylglycerol hydrolase. Protein expression and purification 24, 33–42, https://doi.org/10.1006/prep.2001.1553 (2002).
Zhao, B. et al. Macrophage-specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr mice. J Clin Invest 117, 2983–2992, https://doi.org/10.1172/JCI30485 (2007).
Zhao, B., Song, J. & Ghosh, S. Hepatic overexpression of cholesteryl ester hydrolase enhances cholesterol elimination and in vivo reverse cholesterol transport. J Lipid Res 49, 2212–2217, https://doi.org/10.1194/jlr.M800277-JLR200 (2008).
Bie, J. et al. Liver-specific transgenic expression of cholesteryl ester hydrolase reduces atherosclerosis in Ldlr-/- mice. J Lipid Res 55, 729–738, https://doi.org/10.1194/jlr.M046524 (2014).
Buchebner, M. et al. Cholesteryl ester hydrolase activity is abolished in HSL-/- macrophages but unchanged in macrophages lacking KIAA1363. J Lipid Res 51, 2896–2908, https://doi.org/10.1194/jlr.M004259 (2010).
Igarashi, M. et al. The critical role of neutral cholesterol ester hydrolase 1 in cholesterol removal from human macrophages. Circulation research 107, 1387–1395, https://doi.org/10.1161/CIRCRESAHA.110.226613 (2010).
Holmes, R. S. et al. Recommended nomenclature for five mammalian carboxylesterase gene families: human, mouse, and rat genes and proteins. Mamm Genome 21, 427–441, https://doi.org/10.1007/s00335-010-9284-4.
Quiroga, A. D. et al. Deficiency of carboxylesterase 1/esterase-x results in obesity, hepatic steatosis, and hyperlipidemia. Hepatology 56, 2188–2198, https://doi.org/10.1002/hep.25961 (2012).
Xu, J. et al. Hepatic carboxylesterase 1 is essential for both normal and farnesoid X receptor-controlled lipid homeostasis. Hepatology 59, 1761–1771, https://doi.org/10.1002/hep.26714 (2014).
Bie, J. et al. Liver-specific cholesteryl ester hydrolase deficiency attenuates sterol elimination in the feces and increases atherosclerosis in ldlr-/- mice. Arterioscler Thromb Vasc Biol 33, 1795–1802, https://doi.org/10.1161/ATVBAHA.113.301634 (2013).
Lian, J., Quiroga, A. D., Li, L. & Lehner, R. Ces3/TGH deficiency improves dyslipidemia and reduces atherosclerosis in Ldlr(-/-) mice. Circulation research 111, 982–990, https://doi.org/10.1161/CIRCRESAHA.112.267468 (2012).
Miyake, J. H. et al. Increased production of apolipoprotein B-containing lipoproteins in the absence of hyperlipidemia in transgenic mice expressing cholesterol 7alpha-hydroxylase. The Journal of biological chemistry 276, 23304–23311, https://doi.org/10.1074/jbc.M101853200 (2001).
Ratliff, E. P., Gutierrez, A. & Davis, R. A. Transgenic expression of CYP7A1 in LDL receptor-deficient mice blocks diet-induced hypercholesterolemia. J Lipid Res 47, 1513–1520, https://doi.org/10.1194/jlr.M600120-JLR200 (2006).
Inagaki, T. et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2, 217–225, https://doi.org/10.1016/j.cmet.2005.09.001 (2005).
Kong, B. et al. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 56, 1034–1043, https://doi.org/10.1002/hep.25740 (2012).
Escary, J. L., Choy, H. A., Reue, K. & Schotz, M. C. Hormone-sensitive lipase overexpression increases cholesteryl ester hydrolysis in macrophage foam cells. Arterioscler Thromb Vasc Biol 18, 991–998, https://doi.org/10.1161/01.ATV.18.6.991 (1998).
Tall, A. R. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J Intern Med 263, 256–273, https://doi.org/10.1111/j.1365-2796.2007.01898.x (2008).
Peled, M. & Fisher, E. A. Dynamic Aspects of Macrophage Polarization during Atherosclerosis Progression and Regression. Front Immunol 5, 579, https://doi.org/10.3389/fimmu.2014.00579 (2014).
Lian, J., Nelson, R. & Lehner, R. Carboxylesterases in lipid metabolism: from mouse to human. Protein & cell. https://doi.org/10.1007/s13238-017-0437-z (2017).
Lian, J. et al. Liver specific inactivation of carboxylesterase 3/triacylglycerol hydrolase decreases blood lipids without causing severe steatosis in mice. Hepatology 56, 2154–2162, https://doi.org/10.1002/hep.25881 (2012).
Wei, E. et al. Loss of TGH/Ces3 in mice decreases blood lipids, improves glucose tolerance, and increases energy expenditure. Cell Metab 11, 183–193, https://doi.org/10.1016/j.cmet.2010.02.005 (2010).
Quiroga, A. D., Lian, J. & Lehner, R. Carboxylesterase1/Esterase-x regulates chylomicron production in mice. PloS one 7, e49515, https://doi.org/10.1371/journal.pone.0049515 (2012).
Bahitham, W., Watts, R., Nelson, R., Lian, J. & Lehner, R. Liver-specific expression of carboxylesterase 1g/esterase-x reduces hepatic steatosis, counteracts dyslipidemia and improves insulin signaling. Biochimica et biophysica acta 1861, 482–490, https://doi.org/10.1016/j.bbalip.2016.03.009 (2016).
Southern, E. M. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98, 503–517, https://doi.org/10.1016/S0022-2836 (1975).
Zilversmit, D. B. A single blood sample dual isotope method for the measurement of cholesterol absorption in rats. Proc Soc Exp Biol Med 140, 862–865, https://doi.org/10.3181/00379727-140-36568 (1972).
Sekiya, M. et al. Ablation of neutral cholesterol ester hydrolase 1 accelerates atherosclerosis. Cell Metab 10, 219–228, https://doi.org/10.1016/j.cmet.2009.08.004 (2009).
Roberts, D. C. K. et al. An Alternative Procedure for Incorporating Radiolabeled Cholesteryl Ester into Human-Plasma Lipoproteins Invitro. Biochem J 226, 319–322, https://doi.org/10.1042/bj2260319 (1985).
Zhang, Y. et al. FXR deficiency causes reduced atherosclerosis in Ldlr-/- mice. Arterioscler Thromb Vasc Biol 26, 2316–2321, https://doi.org/10.1161/01.ATV.0000235697.35431.05 (2006).
Paigen, B., Morrow, A., Holmes, P. A., Mitchell, D. & Williams, R. A. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis 68, 231–240, https://doi.org/10.1016/0021-9150 (1987).
Acknowledgements
This work was supported by NIH grants R01HL103227, R01DK095895 and R01DK102619 to Y.Z.
Author information
Authors and Affiliations
Contributions
J.X., Y.X., Y.Y.X., L.Y. and Y.Z. designed and carried out the studies and analyzed the data. J.X. and Y.Z. wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, J., Xu, Y., Xu, Y. et al. Global inactivation of carboxylesterase 1 (Ces1/Ces1g) protects against atherosclerosis in Ldlr −/− mice. Sci Rep 7, 17845 (2017). https://doi.org/10.1038/s41598-017-18232-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18232-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
