
Abstract
Huntington’s disease (HD) is an inherited neurodegenerative disorder of which skeletal muscle atrophy is a common feature, and multiple lines of evidence support a muscle-based pathophysiology in HD mouse models. Inhibition of myostatin signaling increases muscle mass, and therapeutic approaches based on this are in clinical development. We have used a soluble ActRIIB decoy receptor (ACVR2B/Fc) to test the effects of myostatin/activin A inhibition in the R6/2 mouse model of HD. Weekly administration from 5 to 11 weeks of age prevented body weight loss, skeletal muscle atrophy, muscle weakness, contractile abnormalities, the loss of functional motor units in EDL muscles and delayed end-stage disease. Inhibition of myostatin/activin A signaling activated transcriptional profiles to increase muscle mass in wild type and R6/2 mice but did little to modulate the extensive Huntington’s disease-associated transcriptional dysregulation, consistent with treatment having little impact on HTT aggregation levels. Modalities that inhibit myostatin signaling are currently in clinical trials for a variety of indications, the outcomes of which will present the opportunity to assess the potential benefits of targeting this pathway in HD patients.
Similar content being viewed by others
Introduction
Huntington’s disease (HD) is an inherited progressive neurodegenerative disorder for which the age of onset is generally in midlife. It is caused by a CAG repeat expansion in the first exon of the huntingtin (HTT) gene that results in an abnormally long polyglutamine tract in the huntingtin protein (HTT)1. Symptoms include problems with motor coordination, neuropsychiatric symptoms and cognitive decline; and there are no disease-modifying treatments2. The mutant form of the HTT protein aggregates into oligomeric and fibrillary structures which are deposited in the brains of HD patients3,4 and neurodegeneration occurs in the striatum, cortex, hypothalamus and other brain regions5. There is increasing evidence to indicate that Huntington’s disease is also associated with pathological processes that occur in peripheral tissues, including skeletal muscle6. Treatments targeted to tissues and organs outside the CNS have the potential to substantially improve the quality of life of HD patients, either in the absence of disease modifying treatments or combined with CNS-targeted therapies7.
Mouse models of HD include mice that are transgenic for the 5′ region of the human HTT gene (e.g. R6/2), those transgenic for the entire human HTT gene (e.g. BACHD and YAC128) and a range of knock-in models (e.g. HdhQ150 and zQ175)8. In recent years, we have performed detailed comparative analyses of the R6/2 mouse model9 and HdhQ150 knock-in mice10 and found that at late stage disease (14 weeks for R6/2 and 22 months for homozygous HdhQ150) the phenotypes of these two models are extremely similar11,12,13,14,15,16, the main difference between the lines being the age at phenotype onset and the rate of disease progression. The R6/2 mouse is transgenic for a genomic DNA fragment encoding exon 1 of human HTT and is a model of the incomplete splicing event that occurs in all HD knock-in mouse models, YAC128 and BACHD mice17, and in HD patient tissues18, resulting in an exon 1 – intron 1 mRNA that in all cases produces the highly pathogenic exon 1 HTT protein. Both the R6/2 and HdhQ150 models develop a progressive failure to gain body weight, followed by weight loss11 and a highly comparable skeletal muscle atrophy and underlying muscle pathophysiology19. The skeletal muscle phenotype manifests as a uniform muscle fibre atrophy19,20,21 and functional deficits19,22, associated with the formation of nuclear inclusions13,20, transcriptional dysregulation19,23,24,25, energetic disturbances19,25, ion channel perturbations26, alterations in protein synthesis and degradation pathways27 and an apparent dissociation of trophic signaling between motor neurons and skeletal muscle21. The investigation of the mechanisms underlying muscle pathology in HD mouse models is likely to lead to insights into the muscle atrophy and loss of muscle strength that has been reported in HD patients (for review, see28).
Myostatin is a member of the transforming growth factor β super family of secreted growth factors that is synthesized as a full length precursor and becomes cleaved into an amino-terminal propeptide and carboxy terminal mature region29. It has been shown to be a muscle specific regulator of muscle size, and the knock-out of myostatin results in muscle hypertrophy and a reduction in adipose tissue mass30,31. Binding of myostatin and activin A to the activin A receptor type IIB (ActRIIB) on muscle cell membranes, activates SMAD 2/3-mediated transcription, which stimulates FOXO-dependent transcription increasing muscle protein degradation. SMAD activation also inhibits muscle protein synthesis by suppressing AKT signaling (for review see32). A soluble form of the activin type IIB receptor (ACVR2B/Fc) has been developed to compete with the natural receptor for circulating agonists and this caused a dramatic increase in muscle mass when injected into wild type (WT) mice33. Therefore, agents that block the myostatin signaling pathway have the potential to treat human disease associated with cachexia e.g. cancer, chronic kidney disease and chronic heart failure, as well as neuromuscular diseases32.
We have used the ACVR2B/Fc soluble receptor to test whether the inhibition of myostatin/activin A signaling might prevent or decrease skeletal muscle atrophy and restore muscle function in mouse models of HD. Administration of ACVR2B/Fc to R6/2 mice from 5 weeks of age, completely prevented deficits in body weight gain, muscle atrophy, grip strength, muscle function and motor unit loss and also delayed end-stage disease. A range of therapeutic agents that inhibit myostatin signaling are currently being tested in clinical trials for a number of indications34, the outcomes of which will provide the opportunity to design clinical trials to treat muscle atrophy in people affected with HD.
Results
Treatment with ACVR2B/Fc prevents body weight loss, grip strength deficits and muscle atrophy in R6/2 mice
We first set out to determine whether the inhibition of myostatin/activin A signaling might reduce the extent of muscle atrophy that occurs in the R6/2 mouse model of HD. The soluble ActRIIB receptor (ACVR2B/Fc) was administered by a subcutaneous weekly 10 mg/kg injection to both WT and R6/2 male and female mice (n = 5 per gender per genotype) from 5 to 11 weeks of age, and mice were sacrificed one week later at 12 weeks. Both male and female R6/2 mice developed a progressive failure to gain weight (Fig. 1A). Treatment with ACVR2B/Fc increased the body mass of both wild type (WT) and R6/2 mice as compared to their vehicle treated littermates and in the case of R6/2 mice, this completely prevented the decreased body weight phenotype (Fig. 1A). Similarly, fore limb grip strength was progressively impaired in both male and female R6/2 mice (Fig. 1B). Treatment with the receptor decoy resulted in an increase in fore limb grip strength in both WT and R6/2 mice as compared to vehicle treated littermates, and again completely prevented the R6/2 deficits (Fig. 1B). At sacrifice, the tibialis anterior (TA), quadriceps and gastrocnemius skeletal hind limb muscles were weighed. All three muscles showed a marked degree of atrophy in R6/2 mice (Fig. 1C). Treatment with ACVR2B/Fc increased muscle mass in both WT and R6/2 mice and, once more, in the case of R6/2, the weight of all three muscles had been restored to WT levels (Fig. 1C). In contrast, there was no effect on brain atrophy (Fig. S1A) or disease-associated changes in heart weight (Fig. S1A). Treatment with ACVR2B/Fc had been equally effective in male and female mice and, therefore, all further experiments were performed on female mice only.

Treatment with ACVR2B/Fc restores deficits in body weight, grip strength and muscle mass in R6/2 mice. (A) ACVR2B/Fc treatment resulted in a progressive weight gain in both WT and R6/2 mice and prevented body weight loss in R6/2 mice. (B) ACVR2B/Fc treatment resulted in a progressive increase in fore-limb grip strength in both WT and R6/2 mice and prevented grip strength deficits in R6/2 mice. (C) Treatment with ACVR2B/Fc increased muscle mass in both WT and R6/2 mice with the consequence that the R6/2 muscle mass at 12 weeks of age (genders combined) is equivalent to that of wild type mice for quadriceps, gastrocnemius and tibialis anterior hind limb skeletal muscles. Statistical analysis was two-way ANOVA with post-hoc Bonferroni correction (see Table S8 for main effects and Table S9 for multiple comparisons). The statistical significance between values for ACVR2B/Fc treated and vehicle treated R6/2 mice is depicted: *p < 0.05; **p < 0.01; ***p < 0.001. n = 5 mice per gender per genotype. All data presented as means ± SEM.
To determine whether ACVR2B/Fc treatment had had an impact on body fat levels, we acquired magnetic resonance imaging (MRI) data using a 3-point Dixon technique to separate fat and water images. However, this approach did not detect a statistically significant difference between the treatment groups (Table S1).
Treatment with ACVR2B/Fc prevents muscle fibre atrophy
The skeletal muscle atrophy that occurs in R6/2 mice has previously been studied in detail and no evidence was found of changes typically associated with muscle pathology other than a diffuse atrophy20,21. The adult neuromuscular innervation pattern develops normally and the incidence of abnormalities at the neuromuscular junction was very low and only occurred in animals close to end stage disease21. The only prominent morphological change was a severe generalized atrophy of all muscle fibres that occurred in both fibre types20,21. Therefore, to investigate the effect of ACVR2B/Fc treatment on muscle pathology, we measured the lesser diameters of fibres in the TA and quadriceps muscles of WT and R6/2 mice that had been treated with ACVR2B/Fc or vehicle. We found that there was a statistically significant difference in the fibre diameters between WT and R6/2 quadriceps and TA muscles (Fig. 2) and that ACVR2B/Fc treatment resulted in a statistically significant increase in the fibre diameter in both cases. Representative plots for the range of fibre diameters for the WT and R6/2 quadriceps and TA muscle treated with either vehicle or ACVR2B/Fc are illustrated in Fig. 2. ACVR2B/Fc treatment increased fibre diameter in the quadriceps (p < 0.001) and the TA (p = 0.061) of R6/2 mice.

ACVR2B/Fc treatment prevents muscle fibre atrophy. (A) the lesser fibre diameter of myofibres in the TA and quadriceps of WT and R6/2 mice treated with ACVR2B/Fc or vehicle. Fibre counts were obtained from between 3 and 8 sections per mouse, n = 3 or 4 mice/treatment group. The WT muscle fibre diameters were greater than those for R6/2: quadriceps (F(13,10) = 28.82, p ≤ 0.001), TA (F(15,12) = 10.29, p = 0.008). ACVR2B/Fc treatment increased the fibre diameter in both cases: quadriceps (F(13,10) = 92.8, p ≤ 0.001) and TA (F(15,12) = 12.73, p = 0.004). ACVR2B/Fc treatment increased fibre diameter in the quadriceps (p < 0.001) and the TA (p = 0.061) of R6/2 mice. Representative traces from individual mice are depicted. Statistical analysis was two-way ANOVA with post-hoc Bonferroni correction.
Treatment with ACVR2B/Fc improved muscle function in R6/2 mice
Neuromuscular function was assessed for the extensor digitorum longus (EDL) and TA muscles after treatment of female mice with either vehicle or ACVR2B/Fc at 12 weeks of age as compared to their littermate controls. We began by measuring the isometric muscle tension of these two muscles to assess the effect of ACVR2B/Fc on the contractile abnormalities that we have previously described in R6/2 mice19,22. EDL is normally a fast twitch muscle that contracts and relaxes rapidly. In R6/2 mice, EDL took longer than WT EDL to exert a maximum twitch and then longer to relax (Fig. 3A and C). Upon treatment, the time to reach maximum twitch force was restored (Fig. 3A). In R6/2 mice TA muscles take the equivalent time as WT TA muscles to reach the peak force but take longer to relax (Fig. 3B and D). ActRIIB treatment completely restored the contractile dysfunction in the R6/2 TA (Fig. 3D).

Treatment with ACVR2B/Fc improves muscle function in R6/2 mice. (A–D) Contractile dysfunction of the R6/2 EDL muscles was assessed by measuring twitch tension: the time to peak and half relaxation time and this was corrected by ACVR2B/Fc treatment in both EDL (A,C) and TA (B,D) muscles. (E,F) The maximum tetanic force in mice treated with ACVR2B/Fc and vehicle. The maximum EDL (E) and TA (F) forces were decreased in R6/2 mice and completely rescued in mice treated with ACVR2B/Fc. (G) Examples of motor unit traces and the quantification of functional motor units. The number of functional motor units was reduced in R6/2 EDL muscles and this was restored in treated mice. Statistical analysis was two-way ANOVA with post-hoc Bonferroni correction (see Table S8 for main effects and Table S9 for multiple comparisons). Statistically significant differences between vehicle treated WT and vehicle treated R6/2: # p < 0.05, ### p < 0.001; statistically significant differences between ACVR2B/Fc treated R6/2 and vehicle treated R6/2: *p < 0.05; **p < 0.01; ***p < 0.001. n = 11 WT vehicle (EDL), n = 12 WT vehicle (TA), n = 8 WT ACVR2B/Fc (EDL and TA), n = 14 R6/2 vehicle (EDL and TA), n = 12 R6/2 ACVR2B/Fc (EDL and TA). All data presented as means ± SEM. The baseline WT and R6/2 vehicle treated phenotype data were previously published10.
Next, we measured the maximum tetanic force exhibited by the EDL and TA muscles in ACVR2B/Fc and vehicle treated WT and R6/2 mice. The EDL and TA muscle of R6/2 mice were weaker as compared to those from WT littermates (Fig. 3E and F); ACVR2B/Fc treatment increased R6/2 EDL strength to WT levels and there was a slight increase in the strength of the R6/2 TA muscles (87.2 ± 8.2 g as compared to 68.7 ± 4.5 g, p < 0.1) (Fig. 3E and F).
EDL muscles in WT mice are innervated by approximately 30 functional motor units35 and we have previously shown that this is reduced to approximately 20 motor units in the R6/2 EDL at 12 weeks of age and then further decreased to approximately 10 units by 14 weeks19. The number of functionally active motor neurons that innervate the EDL was assessed in vehicle and ACVR2B/Fc treated 12 week old WT and R6/2 mice. Representative examples of motor unit recordings and the mean motor unit number are illustrated in Fig. 3G. ACVR2B/Fc treatment completely prevented the loss of functional motor units in R6/2 mice and had no effect on motor unit number in WT mice.
Treatment with ACVR2B/Fc delays end-stage disease but does not improve motor impairments
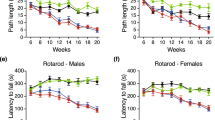
The progressive impairment of motor function is a cardinal feature of HD mouse models, to which both central nervous system (CNS)-driven and skeletal muscle-driven pathogenic mechanisms might contribute. Given that ACVR2B/Fc treatment completely rescued muscle atrophy and weakness, this provided an opportunity to dissect the extent to which a muscle-based pathology might contribute to motor related tasks. To address this, we dosed female R6/2 mice with either vehicle or ACVR2B/Fc from five weeks of age. A vehicle treated WT group was included to allow an estimation of effect size and to provide a control for any adverse effects (Fig. 4). Body weight and fore limb and hind limb grip strength were measured until 15 weeks of age. Mice were assessed for performance on an accelerating rotarod until 12 weeks of age and activity measures until 15 weeks. As before, ACVR2B/Fc treatment rescued deficits in body weight (Fig. 4A), and this rescue was maintained until 15 weeks of age. End-stage disease, as defined by the criteria in Table S2, was statistically significantly delayed in treated mice (Fig. 4B). Due to the development of an anal prolapse, two ACVR2B/Fc treated R6/2 mice were culled (on days 106 and 122) and these were not included in the final analysis. Although the deficits in grip strength were completely restored (Fig. 4C), there was no improvement in the decline in rotarod performance or the ensuing hypoactivity in the open field (Fig. 4D). Therefore, the restoration in muscle mass and strength did not improve the motor-related tasks measured here, suggesting that the pathogenic basis of these impairments is driven by CNS-related mechanisms.

Treatment with ACVR2B/Fc delays end-stage disease, but has no effect on rotarod performance or activity measures. (A) ACVR2B/Fc treatment completely prevented the progressive loss in body weight loss that occurs in R6/2 mice. The effect was such that R6/2 mice were significantly heavier than WT mice at some ages (Table S9). (B) Kaplan-Meier curve showing that ACVR2B/Fc treatment delays end-stage disease in R6/2 mice (Chi = 8.764, p < 0.01 Mantel-Cox log-rank test). (C) ACVR2B/Fc treatment completely prevented the progressive loss in fore-limb as well as the combined fore- and hind-limb grip strength that occurs in R6/2 mice. The effect was such that R6/2 mice were significantly stronger than WT mice at some ages (Table S9) (D) ACVR2B/Fc had no effect on the impairment in R6/2 rotarod performance or hypoactivity. Statistical analysis was two-way ANOVA with post-hoc Bonferroni correction (see Table S8 for main effects and Table S9 for multiple comparisons). The statistical significance between values for ACVR2B/Fc treated and vehicle treated R6/2 mice is depicted: *p < 0.05; **p < 0.01; ***p < 0.001. WT vehicle, n = 13; R6/2 vehicle, n = 17; R6/2 ACVR2B/Fc, n = 14. All data presented as means ± SEM.
ACVR2B/Fc treatment and huntingtin aggregation
HTT aggregation in the form of nuclear inclusions has been detected in the skeletal muscle of both R6/2 and the Hdh150 knock-in HD models13,20,21. Quantification of the aggregate load using the Seprion ELISA has demonstrated that statistically significant levels of aggregated HTT can be detected in R6/2 quadriceps by 8 weeks of age36 and that the aggregate load is higher in the TA than in the quadriceps or gastrocnemius/plantaris19.
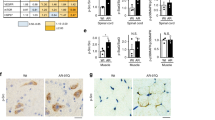
We began by applying the Seprion ELISA to investigate the effect of ACVR2B/Fc treatment on HTT aggregation in R6/2 quadriceps and TA muscles from 12 week old mice that had been treated with ACVR2B/Fc or vehicle. This suggested that treatment had led to a reduction in the aggregate load in both TA (p < 0.01) and quadriceps (p = 0.05) (Fig. 5A). HTT aggregation is the earliest phenotype to have been detected in skeletal muscle, and therefore, this result suggested that ACVR2B/Fc treatment might have had disease modifying properties. However, alternatively, the ACVR2B/Fc induced hypertrophy might have diluted the level of HTT aggregation in a given tissue mass, in which case, the degree of HTT aggregation could have remained unchanged, and our Seprion ELISA results might have been misleading. To investigate this further, TA sections were immunostained with antibodies to HTT (S830), and laminin, counterstained with DAPI (Fig. S2A and C), and immunofluorescence signals were quantified as illustrated in the work flow in Fig. S2B. The number of DAPI-stained nuclei in the regions of interest (ROI) was increased in R6/2 TA as compared with WT, which is consistent with the myofibre atrophy that had occurred in this R6/2 muscle (Figs 5B and S3A). Conversely, the number of nuclei had decreased in both WT and R6/2 TA in response to ACVR2B/Fc treatment, consistent with the hypertrophic response (Figs 5B and S3A). The average size of the nuclei, as determined by DAPI pixels, was unchanged between WT and R6/2 TA and did not change upon treatment in either case (Figs 5B and S3A).

ACVR2B/Fc treatment and HTT aggregation. (A) ACVR2B/Fc treatment results in a decrease in the aggregate load in R6/2 TA and quadriceps muscles as assessed by the Seprion ELISA. WT vehicle, n = 5; WT ACVR2B/Fc, n = 10; R6/2 vehicle n = 10; R6/2 ACVR2B/Fc, n = 9. WT background signal derives from the substrate. (B) Number of DAPI stained nuclei per ROI and average nucleus size in DAPI pixels. (C) Number of S830 inclusions per ROI and average inclusion size in pixels. (D) Percentage of S830 signal co-localized with DAPI and percentage of inclusions localized to the nucleus. (E) Number of nuclear S830 inclusions per ROI, the percentage of nuclei with inclusions and average size of nuclear inclusions in pixels (F) Level of expression of Pax7 as fold change from WT. Taqman qPCR values were normalized to the geometric mean of Atp5b, Actb and Sdha. WT vehicle, n = 8; WT ACVR2B/Fc, n = 7; R6/2 vehicle n = 6; R6/2 ACVR2B/Fc, n = 8. Statistical analysis for (B) and (D) was two-way ANOVA with post-hoc Bonferroni correction (see Table S8 for main effects) and for (A), (C), (D) and (E) was Student’s t-test (n = 4/treatment group). Statistical significance for R6/2 vehicle vs R6/2 ACVR2B/Fc is depicted by *p < 0.05; **p < 0.01; ***p < 0.001 and for WT vehicle vs R6/2 vehicle by ###p < 0.001. All data presented ± SEM. WT = wild type, ROI = regions of interest.
To investigate the pattern and level of aggregation we quantified the level of S830 immunofluorescence. There were an equivalent number of S830 reactive inclusions in the ROI in R6/2 TA muscle that had been treated with either vehicle or with ACVR2B/Fc (Fig. 5C) although the average size of the inclusions (as measured in red pixels) was greater in the ACVR2B treated muscle (Fig. 5C). Previous studies have documented the formation of nuclear inclusions in the skeletal muscle of HD mouse models13,20, however, our immunostaining clearly demonstrated that cytoplasmic inclusions were also present (Fig. S2C). To compare the level of aggregated HTT between the nucleus and cytoplasm, we performed an unbiased examination of the subcellular localization of the aggregates in the vehicle and ACVR2B/Fc treated R6/2 TA muscle. Approximately 70% of the S830 immunoreactivity co-localized with DAPI staining (Figs 5D and S3B) for both treatment groups, corresponding to approximately 55% of inclusions occurring in the nucleus (Fig. 5D). This is consistent with our observation that the nuclear inclusions tended to be larger than those in the cytoplasm.
We next examined the frequency and size of nuclear inclusions between R6/2 TA muscle that had been treated with ACVR2B/Fc and vehicle. Given that the average size of all inclusions per ROI was larger in ACVR2B/Fc treated muscle (Fig. 5C), that the total level of S830 signal in the nucleus was equivalent between treatment groups (Figs 5D and S3C) and that there were fewer nuclei in ACVR2B/Fc treated muscle (Fig. 5B), we asked whether the nuclear inclusions were larger in the ACVR2B/Fc treated TA. We found that the number of inclusions that co-localised with the DAPI staining was equivalent between the two treatment groups (Fig. 5E); that there was a trend toward an increase in the percentage of nuclei containing inclusions in the ACVR2B/Fc treated TA (p = 0.097) (Fig. 5E) and that the size of the nuclear inclusions was greater in the ACVR2B/Fc treated TA (Fig. 5E). These data are consistent with the ACVR2B/Fc treated TA having fewer nuclei per ROI (Fig. 5B), of which a higher percentage have larger inclusions than in the vehicle treated TA.
Skeletal muscle regenerates in response to injury and Pax7 expressing satellite cells are the major or only mediators of myofibre regeneration in the adult mouse37. After multiple rounds of injury, the satellite pool is maintained through a process of self-renewal37. We found an increased level of Pax7 expression in R6/2 TA as compared to WT at 12 weeks of age suggesting that the satellite cell pool comprises a greater percentage of total nuclei in R6/2 than in WT mice. Treatment with ACVR2B/Fc brought the level of Pax7 expression back toward WT levels, indicating that the percentage of nuclei expressing Pax7 is lower in ACVR2B/Fc treated than in vehicle treated R6/2 mice (Fig. 5F).
ACVR2B/Fc activates a transcriptional program to increase muscle mass, with little impact on HD-related transcriptional dysregulation
Transcriptional dysregulation is a well-established pathogenic process in HD38. Microarray profiles have been used to identify dysregulated transcripts in specific regions of HD post mortem brains39,40 and in quadricep biopsies from HD patients23 as compared to control subjects. Given that the transcriptome is highly dysregulated in the brain12 and skeletal muscle23 of R6/2 transgenic mice, we used quantitative real time PCR (qPCR) to investigate the level of expression the ActRIIB receptor (Acvr2b) and myostatin (Mstn), and found that the receptor and ligand were expressed at comparable levels in WT and R6/2 muscle, hence their transcriptional dysregulation does not contribute to the muscle wasting in R6/2 mice (Fig. S4). The expression levels of Mstn and Acvr2b were not affected by treatment in either case.
Inhibition of myostatin signaling activates pathways that directly stimulate protein synthesis and inhibit protein breakdown in the muscle fibre32. In order to uncover the transcriptional changes that underlie the phenotypic rescue by ACVR2B/Fc, RNA from the quadriceps and TA of WT and R6/2 mice at 12 weeks of age that had been treated with either vehicle or ACVR2B/Fc was sequenced (n = 10/treatment group). Dysregulated gene analysis (DESeq. 2) was applied to identify genes for which there was a statistically significant change in expression (Benjamini-Hochberg adjusted p < 0.05). The transcriptional response of WT and R6/2 muscles to ACVR2B/Fc treatment was relatively comparable (Table S3). We did not see changes in the expression of genes controlled by SMAD 2/3-signaling. This is most likely because the RNAseq analysis was performed on tissue harvested one week after the last ACVR2B/Fc dose, by which time this response had diminished. The gene ontology (GO) terms associated with the few hundred transcripts whose expression levels were altered in response to treatment, in both WT and R6/2 mice, were broadly related to muscle function and energy metabolism (Tables S3 and S4).
The comparison of gene expression levels in the quadriceps and TA of R6/2 as compared to WT mice indicated that approximately a half of all genes were dysregulated in these muscles (Table S3). We next analysed the effect of ACVR2B/Fc treatment on the extent of transcriptional dysregulation in R6/2 TA and quadriceps. We found that the expression level of only a very small number of R6/2-dysregulated genes was changed (cut off of 1.5 fold change and adjusted to p < 0.05) (detailed in Fig. 6 and Table S4). In some cases, administration of ACVR2B/Fc restored the expression levels of dysregulated genes toward WT levels, but in others, the extent to which genes were dysregulated was increased (Fig. 6, Table S4). Therefore, inhibition of signaling through the ActRIIB receptor had very little impact on the disease-related transcriptional dysregulation in these two muscles. We applied weighted gene correlation network analysis (WGCNA), in an attempt to uncover subtle changes in transcriptional regulation, which might not have been picked up by DESeq. 2, however, this did not reveal any additional GO terms (Tables S5 and S6).

The effect of ACVR2B/Fc treatment on genes that are dysregulated in R6/2 muscle. The first column in each panel indicates the change in gene expression in R6/2 muscle as compared to WT. Red squares represent an increase and blue squares, a decrease in expression levels. The second column represents the change in gene expression levels in R6/2 muscle in response to ACVR2B/Fc treatment and the third column indicates when a gene in WT muscle was also significantly changed in response to treatment. All of the depicted genes were changed to statistically significant levels (adjusted p < 0.05) with a fold change greater than 1.5. Genes in bold were changed in both quadriceps and tibialis anterior. The panels represent genes that are (A) increased or (B) decreased in the quadriceps and (C) increased or (D) decreased in the tibialis anterior of R6/2 mice as compared to WT. RNAseq was performed on RNA extracted from n = 10 mice/treatment group.
Discussion
Pathophysiological alterations in skeletal muscle contribute to the phenotypes exhibited by mouse models of HD (for review see28). Here we have shown that treatment with a soluble ActRIIB receptor (ACVR2B/Fc) completely prevented skeletal muscle atrophy and body weight loss from occurring in the R6/2 HD mouse model. Treatment prevented the onset of skeletal muscle weakness, contractile abnormalities and the loss of functional motor units in EDL muscles. Remarkably, ACVR2B/Fc treatment also delayed the age at which R6/2 mice reached end-stage disease.
The deposition of aggregated forms of the HTT protein is the earliest phenotype to have been reported in the skeletal muscle of HD mouse models36, and therefore it was important to determine whether treatment with ACVR2B/Fc might have modulated this process, which would potentially be indicative of disease modifying properties. However, our assessment of the comparative levels of HTT aggregation turned out to be more complicated than anticipated. Our routine approach to measuring HTT aggregation, using the Seprion ELISA, suggested that ACVR2B/Fc treatment might have decreased the HTT aggregate load. However, this interpretation was complicated by two factors: the ACVR2B/Fc induced hypertrophy resulted in a lower density of nuclei in the treated muscle, potentially diluting the levels of both nuclear and cytoplasmic aggregates; second, the satellite cell pool was greater in vehicle than in ACVR2B/Fc treated muscle reflecting attempts by the R6/2 muscle at regeneration in response to mutation-induced pathogenic processes. In fact, our immunohistochemical analyses indicated that the levels of aggregation in the vehicle and ACVR2B/Fc treated muscles were relatively comparable, although the size and frequency of nuclear inclusions differed. The ACVR2B/Fc treated muscle had a lower density of nuclei, of which a higher percentage contained larger nuclear inclusions than in the vehicle treated samples. It is possible that replenishment of the satellite cell pool in vehicle-treated R6/2 muscle provided a source of ‘younger’ nuclei in which the aggregation process was less well advanced. A more detailed analysis of the effect of ACVR2B/Fc on HTT aggregation requires a much better baseline understanding of this process in skeletal muscle and is beyond the scope of this study at the present time.
Inhibition of myostatin/activin A signaling activated a relatively comparable transcriptional program in both WT and R6/2 mice to increase muscle protein synthesis and decrease the degradation of muscle proteins. The gene ontology terms associated with these changes were broadly associated with muscle function and energetics. Our RNAseq transcriptome analysis showed that approximately one half of all genes were dysregulated in the R6/2 TA and quadriceps at 12 weeks of age as compared to wild type. In light of the complete restoration of muscle mass and strength, it was surprising that we found such little evidence of an improvement in this HD-associated transcriptional dysregulation. However, this result was compatible with the fact that ACVR2B/Fc treatment did not prevent HTT aggregation in nuclei.
The mouse models in which skeletal muscle phenotypes have been described all carry highly expanded CAG repeats and represent the severe forms of HD with symptom onset occurring in childhood or adolescence. Therefore, is there any evidence that these preclinical findings will be applicable to HD patients with CAG repeat expansions that cause the adult onset form of the disease? Although studies are limited, impairments in skeletal muscle function have been reported in this patient group. Muscle strength has only been assessed in one study. This used a hand-held dynamometer to measure the isometric muscle strength in six lower limb muscle groups and found that people with HD had, on average, half the strength of healthy matched controls and that muscle strength significantly correlated with stage of disease as measured by UHDRS (unified Huntington’s disease rating scale) scores41. Strikingly, myopathy was reported to be the first HD-related symptom in a semi-professional marathon runner, which occurred years before the appearance of other neurological symptoms42. Most mechanistic studies of the skeletal muscle pathophysiology of HD patients have focused on mitochondrial function and energy metabolism. These have revealed a decreased oxidative capacity43,44 and lower anaerobic threshold45 than in control subjects and deficits in complex II/III activity of mitochondrial respiratory chain46, although normal mitochondrial function has recently been reported in presymptomatic mutation carriers47. Gene expression profiling of the RNA from HD patient skeletal muscle biopsies revealed the beginnings of a transition from fast twitch to slow twitch fibres23, consistent with that observed in the mouse models19,23. Histological studies have been very limited, often being restricted to the analysis of a single patient, and have reported: multiple centralized nuclei44, non-specific myopathic changes48, an abnormal pattern of HTT staining44 and the presence of nuclear inclusions46. A systematic longitudinal study of muscle function and pathology at all clinical stages of HD is clearly warranted.
In general, systematic muscle wasting is associated with weakness, fatigue, frailty, insulin resistance, bone fracture, disability and death32. In HD, muscle atrophy and weakness would be expected to exacerbate physical inactivity, which could in turn increase the rate of atrophy41. It may contribute to the frequency of falls and accelerate the loss of independent mobility and the use of a wheelchair. It could also lead to an increase in HD-associated apathy thereby contributing to cognitive decline. Myostatin inhibition in muscle has been shown to improve metabolism, effects that may also be beneficial to individuals with HD49. Therefore, although our data suggest that inhibition of myostatin/activin A signalling would provide a symptomatic treatment rather than one targeting the underlying pathogenic basis of the disease, this may have considerable impact on the overall quality of life for HD patients in terms of increased mobility and a reduction in falls, as well as the cognitive and mood benefits that could arise through an increased exercise capacity50,51. Consistent with this approach, myostatin inhibitors are currently being tested in clinical trials as a symptomatic treatment for inclusion body myositis34,52 and have been proposed as treatments to target the periphery in amyotrophic lateral sclerosis53.
The development of agents that inhibit myostatin signalling has been an active area of research since the knock-out of myostatin was shown to induce muscle hypertrophy in mice30. These approaches fall into two main categories, those that specifically target myostatin e.g. anti-myostatin antibodies, or those that, like the ACVR2B/Fc soluble receptor used here, also inhibit signaling via its alternative ligands: activin A, growth differentiation factor 11 (GDF11) and bone morphogenic proteins (BMPs)32. Therapeutics that inhibit myostatin/activin A signaling are currently undergoing clinical trials for a wide range of indications which include sarcopenia, cancer cachexia, muscular dystrophy, sporadic inclusion body myositis and rehabilitation post-orthopedic surgery (for reviews, see34,54). The comparative safety, tolerability and efficacy of these modalities will soon have been established34 paving the way to the design of clinical trials to assess the benefits of myostatin inhibition in individuals with HD.
Methods
Mouse maintenance, breeding and genotyping
All animal care and procedures were performed in compliance with the regulation on the use of Animals in Research (UK Animals and Scientific Procedures Act of 1996 and the EU Directive of 2010/63/EU) with approval by the King’s College London Ethical Review Process Committee. Hemizygous R6/2 mice were bred by backcrossing R6/2 males to (CBA x C57BL/6) F1 females (B6CBAF1/OlaHsd, Harlan Olac, Bicester, UK). All animals had unlimited access to water and breeding chow (Special Diet Services, Witham, UK), and housing conditions and environmental enrichment were as previously described55. Mice were subject to a 12-h light/dark cycle. Genotyping of tail DNA by PCR and measurement of the CAG repeat length were performed as previously described36. Dissected tissues were weighed before being snap frozen in liquid nitrogen for molecular analyses and stored at −80 °C until further analysis.
ACRB2R/Fc treatment
ACVR2B/Fc was prepared as previously described56 in a series of batches and stored at 4 °C in PBS as vehicle. It was administered weekly by subcutaneous injection at 10 mg/kg starting at 5 weeks of age. For most experiments, the last dose was given at 11 weeks of age and mice were sacrificed one week later at 12 weeks. Mice were randomized to treatment groups based on body weight, litter of origin and CAG repeat size and investigators were blind to genotype and treatment group throughout each trial. Body weight and/or grip strength were measured longitudinally for all experiments to ensure that all batches of ACVR2B/Fc had comparable efficacious effects (Table S7). A reduction in body temperature can be indicative of an adverse response to a treatment. Body temperature was monitored weekly using an infra-red temperature reader (ThermoScan Instant Thermometer, Braun) that was reproducibly positioned under the thorax. There was no change in body temperature (Fig. S1B), which taken together with other measures of appearance, indicated that the treatment was well-tolerated. For the assessment of ACVR2B/Fc treatment on end-stage disease, dosing continued weekly after 11 weeks of age until it was judged that a mouse had reached its humane end-point (Table S2). A summary of the mice used in each experiment and their CAG repeat size is listed in Table S7.
Assessment of motor-related tasks
Grip strength was measured as previously described57. For the initial pilot study, fore-limb grip strength was measured using a San Diego Instruments Grip Strength Meter (San Diego, CA, USA) and for subsequent experiments, fore-limb grip strength and also the combined fore- and hind-limb grip strength were measured using a grip strength meter from Bioseb in Vivo Research Instruments. Mice were tested for motor co-ordination as previously described58 on an Ugo Basile accelerating rotarod (Linton Instruments, UK) that had been modified with a smooth rubber coating over the beam55. General ambulation was probed in square, plain white arenas (50 × 50 × 50 cm, Engineering Design Plastics Ltd, Cambridge, UK) for 30 min and behaviour was recorded through a video camera positioned above the apparatus. Activity (distance moved) was tracked and then analysed using Ethovision 7XT software (Noldus, Netherlands) as described57.
Assessment of muscle function in vivo
Twelve week old female WT and R6/2 mice that had been treated with vehicle or ACVR2B/Fc were deeply anesthetized with isoflurane. The distal tendons of the tibialis anterior (TA) and extensor digitorum longus (EDL) muscles in both hindlimbs were dissected free and the isometric tension was recorded in vivo as described in detail elsewhere35. The contractile characteristics were determined by measuring the time taken (ms) for the muscle to elicit peak twitch tension (time to peak, TTP) and the time taken for the muscle to reach half relaxation from peak contraction (half relaxation time). Tetanic contractions were measured as previously described35 and recorded on a Lectromed Multitrace 2 recorder (Lectromed Ltd, UK). All parameters were measured using the Picoscope v5 and v6 software (Pico Technology, Cambridgeshire, UK). The number of functional motor units innervating the EDL muscles was determined as described previously35.
Immunohistochemistry and lesser fibre diameter measurements
Muscles were dissected, embedded, frozen in isopentane and sectioned as previously described59. Immunohistochemical staining was performed using the Ventana Discovery XT instrument, using the Ventana DAB Map Kit (760–124). Anti-laminin (Sigma L9393) primary antibody incubation was for 1 h using a 1:1000 dilution. Swine anti- Rabbit (Dako E0353) secondary antibody incubation was for 32 min, using a 1:200 dilution. Slides were haematoxylin counterstained.
Analysis of laminin stained muscle sections was performed on whole slide images, generated using a Leica SCN400F, using Definiens Developer XD (Definiens, Munich, Germany) and between 3 and 8 tissue sections were analysed per mouse. An automatic threshold method was used to separate the tissue from the background area, then again to identify sarcolemma within the tissue. A series of shape based manipulations and edge smoothing operations were then performed to separate joined fibres, combine split fibres and clarify the definition of muscle fibres. Finally muscle fibres which do not satisfy strict morphometric rules were excluded to provide a reliable set of transversely sectioned muscle fibres; width and shape measurements were then exported. The fibre diameter profile each tissue section was generated in excel.
MR Imaging and sample preparation
The methods for sample preparation, scanning and data processing are described in detail in the Supplemental Information.
HTT aggregation analysis by ELISA and immunohistochemistry
Aggregates were captured in Seprion ligand coated plates (Microsens, PADPCB1 SEP1–01) and detected using the MW8 mouse monoclonal antibody60 (1:2000) as previously documented36. For immunohistochemistry, muscles were dissected, embedded, frozen in isopentane and sectioned as described59 and slides were stored −20 °C until processing for immunohistochemistry. Prior to staining, slides were equilibrated to 50 °C to remove moisture and a wax pen was used to create a hydrophobic barrier around the sections. After washing with PBS, sections were blocked using 10% normal donkey serum and 0.3% triton X-100 in PBS. Antibodies against laminin (Sigma L9393, 1:1000) and HTT (S83061, 1:1000) were applied overnight at 4 °C prior to the application of appropriate secondary antibodies (Molecular Probes, 1:500) at RT for 2 h followed by the DAPI nuclear stain for 15 min.
Confocal focal images were taken using the Nikon AR1 Confocal microscope (Nikon Instruments using a 40x objective (Plan Apo 40x Ph2, NA 0.95) and NIS-Elements C software (Nikon Instruments). A grid was applied to capture nine regions of interest (ROI) per section through an automated unbiased process from two muscle sections per mouse (n = 4 mice per treatment group) to generate 18 captured ROIs per mouse. Images were exported as TIFFs and analysed using threshold fluorescence levels in ImageJ (U. S. National Institutes of Health, http://imagej.nih.gov/ij/) (Fig. S2A and C). A threshold intensity value of 90 was applied to DAPI images and of 50 to S830 images, and pixels below these thresholds were excluded. The S830 signal in the sections from WT mice treated with either vehicle or with ACVR2B/Fc was negligible (Fig. S3C). Objects were identified as groups of adjacent pixels. Objects with less than 25 DAPI pixels were considered debris and not counted as nuclei. DAPI threshold images were used to mask the S830 images and co-localised pixels were counted as intra-nuclear inclusions and those not co-localising were considered to be extra-nuclear inclusions (Fig. S3B). The percentage of nuclei containing inclusions was counted manually.
RNA extraction and Taqman real-time PCR expression analysis
Total RNA from skeletal muscles was extracted with the mini-RNA kit according to the manufacturer instructions (Qiagen). The reverse transcription reaction (RT) was performed using MMLV superscript reverse transcriptase (Invitrogen) and random hexamers (Operon) as described elsewhere19,62. The final RT reaction was diluted 10-fold in nuclease free water (Sigma). All Taqman qPCR reactions were performed as described previously62 using the CFX96 Real-Time PCR Detector (BioRad). Stable housekeeping genes for qPCR profiling of various skeletal muscles for HD mouse models were determined using the Primer Design geNorm Housekeeping Gene Selection Mouse Kit with PerfectProbe software. Estimation of mRNA copy number was determined in triplicate for each RNA sample by comparison to the geometric mean of three endogenous housekeeping genes (Primer Design) as described62. Primer and probe sets for genes of interest were purchased from Thermo Fisher Scientific.
RNA sequencing and data analysis
RNA was prepared from the quadriceps and tibialis from n = 10 mice per treatment group and sequencing was performed by Expression Analysis on an Illumina Hi-seq. 2000. Paired-end sequencing was obtained, 4-plexed across lanes for a minimum of 38 million 50mer paired reads per sample. Alignment and QC was conducted in Omicsoft using the OSA algorithm63 against the mouse genome version B38 with EMSEMBL gene models version R75. FPKMs were then calculated following standard formulas. QC assessment found 79 of 80 samples of high quality both at the RNA quality and alignment mapping levels. One sample (quadriceps mouse 93) was found to be a strong outlier by principal component analysis and omitted from the analysis. The RNAseq data have been deposited in the GEO database under the accession number GSE81367. Rounded counts to the next integer were used as input data for dysregulated gene analysis (DESeq. 2)64. Only genes with a count of 2 or more in at least 10 out of the 79 samples were used for the analysis which left 18558 genes in the dataset. For weighted gene co-expression network analysis (WGCNA) we used variance stabilizing transformed counts from the DESeq. 2 output (function varianceStabilizingTransformation) as input. The subsequent WGCNA analysis was carried out as previously described40. Soft-threshold power for all networks was 30. Modules with highly correlated module eigengenes were merged (r > 0.7). Enrichment analysis was carried out using Enrichr http://amp.pharm.mssm.edu/Enrichr/index.html 65. We used the gene lists of dysregulated genes or the identified network modules as input. We summarized all gene ontology terms (GO-term) of similar sub-terms into an overarching term. Only GO-terms, regulators, pathways, etc. with a combined score larger than 5 were considered.
Statistics
Data were graphed using Prism Ver.5.0b (Graphpad software, California USA). Statistical analyses were calculated using SPSS Statistics Ver. 22 (IBM Portsmouth, UK). Data were screened for statistical outliers using Grubb’s test (GraphPad software, California, USA). The total number of mice used in each of the experiments is summarised in Table S7. The datasets can be separated into those that were collected longitudinally and those that were collected at a single time point.
Longitudinal datasets
These include assessment of body weight, body temperature, grip strength, locomotor activity and rotarod performance. To probe the influence of treatment across time, a two-way ANOVA was employed (Treatment group and Age as between-subject factors). All main effects from ANOVAs can be found in Table S8. Post-hoc tests with a Bonferroni correction for multiple comparisons were applied where appropriate and represented in the figures and in Table S9.
Datasets takes at a single time point
These include muscle mass, fibre diameters, muscle physiological measures, assessment of aggregate load by Seprion ELISA and qPCR. To probe the influence of genotype and treatment on these measures, either a two-way ANOVA was applied (Genotype and Treatment as between-subject factors) or a one way ANOVA to compare Treatment Groups. All main effects from ANOVAs can be found in Table S8 or in the text. Post-hoc tests with a Bonferroni correction for multiple comparisons were applied where appropriate and represented in the figures and in Table S9.
End-stage disease was determined using a scoring system that combined an assessment of appearance, body weight, disease score and unprovoked behaviour (Table S2). The data were represented by a Kaplan-Meier cumulative survival curve and statistical analysis was performed in SPSS using the log-rank (Mantel-Cox) test.
References
HDCRG. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–983 (1993).
Ross, C. A. et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 10, 204–216, https://doi.org/10.1038/nrneurol.2014.24 (2014).
Scherzinger, E. et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558 (1997).
DiFiglia, M. et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 (1997).
Waldvogel, H. J., Kim, E. H., Tippett, L. J., Vonsattel, J. P. & Faull, R. L. In Huntington’s Disease (eds G. P. Bates, S. J. Tabrizi, & L. Jones) Ch. 9, 185–217 (Oxford University Press, 2014).
van der Burg, J. M., Aziz, N. A. & Bjorkqvist, M. In Huntington’s disease (eds G. P. Bates, Tabrizi S. J., & L. Jones) Ch. 14, 370–392 (Oxford Univesity Press, 2014).
Carroll, J. B., Bates, G. P., Steffan, J., Saft, C. & Tabrizi, S. J. Treating the whole body in Huntington’s disease. Lancet Neurol 14, 1135–1142, https://doi.org/10.1016/S1474-4422(15)00177-5 (2015).
Bates, G. P. & Landles, C. In Huntington’s Disease (eds G. P. Bates, S. J. Tabrizi, & L. Jones) Ch. 16, 41–461 (Oxford University Press, 2014).
Mangiarini, L. et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506 (1996).
Lin, C. H. et al. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum Mol Genet 10, 137–144 (2001).
Woodman, B. et al. The Hdh(Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res Bull 72, 83–97 (2007).
Kuhn, A. et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet 16, 1845–1861 (2007).
Moffitt, H., McPhail, G. D., Woodman, B., Hobbs, C. & Bates, G. P. Formation of polyglutamine inclusions in a wide range of non-CNS tissues in the HdhQ150 knock-in mouse model of Huntington’s disease. PLoS One 4, e8025, https://doi.org/10.1371/journal.pone.0008025 (2009).
Labbadia, J. et al. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest 121, 3306–3319, doi:57413 [pii] https://doi.org/10.1172/JCI57413 (2011).
Mielcarek, M. et al. Dysfunction of the CNS-heart axis in mouse models of Huntington’s disease. PLoS Genet 10, e1004550, https://doi.org/10.1371/journal.pgen.1004550 (2014).
Trager, U. et al. Characterisation of immune cell function in fragment and full-length Huntington’s disease mouse models. Neurobiol Dis 73, 388–398, https://doi.org/10.1016/j.nbd.2014.10.012 (2015).
Sathasivam, K. et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci USA 110, 2366–2370, https://doi.org/10.1073/pnas.1221891110 (2013).
Neueder, A. et al. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci Rep 7, 1307, https://doi.org/10.1038/s41598-017-01510-z (2017).
Mielcarek, M. et al. HDAC4-myogenin axis as an important marker of HD-related skeletal muscle atrophy. PLoS Genet 11, e1005021, https://doi.org/10.1371/journal.pgen.1005021 (2015).
Sathasivam, K. et al. Formation of polyglutamine inclusions in non-CNS tissue. Hum Mol Genet 8, 813–822 (1999).
Ribchester, R. R. et al. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur J Neurosci 20, 3092–30114, doi:EJN3783 [pii] https://doi.org/10.1111/j.1460-9568.2004.03783.x (2004).
Hering, T., Braubach, P., Landwehrmeyer, G. B., Lindenberg, K. S. & Melzer, W. Fast-to-Slow Transition of Skeletal Muscle Contractile Function and Corresponding Changes in Myosin Heavy and Light Chain Formation in the R6/2 Mouse Model of Huntington’s Disease. PLoS One 11, e0166106, https://doi.org/10.1371/journal.pone.0166106 (2016).
Strand, A. D. et al. Gene expression in Huntington’s disease skeletal muscle: a potential biomarker. Hum Mol Genet 14, 1863–1876 (2005).
Magnusson-Lind, A. et al. Skeletal muscle atrophy in R6/2 mice - altered circulating skeletal muscle markers and gene expression profile changes. J Huntingtons Dis 3, 13–24, https://doi.org/10.3233/JHD-130075 (2014).
Chaturvedi, R. K. et al. Impaired PGC-1alpha function in muscle in Huntington’s disease. Hum Mol Genet 18, 3048–3065, doi:ddp243 [pii] https://doi.org/10.1093/hmg/ddp243 (2009).
Waters, C. W., Varuzhanyan, G., Talmadge, R. J. & Voss, A. A. Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc Natl Acad Sci USA 110, 9160–9165, https://doi.org/10.1073/pnas.1220068110 (2013).
She, P. et al. Molecular characterization of skeletal muscle atrophy in the R6/2 mouse model of Huntington’s disease. Am J Physiol Endocrinol Metab 301, E49–61, https://doi.org/10.1152/ajpendo.00630.2010 (2011).
Zielonka, D., Piotrowska, I., Marcinkowski, J. T. & Mielcarek, M. Skeletal muscle pathology in Huntington’s disease. Front Physiol 5, 380, https://doi.org/10.3389/fphys.2014.00380 (2014).
Lee, S. J. & McPherron, A. C. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA 98, 9306–9311, https://doi.org/10.1073/pnas.151270098 (2001).
McPherron, A. C., Lawler, A. M. & Lee, S. J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387, 83–90, https://doi.org/10.1038/387083a0 (1997).
McPherron, A. C. & Lee, S. J. Suppression of body fat accumulation in myostatin-deficient mice. J Clin Invest 109, 595–601, https://doi.org/10.1172/JCI13562 (2002).
Han, H. Q., Zhou, X., Mitch, W. E. & Goldberg, A. L. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol 45, 2333–2347, https://doi.org/10.1016/j.biocel.2013.05.019 (2013).
Lee, S. J. et al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA 102, 18117–18122, https://doi.org/10.1073/pnas.0505996102 (2005).
Smith, R. C. & Lin, B. K. Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr Opin Support Palliat Care 7, 352–360, https://doi.org/10.1097/SPC.0000000000000013 (2013).
Kalmar, B. et al. Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J Neurochem 107, 339–350, https://doi.org/10.1111/j.1471-4159.2008.05595.x (2008).
Sathasivam, K. et al. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Hum Mol Genet 19, 65–78, doi:ddp467 [pii] https://doi.org/10.1093/hmg/ddp467 (2010).
Bentzinger, C. F., Wang, Y. X. & Rudnicki, M. A. Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol 4, doi:https://doi.org/10.1101/cshperspect.a008342 (2012).
Cha, J. H. Transcriptional signatures in Huntington’s disease. Prog Neurobiol 83, 228–248, doi:S0301-0082(07)00071-8 [pii] https://doi.org/10.1016/j.pneurobio.2007.03.004 (2007).
Hodges, A. et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet 15, 965–977 (2006).
Neueder, A. & Bates, G. P. A common gene expression signature in Huntington’s disease patient brain regions. BMC Med Genomics 7, 60, https://doi.org/10.1186/s12920-014-0060-2 (2014).
Busse, M. E., Hughes, G., Wiles, C. M. & Rosser, A. E. Use of hand-held dynamometry in the evaluation of lower limb muscle strength in people with Huntington’s disease. J Neurol 255, 1534–1540, https://doi.org/10.1007/s00415-008-0964-x (2008).
Kosinski, C. M. et al. Myopathy as a first symptom of Huntington’s disease in a Marathon runner. Mov Disord 22, 1637–1640, https://doi.org/10.1002/mds.21550 (2007).
Lodi, R. et al. Abnormal in vivo skeletal muscle energy metabolism in Huntington’s disease and dentatorubropallidoluysian atrophy. Ann Neurol 48, 72–76 (2000).
Saft, C. et al. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington’s disease. Mov Disord 20, 674–679, https://doi.org/10.1002/mds.20373 (2005).
Ciammola, A. et al. Low anaerobic threshold and increased skeletal muscle lactate production in subjects with Huntington’s disease. Mov Disord 26, 130–137, https://doi.org/10.1002/mds.23258 (2011).
Turner, C., Cooper, J. M. & Schapira, A. H. Clinical correlates of mitochondrial function in Huntington’s disease muscle. Mov Disord 22, 1715–1721, https://doi.org/10.1002/mds.21540 (2007).
Buck, E. et al. High-resolution respirometry of fine-needle muscle biopsies in pre-manifest Huntington’s disease expansion mutation carriers shows normal mitochondrial respiratory function. PLoS One 12, e0175248, https://doi.org/10.1371/journal.pone.0175248 (2017).
Arenas, J. et al. Complex I defect in muscle from patients with Huntington’s disease. Ann Neurol 43, 397–400, https://doi.org/10.1002/ana.410430321 (1998).
McPherron, A. C., Guo, T., Bond, N. D. & Gavrilova, O. Increasing muscle mass to improve metabolism. Adipocyte 2, 92–98, https://doi.org/10.4161/adip.22500 (2013).
Groot, C. et al. The effect of physical activity on cognitive function in patients with dementia: A meta-analysis of randomized control trials. Ageing Res Rev 25, 13–23, https://doi.org/10.1016/j.arr.2015.11.005 (2016).
Paillard, T. Preventive effects of regular physical exercise against cognitive decline and the risk of dementia with age advancement. Sports Med Open 1, 4, https://doi.org/10.1186/s40798-015-0016-x (2015).
Machado, P. M. et al. Ongoing developments in sporadic inclusion body myositis. Curr Rheumatol Rep 16, 477, https://doi.org/10.1007/s11926-014-0477-9 (2014).
Holzbaur, E. L. et al. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiol Dis 23, 697–707, https://doi.org/10.1016/j.nbd.2006.05.009 (2006).
Cohen, S., Nathan, J. A. & Goldberg, A. L. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 14, 58–74, https://doi.org/10.1038/nrd4467 (2015).
Hockly, E., Woodman, B., Mahal, A., Lewis, C. M. & Bates, G. Standardization and statistical approaches to therapeutic trials in the R6/2 mouse. Brain Res Bull 61, 469–479 (2003).
Lee, S. J. et al. Role of satellite cells versus myofibers in muscle hypertrophy induced by inhibition of the myostatin/activin signaling pathway. Proc Natl Acad Sci USA 109, E2353–2360, https://doi.org/10.1073/pnas.1206410109 (2012).
Rattray, I. et al. Correlations of behavioral deficits with brain pathology assessed through longitudinal MRI and histopathology in the R6/1 mouse model of Huntington’s disease. PLoS One 8, e84726, https://doi.org/10.1371/journal.pone.0084726 (2013).
Rattray, I. et al. Correlations of behavioral deficits with brain pathology assessed through longitudinal MRI and histopathology in the R6/2 mouse model of HD. PLoS One 8, e60012, https://doi.org/10.1371/journal.pone.0060012 (2013).
Davies, S. W. et al. Detection of polyglutamine aggregation in mouse models. Methods Enzymol 309, 687–701 (1999).
Ko, J., Ou, S. & Patterson, P. H. New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res Bull 56, 319–329 (2001).
Sathasivam, K. et al. Centrosome disorganization in fibroblast cultures derived from R6/2 Huntington’s disease (HD) transgenic mice and HD patients. Hum Mol Genet 10, 2425–2435 (2001).
Benn, C. L., Fox, H. & Bates, G. P. Optimisation of region-specific reference gene selection and relative gene expression analysis methods for pre-clinical trials of Huntington’s disease. Mol Neurodegener 3, 17, doi:1750-1326-3-17 [pii]https://doi.org/10.1186/1750-1326-3-17 (2008).
Hu, J. et al. OSA: a fast and accurate alignment tool for RNA-Seq. Bioinformatics 28, 1933–1934, https://doi.org/10.1093/bioinformatics/bts294 (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128, https://doi.org/10.1186/1471-2105-14-128 (2013).
Acknowledgements
We wish to thank Rainer Kuhn, Andreas Weiss and Michal Mielcarek for helpful discussions and Michal Mielcarek for experimental work. This work was supported by the CHDI Foundation, the Medical Research Council (MR/L003627/1) to GPB and NIH to S.J.-L. (R01AR060636). LG is the Graham Watts Senior Research Fellow and LG and JRT Dick are supported by the Brain Research Trust. SB was supported by the Department of Health’s NIHR Biomedical Research Centre’s funding scheme.
Author information
Authors and Affiliations
Contributions
M.K.B., N.J., G.F.O., E.J.S., I.R., A.N., K.S., M.A., N.A., A.C.B., X.C., M.E., S.A.F., D.G., L.I., H.L., A.L., A.R.-L., D.L.S., T.W., performed the experiments. E.J.S., I.R., A.N., K.S., J.R.T.D., J.R., T.W., S.B., L.G., S.-J.L., G.P.B. designed the experiments, M.K.B., N.J., G.F.O., E.J.S., I.R., A.N., K.S., M.A., N.A., A.C.B., J.R.T.D., M.E., T.W., L.G., S.-J.L., G.P.B. analyzed the data. D.H., I.M.-S. facilitated reagents. D.H., I.M.-S. S.-J.L., G.P.B. designed the study. S.J.T., G.P.B. conceived the study. E.J.S., A.N., L.G., G.P.B. wrote the manuscript. All authors reviewed the manuscript before submission.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bondulich, M.K., Jolinon, N., Osborne, G.F. et al. Myostatin inhibition prevents skeletal muscle pathophysiology in Huntington’s disease mice. Sci Rep 7, 14275 (2017). https://doi.org/10.1038/s41598-017-14290-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-14290-3
This article is cited by
-
An exploratory metabolomic comparison of participants with fast or absent functional progression from 2CARE, a randomized, double-blind clinical trial in Huntington’s disease
Scientific Reports (2024)
-
A causative role for periarticular skeletal muscle weakness in the progression of joint damage and pain in OA
Scientific Reports (2023)
-
Abnormal molecular signatures of inflammation, energy metabolism, and vesicle biology in human Huntington disease peripheral tissues
Genome Biology (2022)
-
Cross-sectional analysis of plasma and CSF metabolomic markers in Huntington’s disease for participants of varying functional disability: a pilot study
Scientific Reports (2020)
-
MEF2 impairment underlies skeletal muscle atrophy in polyglutamine disease
Acta Neuropathologica (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
