
Abstract
The mechanisms underlying insecticide and acaricide resistance in insects and mites are often complex, including additive effects of target-site insensitivity, increased metabolism and transport. The extent to which target-site resistance mutations contribute to the resistance phenotype is, however, not well studied. Here, we used marker-assisted backcrossing to create 30 congenic lines carrying nine mutations (alone, or in combination in a few cases) associated with resistance to avermectins, pyrethroids, mite growth inhibitors and mitochondrial complex III inhibitors (QoI) in a polyphagous arthropod pest, the spider mite Tetranychus urticae. Toxicity tests revealed that mutations in the voltage-gated sodium channel, chitin synthase 1 and cytochrome b confer high levels of resistance and, when fixed in a population, these mutations alone can result in field failure of acaricide treatment. In contrast, although we confirmed the implication of mutations in glutamate-gated chloride channels in abamectin and milbemectin insensitivity, these mutations do not lead to the high resistance levels that are often reported in abamectin resistant strains of T. urticae. Overall, this study functionally validates reported target-site resistance mutations in T. urticae, by uncoupling them from additional mechanisms, allowing to finally investigate the strength of the conferred phenotype in vivo.
Similar content being viewed by others

Introduction
Insecticide resistance is a major threat for the chemical control of insects and mites in public health and agriculture. At present, the Insecticide Resistance Action Committee (IRAC) distinguishes between at least fifty-five different chemical classes and more than twenty-five distinct mode of action (MoA) groups1. MoA diversity is of key importance for effective Insecticide Resistance Management (IRM). However, the costs involved in the discovery, development and marketing of chemicals with new properties, increased immensely and slow down the development of compounds with new MoA. In addition, concerns about the environment and human health, integrated in new regulations, demand molecules with better selectivity2. To preserve the utility and diversity of available and newly developed insecticides/acaricides, it is of utmost importance to understand the resistance mechanisms against these compounds1 and develop diagnostic tools that support monitoring activities and resistance management.
A number of mechanisms have been shown to underlie insecticide resistance, most often quantitative or qualitative changes in major detoxification enzymes and transporters (pharmacokinetic mechanisms) and/or target-site mutations (pharmacodynamic mechanisms)3,4,5. When resistance is caused by a combination of factors (polygenic resistance), the overall resistance levels may be the sum of contribution of each individual factor6, 7 but synergistic or antagonistic interactions between resistance loci also occur8,9,10. The relative contribution of each individual resistance locus to complex insecticide/acaricide resistance phenotypes has only been sporadically investigated11. In particular, the relative importance and strength of target-site mutations is often hard to assess by merely associating a phenotype with mutation frequency in field populations, where prolonged selection may have led to the accumulation of additional resistance mechanisms. Furthermore, the majority of studies that look into epistatic interactions and/or resistance levels confirmed by a single genetic factor, are sometimes difficult to interpret if resistance alleles are not investigated in a common genetic background9, 12,13,14,15. Therefore, analysis of a resistance trait requires the studied strains to be identical, except for its causal gene16, 17. Functional validation of resistance mutations has been reported after recombinant expression. Inhibitor-protein interactions are then quantified via enzymatic reactions or ligand binding assays such as voltage-clamp electrophysiology. Although they provide strong evidence of the effect of a mutation on the affinity for the compound to the target-site, they are less suitable to assess the relative phenotypic consequences in vivo 18, 19. A more precise way to determine the effect of a mutation in vivo is to introduce it in a defined susceptible genetic background, by utilizing genome editing techniques, such as CRISPR-Cas920, 21, in species where this approach is applicable. In species where genome editing tools are not yet available, a more feasible alternative is to repeatedly backcross resistant individuals with susceptible ones16, 22, 23. Marker-assisted backcrossing provides a straight-forward and relatively precise method to untangle a mutation of interest from other mechanisms that might have been co-selected. The impact of a modifier or interactions between modifiers can be then analyzed by comparing the genetically identical strains that differ only in a small region on the chromosome, which harbors the resistant locus of interest24, 25.
The two-spotted spider mite, Tetranychus urticae (Chelicerata: Acari: Acariformes) is an important agricultural pest, that thrives on more than a 1,000 plant species26, 27. Its short life cycle, high fecundity and haplo-diploid system facilitates a rapid evolution of acaricide resistance. Today, T. urticae has developed resistance to more than 90 different chemical compounds, including major groups of currently used acaricides1, 28, 29. In T. urticae and other related spider mites, very high resistance ratios (RRs) have been reported for a number of compounds (RR > 10,000)28, 30 with cases of cross-resistance to newly introduced acaricides, for example, Khalighi, et al.31. Several target-site mutations have been uncovered and were associated with acaricide resistance in populations of T. urticae, recently summarized in Van Leeuwen and Dermauw4. These include mutations leading to amino acid substitutions in acetylcholinesterase (AChE) (G119S, A201S, T280A, G328A and F331W) that are associated with resistance to organophosphates and carbamates32. The L1024V and A1215D + F1538I substitutions in the voltage-gated sodium channel (VGSC) have been linked to resistance to Type I (absence of α-cyano group) and Type II (presence of α-cyano group) pyrethroids33, 34. Six orthologous glutamate–gated chloride channel (GluCl) genes have been reported in spider mites and the substitutions G314D and G326E in GluCl1 and GluCl3, respectively, were associated with resistance to abamectin35, 36. The G126S, I136T, S141F, D161G, P262T substitutions (in different combinations) identified in the cytochrome b (cytb) cause strong bifenazate resistance (Mitochondrial Qo inhibitors: QoI)37. A substitution I1017F in the chitin synthase 1 gene (CHS1) has been linked with high levels of resistance to mite growth inhibitors, etoxazole, clofentezine and hexythiazox38, 39. Most recently, an H92R substitution in the PSST subunit of the Mitochondrial Respiratory Complex I, has been associated with resistance to pyridaben, tebufenpyrad and fenpyroximate (Mitochondrial Electron Transport Inhibitors, site I, METI-I)25. As resistance in spider mites often has a polygenic basis, the relative contribution of target-site resistance to the overall resistance levels is currently unknown. One notable exception for T. urticae is the H92R mutation in the PSST subunit, which was introduced into a susceptible background by repeated backcrossing and shown to confer moderate levels of METI resistance25.
In this study, we investigated the relative contribution of nine known target-site mutations conferring resistance to abamectin, pyrethroids, bifenazate and mite growth inhibitors. We adopted the method of Bajda, et al.25 and succeeded in generating 30 congenic resistant and susceptible lines of T. urticae. When a combination of mutations in homologous genes was reported, the phenotypic levels of resistance were examined for both the single mutations, as well as their combination.
Materials and Methods
Acaricides
Acaricides used in this study were commercial formulations of bifenazate (Floramite, 240 g l−1 SC) and acequinocyl (Cantack 164 g l−1 SC), etoxazole (Borneo, 120 g l−1 SC), hexythiazox (Nissorun, 250 g l−1 SC) and clofentezine (Apollo, 500 g l−1 SC), abamectin (Vertimec 18 g l−1 EC), milbemectin (Milbeknock 10 g l−1 EC), bifenthrin (Talstar 100 g l−1 EC), fluvalinate (Mavrik 240 g l−1 EW) and analytical grade fenpropathrin (Sigma Aldrich).
Spider mite strains
The susceptible Wasatch strain is an inbred line, originally collected from tomato in a greenhouse near Salt Lake City, Utah, USA. The pyrethroid susceptible strain KOP8 is an inbred line derived from the Houten strain40. Wasatch does not contain any of the so far described mutations. KOP8 harbors the A1215D substitution, potentially associated with pyrethroid resistance. The GH strain carries the L1024V genotype (Musca domestica numbering) of the VGSC gene and was collected from greenhouse grown maize in Utah USA. The TuSB9 strain carrying the A1215D and F1538I mutations (Musca domestica numbering) in VGSC was previously described33. The MAR-AB strain carrying the G314D and G326E substitutions (Tetranychus urticae numbering) in GluCl1 and GluCl3, respectively, was previously described in Dermauw, et al.35. Strains with mutations associated with bifenazate resistance, HOL3 (cytb, P262T - Tetranychus urticae numbering) and BR-VL (cytb, G126S and S141F – Tetranychus urticae numbering) were described in Van Leeuwen, et al.37 and Van Leeuwen, et al.41 respectively. The EtoxR strain carrying the I1017F mutation (Tetranychus urticae numbering) in the chitin synthase (CHS1) gene was previously described38. An overview of strains is presented in Table 1. All T. urticae strains were maintained on 3-week old potted kidney bean plants (Phaseolus vulgaris L.) in a climatically controlled room or incubator at 25 ± 1 °C, 60% relative humidity, and 16:8 light: dark photoperiod.
Backcrossing experiments
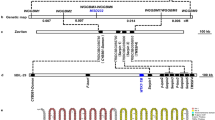
To assess the relative resistance levels associated with mutations, we used a marker assisted backcrossing approach to produce near-isogenic sister lines (Fig. 1 and Table 1). The crossing procedure was previously outlined in Bajda, et al.25. In short, a haploid male of the resistant strain was crossed with a virgin female of the susceptible strain. The resulting heterozygous virgin females were backcrossed to susceptible males and heterozygote genotypes were identified by a TaqMan molecular assay or PCR and sequencing as it is described in section Genotyping. This process was repeated for six to nine generations. In the last generation, a cross was carried out between the backcrossed heterozygous virgin females and their first born sons representing either a susceptible (absence of mutation) or the resistant (presence of mutation) genotype. This finally resulted in congenic homozygous lines for the mutation and the wild type allele. The final crosses were performed as follows (see Table 1): For the mutations in GluCls, G314D in GluCl1 and G326E in GluCl3, MAR-AB males were crossed with Wasatch virgin females in order to separate the mutations in different lines, as they are inherited independently35, after which they were introgressed separately: ♀ 314D/314 G x ♂ 314D or ♂ 314 G to generate GluCl1_R1-R3 and GluCl1_C, ♀326E /326 G x ♂ 326E or ♂326 G to produce homozygous congenic GluCl3_R1-R3 and GluCl3_C respectively. Mutations were later joined in a single line by dedicated crosses as follows: ♀GluCl1_R1 x ♂GluCl3_R1, ♀GluCl1_R2 x ♂GluCl3_R2, ♀GluCl1_R3 x ♂GluCl3_R3 and ♀GluCl1_C x ♂GluCl3_C to produce GluCl1 + 3_R1,R2,R3 and C respectively. For the mutations in VGSC; the ♀1024 V/1024 L x ♂1024 V or ♂1024 L were crossed to obtain homozygous congenic lines VGSC_R1-R3 and VGSC_C1 respectively, ♀1215D + 1538I/1215D + 1538 F x ♂1215D + 1538I or ♂1215D + 1538 F to obtain homozygous congenic VGSC_R4-R6 and VGSC_C2 respectively. For the mutation in CHS1; ♀ 1017 F/1017I x ♂ 1017 F or ♂1017I were crossed to generate homozygous congenic CHS1_R1-R3 and CHS1_C, respectively.

Schematic diagram of marker-assisted backcrossing of nuclear and mitochondrial encoded resistance mutations. The susceptible genotype is depicted by white-colored chromosomes (rectangles) and mitochondria (ovals), while those of the resistant genotype are depicted in blue. An orange color indicates whether the resistance mutation is either nuclear or mitochondrial encoded.
For the mitochondrial mutations in cytb (HOL3 and BR-VL) that are inherited completely maternally, simple crosses between resistant females and susceptible males were performed for 7 generations, as to create a line with the nuclear genome of the susceptible parent (Wasatch), but bearing the mitochondrial haplotype of the resistant line (Fig. 1). Three crosses were set up to produce lines carrying P262T mutation in PEWY motif of cytb (cytb_R1-R3) and consequently three lines with G126S + S141F mutations in cd1 region of cytb (cytb_R4-R6).
Single mite DNA extraction
In order to perform single mite genotyping for I1017F (EtoxR), P262T (HOL3) and G126S + S141F (BR-VL) individual T. urticae mites were homogenized in 20 μl STE buffer (100 mM NaCl, 10 mM Tris- HCl and 1 mM EDTA) with 1 mg ml−1 proteinase K (Sigma-Aldrich). Homogenate was incubated at 37 °C for 30 min followed proteinase K inactivation for 5 min at 95 °C. For G314D, G326E (MAR-AB) and F1538I, A1215D (TuSB9), L1024V (GH) single mite DNA was extracted following the CTAB method42. In short, individual mites were homogenized in 200 μl of extraction buffer (2% CTAB, 1.4 M NaCl, 0.2% β-mercaptoethanol, 20 mM EDTA, 100 mM Tris – HCl, pH:8.0) and incubated at 65 °C for 15 min. Equal volume of chloroform: isoamylalcohol (24:1) was used in order to remove proteins. The DNA was precipitated by isopropanol and washed with 75% ethanol. The pellet was air-dried and resuspended in 20 μl DEPC treated water.
Genotyping
Single mite genotyping was performed with standard PCR and sequencing (mutations I1017F, P262T, G126S + S141F and L1024V) and/or TaqMan method43 (mutations F1538I, G314D and G326E). PCRs were conducted in 50 μl final volume with 10 μl 5x Phusion HF Buffer, 0.2 mM of each dNTP, 0.5 μM each primer, 1 μl template, 0,5 μl polymerase with cycling conditions; 30 s at 98 °C followed by 35 cycles 5 s at 98 °C, 10 s at 55 °C, 15 s at 72 °C and 5 min of final extension. Reactions were performed in Bio-Rad T100™ Thermal Cycler. PCR products were purified with E.Z.N.A.® Cycle Pure Kit DNA purification kit (OMEGA bio-tek) and sequenced at Macrogen sequencing facility (Amsterdam). Sequencing data were analyzed using BioEdit 7.0.1 software44. Primers used for the PCR reactions and sequencing are listed in Supplementary Table S1.
TaqMan assay was performed as previously described43. In short, all assays were carried out in 15 μl total volume containing 2 μl of genomic DNA, 7.5 μl TaqMan Universal PCR Master Mix, 0.8 μM of each primer and 0.2 μM of each probe. Samples were run on CFX Connect, Real-Time PCR Detection System (Biorad) using the temperature cycling conditions of: 10 min at 95 °C followed by 40 cycles of 92 °C for 15 sec and 60 °C for 1 min. The increase in VIC and FAM reporter dyes, representing individuals with the resistant and susceptible alleles respectively, was monitored in real time using the CFX Manager software. Positive and negative template controls were included in each run to aid genotype scoring. Primers and probes used for the TaqMan assay are listed in Supplementary Table S1.
Toxicity bioassays
To assess the toxic effects of etoxazole and hexythiazox, larval bioassays (1) were performed as previously described by Van Pottelberge, et al.45. For the ovicide clofentezine, bioassays were performed on eggs (2) instead of larvae. Approximately fifty females were allowed to lay eggs for 5 hours on the upper side of 9 cm2 square-cut kidney bean leaf discs on wet cotton wool. For adulticidal bioassays (3)46, 20–30 young adult female mites were transferred to arenas, prepared as described above. Plates were sprayed with 1 ml of spray fluid at 1 bar pressure with a Potter Spray Tower (Burkard Scientific, UK) to obtain a homogenous spray film (2 mg deposit/cm2). Experiments were then placed in a climatically controlled room at 25 ± 0.5 °C, 60% RH and 16/8 h (light/dark) photoperiod. Three to four replicates of at least five serial dilutions of each acaricide and a control (deionized water or 1:100 dilution of the mixture of N, N-dimethylformamide and emulsifier W, depending on the acaricide used) were tested. Fenpropathrin was of technical grade and formulated in 3:1 v/v mixture of N, N-dimethylformamide and emusulfier W and subsequently diluted in deionized water as previously described47. Mortality was assessed after 24 h for bifenazate and acequinocyl and 48 h for all other acaricides. Mites treated with growth inhibitors, were considered as unaffected, if at the time of scoring displayed the same developmental stage as water treated control. Adult mites were scored as being alive if they could walk twice the distance of their body size after being prodded with a camel’s hair brush48. All mortalities obtained for control treatment were lower than 10%. LC50 values, slopes, RRs and 95% confidence limits were calculated by probit analysis (POLO, LeOra Software, Berkeley, USA)49. In case 5,000 mg l−1 did not cause 50% mortality, no further attempts were made to determine LC50s and RR was calculated by dividing 5,000 mg l−1 by the LC50 of susceptible strain. The effect of the treatment on the susceptible parent and the experimental line was considered significantly different if the hypothesis of equality of slopes and intercepts was rejected (p value = 0.05)50. If a regression line - illustrating dose response - could not be derived (LC50 of the experimental line was found to be higher than 5,000 mg l−1), the effect of treatment was considered different when the LC90 of the susceptible control was lower than 5,000 mg l−1.
Data availability
All data generated or analyzed during this study are included in this manuscript (and its Supplementary Information Files).
Results
Establishment of congenic lines
The initial crosses between parental resistant and susceptible strains are outlined in Table 1. Briefly, the susceptible strain Wasatch, which does not carry any of the mutations studied here, was used for the most of the backcrossing experiments (Table 1). To study the mutations in GluCl1 (G314D) and GluCl3 (G326E) associated with abamectin resistance, virgin females of Wasatch were crossed with males of the abamectin resistant strain MAR-AB carrying both GluCl mutations. Similarly, for the L1024V mutation associated with pyrethroid resistance, Wasatch virgin females were crossed with males of the pyrethroid resistant strain GH that carries L1024V. The effect of A1215D + F1538I mutations in pyrethroid resistance was examined through crossing males of TuSB9 with females of the parental susceptible strain KOP8 (carrying the A1215D only). To study the effect of mutations in the mitochondrial encoded cytb (P262T and G126S + S141F) that confer resistance to bifenazate, virgin females of bifenazate resistant strains HOL3 (P262T) and BR-VL (G126S + S141F), were crossed to males of Wasatch. Last, to introduce the mutation I1017F associated with resistance to mite growth inhibitors, virgin females of Wasatch were crossed with EtoxR males (Table 1).
For the nuclear encoded mutations, the final cross between heterozygous backcrossed females and their sons resulted in congenic homozygous lines with either the mutation fixed or absent (Fig. 1, Table 1, see Backcrossing experiments paragraph for outline of experimental setup). Since mutations in GluCl1 and GluCl3 are not genetically linked35, the impact of each mutation could be assessed separately. Once homozygous backcrossed lines carrying a mutation either in GluCl1 (GluCl1_R1-R3) or in GluCl3 (GluCl3_R1-R3) and their respective congenic control lines (GluCl1_C and GluCl3_C) were generated, the mutations were joined again by dedicated crosses, giving rise to GluCl1 + 3_R1-R3. The susceptible control GluCl1 + 3_C was obtained with the cross GluCl1_C x GluCl3_C. One replicate with genotype A1215D + F1538I (pyrethroid resistance mutations) and one with genotype G126S + S141F (bifenazate resistance mutations) were lost during backcrossing and only two biological replicates VGSC_R4, R5 and cytb_R4 and R5 could be analyzed for each genotype.
Toxicity assays
Parental strains
Abamectin and milbemectin were tested against the parental susceptible strain, Wasatch and the resistant strain, MAR-AB (G314D + G326E), with the latter one exhibiting high resistance levels to abamectin (1,354.9 fold) and moderate resistance to milbemectin (71.7 fold) in comparison to Wasatch (Supplementary Table S2).
The parental susceptible strains, KOP8, which carries only the A1215D VGSC substitution, and Wasatch showed high susceptibility to bifenthrin, fluvalinate and fenpropathrin whereas the GH (L1024V) and TuSB9 (A1215D + F1538I) resistant strains were highly resistant to the aforementioned pyrethroids (Table 2).
The Wasatch strain and the parental resistant strains HOL3 (P262T) and BR-VL (G126S + S141F) were tested against bifenazate. The resistant strains exceeded 2,000 fold of resistance to bifenazate. Additionally, the parental susceptible strain and BR-VL were treated with acequinocyl, with the latter one showing moderate levels of resistance (RR of 28.9 fold) (Fig. 2, Supplementary Table S3).

Susceptibility of backcrossed T. urticae lines cytb_R1-R3 (P262T) and cytb_R4, R5 (G126S + S141F) and the resistant parental strains HOL3 and BR-VL to QoI acaricides bifenazate and acequinocyl. The RRs were calculated as the LC50 values of the backcrossed lines divided by the LC50 of the parental susceptible strain Wasatch. Stars indicate strains for which the LC50 value exceeded 5,000 mg l−1. Error bars represent 95% confidence limits calculated by probit analysis. Letters above bars indicate lines where acaricide treatment had statistically the same (a) or different (b) effect comparing to Wasatch (PoloPlus LeOra Software)49.
Etoxazole, clofentezine and hexythiazox were tested against the EtoxR strain (I1017F) and the susceptible strain Wasatch. The EtoxR strain showed extremely high levels of insensitivity to all three compounds used, with RR values exceeding 40,000 for etoxazole, 4,000 for hexythiazox and 2,000 for clofentezine (Supplementary Table S4, Fig. 3).

Susceptibility of backcrossed T. urticae lines CHS1_R1-R3 (I1017F) and CHS1_C and their susceptible and resistant parental strains, to mite growth inhibitors etoxazole (most left), hexythiazox (middle) and clofentezine (most right). Bars represent the acaricide concentration at which 50% of the individuals are affected. Error bars represent the 95% confidence limit calculated by probit analysis. As LC50 values exceeded 5,000 mg l−1 for all CHS1_R1, R2 and R3 for each mite growth inhibitor tested, only one bar depicts LC50s. Stars indicate lines for which, LC50 value exceeded 5,000 mg l−1. Letters above bars indicate lines where acaricide treatment had statistically the same (a) or different (b) effect comparing to Wasatch (PoloPlus LeOra Software)49.
Backcrossed strains
Abamectin and Milbemectin. The introgressed strains carrying resistance mutation in only one of the GluCls (either GluCl1 or GluCl3) showed minor resistance to abamectin and milbemectin with RR values up to 3.3 and 1.6 folds, respectively (Fig. 4, Supplementary Table S2). However, when mutations were joined by dedicated crosses, individuals carrying both mutations (GluCl1 + 3_R1-3 congenic lines) showed higher resistance levels to both compounds. The RR values obtained for abamectin and milbemectin were up to 19.8 and 13.7 fold, respectively (Fig. 4, Supplementary Table S2).

Susceptibility levels of backcrossed T. urticae lines GluCl1_R1-R3 (G314D), GluCl1_C, GluCl3_R1-R3 (G326E), GluCl3_C, GluCl1 + 3_R1-R3 (G314D + G326E), GluCl1 + 3_C to abamectin and milbemectin. The RRs were calculated as the LC50 values of the backcrossed lines divided by the LC50 of the parental susceptible strain Wasatch. Error bars represent the 95% confidence limit calculated by probit analysis. Letters above bars indicate lines where acaricide treatment had statistically the same (a) or different (b) effect comparing to Wasatch (PoloPlus LeOra Software)49.
Pyrethroids. The backcrossed strains VGSC_R1-3 and VGSC_R4,5 exhibited high levels of resistance to all pyrethroids used in this study (bifenthrin, fluvalinate and fenpropathrin), with RR values being greater than 200 fold in some cases. In contrast, the backcrossed susceptible lines VGSC_C1 and VGSC_C2 were susceptible to all three compounds (Table 2).
Mitochondrial QoI. The backcrossed strains cytb_R1, R2 and R3 carrying the P262T mutation in cytb sequence showed high resistance levels to bifenazate (Fig. 2, Supplementary Table S3). Interestingly, the combination of cytb substitutions; G126S and S141F provided higher level of resistance to bifenazate compared to P262T, since RRs for both cytb_R4 and R5 were higher than 2,000 fold. The importance of G126S and S141F in the observed levels of cross-resistance to acequinocyl in BR-VL was confirmed (Fig. 2, Supplementary Table S3).
Mite growth inhibitors. The backcrossed strains homozygous for I1017F mutation displayed extreme levels of resistance to all three mite growth inhibitors tested (Fig. 3, Supplementary Table S4). RRs estimated for etoxazole, hexythiazox and clofentezine exceeded 40,000, 4,000 and 2,000 fold, respectively (Fig. 3, Supplementary Table S4). In contrast, the backcrossed control line was highly susceptible to the aforementioned compounds (Fig. 3, Supplementary Table S4).
Discussion
Field collected T. urticae strains often exhibit very high levels of resistance to multiple acaricides used for their control. Due to the identification of acaricide target-site sequences29, 51 and implementation of recently developed genetic mapping tools4, 25, 38, a number of mutations has been uncovered in the target-site of frequently used acaricides. However, to what extent these mutations determine the resistant phenotype is mostly unknown. Resistant field strains investigated so far, typically display a broad altered transcriptional response with the putative involvement of many detoxifying enzymes and transporters that might affect acaricide toxicity52,53,54. Crossing experiments have revealed that a complex genetic make-up typically underlies resistance, implying the additive effect of multiple mechanisms35, 55, 56. Moreover, the extent by which resistant alleles confer resistance can also vary according to the genetic background in which they are expressed57, 58.
Several studies have used congenic backcrossed lines to assess insecticide related fitness cost/advantage and pleiotropic effects59,60,61,62,63,64. By substituting phenotypic selection with molecular marker-assisted backcrossing, the potential accumulation of alleles with additive effect can be uncoupled23. Such a setup has been previously used to assess the effects of Aedes aegypti kdr mutations on pyrethroid resistance and its fitness cost24 and recently, to investigate resistance levels to METI-I acaricides caused by a mutation in the PSST subunit of complex I in T. urticae 25.
Here, we analyzed the relative phenotypic contribution of target-site resistance mutations, previously uncovered in highly resistant T. urticae field populations. We adopted a marker-assisted backcrossing procedure described in Bajda, et al.25 to untangle the target-site resistance loci from potential complex additive genetic mechanisms. Although in this study we cannot exclude a possible effect of closely linked loci65, previous research involving resistance gene mapping by means of bulk segregant analysis, revealed a high recombination rate in T. urticae 38, 39 which makes us believe that the procedure performed here, resulted in near-isogenic lines.
Both abamectin and milbemectin resistance has been reported frequently in spider mite populations worldwide48, 66, 67 exhibiting >1000 fold resistance in some cases35. These molecules target both GluCls and GABA gated chloride channels (GABACl), although GluCls are considered the main target68, 69. In contrast to insects with a single copy, the genome of T. urticae harbors six orthologous GluCl genes35. Two non-synonymous mutations have been associated with resistance to abamectin, the G314D in GluCl1 and G326E in GluCl335, 36. When G314D and G326E were introgressed separately, only low levels of resistance remained. However, when both mutations were joined by dedicated crosses, resistance levels increased to 10–20 fold (henceforth, for the schematic visualization of a relative contribution of the mutations in resistance levels, please consult Fig. 5). These resistance levels are comparable with a previous study, where an abamectin resistant strain homozygous for both GluCl mutations was investigated. Resistance levels in that strain reached only 20-fold36, 70, suggesting that target-site mutations were the only factor contributing to resistance. A possible explanation for the relatively low resistance levels conferred by the combination of two GluCl mutations may lie in the number of genes involved in channel assembly. Glutamate-gated chloride channels typically consist of five subunits, which in T. urticae can be encoded by 5 different GluCl genes. Hence, if the channel consists of a combination of subunits carrying the resistance associated substitution (GluCl1 and/or GluCl3) and a GluCl2 subunit (GluCl2 does not carry a resistance associated substitution, while GluCl4 and GluCl5 naturally carry substitutions that interfere with abamectin binding see Dermauw, et al.35), abamectin binding might still be possible. In addition, we cannot exclude the possibility of heteromeric channel assembly, consisting of GluCls and GABACl18, 19. In such case, the existence of mutations in GluCl1 and GluCl3 alone would also not be capable to fully prevent channel blocking. Consequently, our results also reconfirm the importance of additional mechanisms in abamectin resistance69, 71, 72. Studies with synergists and biochemical tests have previously implied the involvement of detoxification enzymes in resistance in many field collected strains worldwide72,73,74. For instance, a P450 (CYP392A16) was reported to be overexpressed in abamectin resistant strains and detoxifies abamectin rapidly71. Therefore very high abamectin resistance levels in the MAR-AB strain (Supplementary Table S2) may be attributed to a joint action of P450 detoxification and decreased sensitivity of the target-site, potentially even acting synergistically.

Schematic representation of the relative contribution of target site resistance mutations in overall resistance levels to acaricides belonging to different mode of action groups. The size of the circle shape reflects the observed levels of resistance (RR vs susceptible parent strain). Only comparisons between the backcrossed lines versus its resistant parent are drawn to scale (Table 1).
Milbemectin belongs to the same insecticidal class as abamectin and acts on the same target-site. Whether cross-resistance may occur between both compounds is therefore of crucial importance, and still a matter of debate. Here, we show that the combination of both GluCl mutations confers resistance levels of about 10-fold, indicating potential cross-resistance risks between milbemectin and abamectin, as has been previously suggested48, 67.
Pyrethroid resistance has been documented globally in T. urticae with resistance levels exceeding 10,000 folds in some cases75, 76. Unlike most other arthropods, spider mites have mutations in unique positions on VGSC33, 34, 77, instead of the known kdr (L1014F) and super-kdr (M918T) mutations (Musca domestica numbering). The super-kdr mutation has been identified only once in a Tetranychus evansi strain78. Three point mutations have been located in the sodium channel of spider mites, L1024V and F1538I in combination with A1215D33, 34. Backcrossing experiments indicated the major effect of both L1024V and A1215D + F1538I mutations in pyrethroid resistance (Fig. 5). Interestingly, the KOP8 strain has the A1215D mutation uncoupled from F1538I and is susceptible to all pyrethroids, indicating that the mutation alone has no effect on pyrethroid toxicity. So far, the mutation F1538I has been studied most thoroughly and its effect in resistance to pyrethroids has been confirmed by electrophysiological studies79. Here, we showed that both L1024V and A1215D + F1538I mutations confer high resistance levels to all pyrethroid compounds, irrespectively of their type, i.e. presence of α-cyano group and/or extended halogenated acidic moiety, suggesting that the sodium channel mutations can cause field failure of the pyrethroids.
QoI with acaricidal properties have been introduced for the control of mite infestations relatively recently80, 81. Nonetheless, high levels of resistance to bifenazate have already been reported in the field37, 82. Backcrossing revealed that the combination of cd1 helix mutations G126S and S141F in cytb confers high levels of bifenazate resistance. In contrast, the backcrossed lines carrying the P262T substitution, showed LC50 values of 960, 1,400.6 and 886.1 mg l−1, respectively, while the parental resistant HOL3 was resistant to bifenazate concentrations up to 5,000 mg l−1 (Fig. 5). The backcrossed lines with cd1 helix mutations, showed similar levels of resistance to acequinocyl compared to the parental strain, confirming that acequinocyl cross-resistance is completely maternally inherited, and thus linked with the mutation82. One of the possible explanations for the discrepancy between bifenazate resistance of the parental strain HOL3 and those of the backcrossed strains is the presence of additional resistance mechanisms. Indeed, although a strong correlation between the P262T frequency and bifenazate resistance has been reported, to what extent resistance is inherited maternally has only been described for acequinocyl82. This is in contrast to cd1 helix mutations, that have been shown to confer resistance levels that are completely maternally inherited41, suggesting that no additional mechanisms are involved or needed to attain very high resistance levels. Another explanation could be that the P262T substitution only confers high resistance levels in combination with specific nuclear encoded protein variants that co-evolved with mitochondrial encoded cytb mutations, and that uncoupling results in a loss of phenotype.
Resistance to clofentezine and hexythiazox has been frequently reported, and more recently, resistance against etoxazole has been also spreading83,84,85. Insensitivity to etoxazole is thought to be monogenic and recessive84, 85, which is in line with target-site resistance as the main mechanism. Screening for I1017F revealed that the mutation has been segregating in populations from different regions of the world for a long period, although etoxazole has been only recently used to control spider mites, especially in Europe38. This lead to the hypothesis that the mutation was selected by other molecules such as hexythiazox or clofentezine, which was later confirmed in a follow-up genetic mapping study39, 86. Here, we provide clear evidence that the I1017F substitution confers very high levels of resistance to etoxazole, hexythiazox and clofentezine (Fig. 5). Our results confirm target-site based cross-resistance, despite the fact that the three mite growth inhibitors belong to chemically diverse classes87,88,89. In a recent study, Douris, et al.20 found a mutation (I1042M) in CHS1 gene of Plutella xylostella resistant to benzoylureas (BPUs), at the position corresponding to I1017F in T. urticae. Using CRISPR/Cas9 approach coupled with homology directed repair (HDR), both the lepidopteran and spider mite mutations (I1056M/F) were introduced in the Drosophila CHS1 gene (kkv). Flies carrying either of two mutations were found highly resistant to etoxazole, but also to a number of BPUs and the hemipteran chitin biosynthesis inhibitor buprofezin. The study, together with the results reported here, provide convincing evidence that chitin synthesis inhibitors BPUs, buprofezin and mite growth inhibitors, etoxazole, hexythiazox and clofentezin all directly interact with CHS1 and share a similar molecular MoA.
Conclusions
Resistance mechanisms in insects and mites can be complex and the relative strength of target-site mutations in resistance phenotypes is often not well known. Here, we have used a marker-assisted backcrossing approach to look at the phenotypic effect of the main and currently relevant target-site mutations reported to confer resistance to abamectin, pyrethroids, mite growth inhibitors and QoI. Mutations in VGSC, CHS and cytb confer high levels of resistance and their presence in populations alone is enough to cause field failure after acaricide treatment. In contrast, although we confirmed the functional importance of GluCl mutations and the cumulative effect of mutations in multiple channels, mutations in only two channels genes does not lead to the high resistance levels that have been reported for abamectin resistance. Overall, our results functionally validate the importance of mutations that have been inferred from correlation analysis and genetic mapping.
References
Sparks, T. C. & Nauen, R. IRAC: Mode of action classification and insecticide resistance management. Pestic Biochem Physiol. 121, 122–128 (2015).
Sparks, T. C. Insecticide discovery: An evaluation and analysis. Pestic Biochem Physiol. 107, 8–17 (2013).
Feyereisen, R., Dermauw, W. & Van Leeuwen, T. Genotype to phenotype, the molecular and physiological dimensions of resistance in arthropods. Pestic Biochem Physiol. 121, 61–77 (2015).
Van Leeuwen, T. & Dermauw, W. The molecular evolution of xenobiotic metabolism and resistance in chelicerate mites. Annu Rev Entomol. 61, 475–498 (2016).
Li, X. C., Schuler, M. A. & Berenbaum, M. R. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu Rev Entomol. 52, 231–253 (2007).
Raymond, M., Heckel, D. G. & Scott, J. G. Interactions between pesticide genes - model and experiment. Genetics. 123, 543–551 (1989).
Bohannan, B. J. M., Travisano, M. & Lenski, R. E. Epistatic interactions can lower the cost of resistance to multiple consumers. Evolution 53, 292–295 (1999).
Williams, T. N. et al. Negative epistasis between the malaria-protective effects of alpha(+)-thalassemia and the sickle cell trait. Nature Genet. 37, 1253–1257 (2005).
Zhang, Y. X. et al. Synergistic and compensatory effects of two point mutations conferring target-site resistance to fipronil in the insect GABA receptor RDL. Sci Rep. 6, doi:10.1038/srep32335 (2016).
Moore, J. H. & Williams, S. M. Traversing the conceptual divide between biological and statistical epistasis: Systems biology and a more modern synthesis. Bioessays 27, 637–646 (2005).
Hardstone, M. C. & Scott, J. G. A review of the interactions between multiple insecticide resistance loci. Pest Biochem Physiol. 97, 123–128 (2010).
Liu, N. N. & Pridgeon, J. W. Metabolic detoxication and the kdr mutation in pyrethroid resistant house flies, Musca domestica (L.). Pest Biochem Physiol. 73, 157–163 (2002).
Peyronnet, O., Pichon, Y., Carton, Y. & Delorme, R. Genetic-analysis of deltamethrin resistance in laboratory-selected strains of Drosophila melanogaster Meig. Pest Biochem Physiol. 50, 207–218 (1994).
McEnroe, W. D. & Naegele, J. A. The coadaptive process in an organophosphorus-resistant strain of the two-spotted spider mite. Tetranychus urticae. Ann Entomol Soc Am. 61, 1055–1059 (1968).
Shi, M. A. et al. Acetylcholinesterase alterations reveal the fitness cost of mutations conferring insecticide resistance. BMC Evol Biol. 4, doi:10.1186/1471-2148-4-5 (2004).
Georghiou, G. P. Genetics of resistance to insecticide in houseflies and mosquitoes. Exp. Parasitol. 26, 224–255 (1969).
Mckenzie, J. A., Whitten, M. J. & Adena, M. A. The effect of genetic background on the fitness of diazinon resistance genotypes of the Australian Sheep Blowfly. Lucilia cuprina. Heredity 49, 1–9 (1982).
Cully, D. F. et al. Cloning of an avermectin-sensitive glutamate-gated chloride channel from Caenorhabditis elegans. Nature 371, 707–711 (1994).
Ludmerer, S. W. et al. Ivermectin and nodulisporic acid receptors in Drosophila melanogaster contain both γ-aminobutyric acid-gated rdl and glutamate-gated GluClα chloride channel subunits. Biochemistry 41, 6548–6560 (2002).
Douris, V. et al. Resistance mutation conserved between insects and mites unravels the benzoylurea insecticide mode of action on chitin biosynthesis. PNAS. 113, 14692–14697 (2016).
Zimmer, C. T. et al. A CRISPR/Cas9 mediated point mutation in the alpha 6 subunit of the nicotinic acetylcholine receptor confers resistance to spinosad in Drosophila melanogaster. Insect Biochem Mol Biol. 73, 62–69 (2016).
McCart, C., Buckling, A. & ffrench-Constant, R. H. DDT resistance in flies carries no cost. Curr Biol. 15, R587–R589 (2005).
Roush, R. T. & Mckenzie, J. A. Ecological genetics of insecticide and acaricide resistance. Annu Rev Entomol. 32, 361–380 (1987).
Brito, L. P. et al. Assessing the effects of Aedes aegypti kdr mutations on pyrethroid resistance and its fitness cost. Plos One 8, doi:10.1371/journal.pone.0060878 (2013).
Bajda, S. et al. A mutation in the PSST homologue of complex I (NADH:ubiquinone oxidoreductase) from Tetranychus urticae is associated with resistance to METI acaricides. Insect Biochem Molec. 80, 79–90 (2017).
Jeppson, L. R., Keifer, H. H. & Baker, E. W. Mites injurious to economic plants. (University of California Press, 1975).
Migeon, A. & Dorkeld, F. Spider Mites Web: a comprehensive database for the Tetranychidae, http://www.montpellier.inra.fr/CBGP/spmweb (2006–2015).
Van Leeuwen, T., Vontas, J., Tsagkarakou, A. & Tirry, L. In Biorational Control of Arthropod Pests: Application and Resistance Management (eds Isaac Ishaaya & A. Rami Horowitz) 347–393 (Springer Netherlands, 2009).
Van Leeuwen, T., Dermauw, W., Grbic, M., Tirry, L. & Feyereisen, R. Spider mite control and resistance management: does a genome help? Pest Manag Sci 69, 156–159 (2013).
Kramer, T. & Nauen, R. Monitoring of spirodiclofen susceptibility in field populations of European red mites, Panonychus ulmi (Koch) (Acari: Tetranychidae), and the cross-resistance pattern of a laboratory-selected strain. Pest Manag Sci 67, 1285–1293 (2011).
Khalighi, M., Tirry, L. & Van Leeuwen, T. Cross-resistance risk of the novel complex II inhibitors cyenopyrafen and cyflumetofen in resistant strains of the two-spotted spider mite Tetranychus urticae. Pest Manag Sci 70, 365–368 (2014).
Khajehali, J. et al. Acetylcholinesterase point mutations in European strains of Tetranychus urticae (Acari: Tetranychidae) resistant to organophosphates. Pest Manag Sci 66, 220–228 (2010).
Tsagkarakou, A. et al. Identification of pyrethroid resistance associated mutations in the para sodium channel of the two-spotted spider mite Tetranychus urticae (Acari: Tetranychidae). Insect Mol Biol. 18, 583–593 (2009).
Kwon, D. H., Clark, J. M. & Lee, S. H. Cloning of a sodium channel gene and identification of mutations putatively associated with fenpropathrin resistance in Tetranychus urticae. Pest Biochem Physiol. 97, 93–100 (2010).
Dermauw, W. et al. The cys-loop ligand-gated ion channel gene family of Tetranychus urticae: Implications for acaricide toxicology and a novel mutation associated with abamectin resistance. Insect Biochem Molec. 42, 455–465 (2012).
Kwon, D. H., Yoon, K. S., Clark, J. M. & Lee, S. H. A point mutation in a glutamate-gated chloride channel confers abamectin resistance in the two-spotted spider mite, Tetranychus urticae Koch. Insect Mol Biol 19, 583–591 (2010).
Van Leeuwen, T. et al. Mitochondrial heteroplasmy and the evolution of insecticide resistance: Non-Mendelian inheritance in action. PNAS. 105, 5980–5985 (2008).
Van Leeuwen, T. et al. Population bulk segregant mapping uncovers resistance mutations and the mode of action of a chitin synthesis inhibitor in arthropods. PNAS. 109, 4407–4412 (2012).
Demaeght, P. et al. High resolution genetic mapping uncovers chitin synthase-1 as the target-site of the structurally diverse mite growth inhibitors clofentezine, hexythiazox and etoxazole in Tetranychus urticae. Insect Biochem Mol Biol. 51, 52–61 (2014).
Chatzivasileiadis, E. A. & Sabelis, M. W. Toxicity of methyl ketones from tomato trichomes to Tetranychus urticae Koch. Exp Appl Acarol 21, 473–484 (1997).
Van Leeuwen, T., Tirry, L. & Nauen, R. Complete maternal inheritance of bifenazate resistance in Tetranychus urticae Koch (Acari: Tetranychidae) and its implications in mode of action considerations. Insect Biochem Mol Biol. 36, 869–877 (2006).
Navajas, M., Lagnel, J., Fauvel, G. & De Moraes, G. Sequence variation of ribosomal internal transcribed spacers (ITS) in commercially important phytoseiidae mites. Exp Appl Acarol 23, 851–859 (1999).
Ilias, A., Vassiliou, V. A., Vontas, J. & Tsagkarakou, A. Molecular diagnostics for detecting pyrethroid and abamectin resistance mutations in Tetranychus urticae. Pest Biochem Physiol. 135, 9–14 (2017).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 41, 95–98 (1999).
Van Pottelberge, S., Khajehali, J., Van Leeuwen, T. & Tirry, L. Effects of spirodiclofen on reproduction in a susceptible and resistant strain of Tetranychus urticae (Acari: Tetranychidae). Exp Appl Acarol 47, 301–309 (2009).
Van Leeuwen, T., Stillatus, V. & Tirry, L. Genetic analysis and cross-resistance spectrum of a laboratory-selected chlorfenapyr resistant strain of two-spotted spider mite (Acari: Tetranychidae). Exp Appl Acarol 32, 249 (2004).
Van Leeuwen, T., Van Pottelberge, S., Nauen, R. & Tirry, L. Organophosphate insecticides and acaricides antagonise bifenazate toxicity through esterase inhibition in Tetranychus urticae. Pest Manag Sci 63, 1172–1177 (2007).
Sato, M. E., Da Silva, M. Z., Raga, A. & De Souza, M. F. Abamectin resistance in Tetranychus urticae Koch (Acari: Tetranychidae): Selection, cross-resistance and stability of resistance. Neotrop Entomol. 34, 991–998 (2005).
Robertson, J. L. P. H. K. in Pesticide bioassays with arthropods 17–34 (1992).
Robertson, J. L., Russel, R. M., Preisler, H. K. & Savin, N. E. Bioassays in arthropods, 2nd Eds, (Taylor and Francis group LLC, CRC press, 2007).
Grbic, M. et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature 479, 487–492 (2011).
Dermauw, W. et al. A link between host plant adaptation and pesticide resistance in the polyphagous spider mite Tetranychus urticae. PNAS. 110, E113–E122 (2013).
Khalighi, M. et al. Molecular analysis of cyenopyrafen resistance in the two-spotted spider mite Tetranychus urticae. Pest Manag Sci 72, 103–112 (2016).
Demaeght, P. et al. Molecular analysis of resistance to acaricidal spirocyclic tetronic acids in Tetranychus urticae: CYP392E10 metabolizes spirodiclofen, but not its corresponding enol. Insect Biochem Molec. 43, 544–554 (2013).
Van Pottelberge, S., Van Leeuwen, T., Nauen, R. & Tirry, L. Resistance mechanisms to mitochondrial electron transport inhibitors in a field-collected strain of Tetranychus urticae Koch (Acari: Tetranychidae). Bull Entomol Res. 99, 23–31 (2009).
Van Pottelberge, S., Van Leeuwen, T., Khajehali, J. & Tirry, L. Genetic and biochemical analysis of a laboratory-selected spirodiclofen-resistant strain of Tetranychus urticae Koch (Acari: Tetranychidae). Pest Manag Sci 65, 358–366 (2009).
McKenzie, J. A., Whitten, M. J. & Adena, M. A. The effect of genetic background on the fitness of diazinon resistance genotypes of the Australian sheep blowfly, Lucilia cuprina. Heredity 49, 1–9 (1982).
Schrag, S. J., Perrot, V. & Levin, B. R. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc R Soc Lond [Biol]. 264, 1287–1291 (1997).
Wang, R. & Wu, Y. Dominant fitness costs of abamectin resistance in Plutella xylostella. Pest Manag Sci 70, 1872–1876 (2014).
Yuan, G. et al. Development of near-isogenic lines in a parthenogenetically reproduced thrips species, Frankliniella occidentalis. Front Physiol. 8, doi:10.3389/fphys.2017.00130 (2017).
Xiao, Y. et al. A single point mutation resulting in cadherin mislocalization underpins resistance against Bacillus thuringiensis toxin in cotton bollworm. J Biol Chem. 292, 2933–2943 (2017).
Helle, W. Genetics of resistance to organophosphorus compounds and its relation to diapause in Tetranychus urticae Koch (Acari). (H. Veenman, 1962).
Arnaud, L., Brostaux, Y., Assie, L. K., Gaspar, C. & Haubruge, E. Increased fecundity of malathion-specific resistant beetles in absence of insecticide pressure. Heredity 89, 425–429 (2002).
ffrench-Constant, R. H. & Bass, C. Does resistance really carry a fitness cost? Curr Opin Insect Sci. 21, 39–46 (2017).
Hospital, F. Size of donor chromosome segments around introgressed loci and reduction of linkage drag in marker-assisted backcross programs. Genetics 158, 1363–1379 (2001).
Yorulmaz, S. & Ay, R. Multiple resistance, detoxifying enzyme activity, and inheritance of abamectin resistance in Tetranychus urticae Koch (Acarina: Tetranychidae). Turk J Agric For. 33, 393–402 (2009).
Nicastro, R. L., Sato, M. E. & Da Silva, M. Z. Milbemectin resistance in Tetranychus urticae (Acari: Tetranychidae): selection, stability and cross-resistance to abamectin. Exp Appl Acarol 50, 231–241 (2010).
Wolstenholme, A. J. Recent progress in understanding the interaction between avermectins and ligand-gated ion channels: putting the pests to sleep. Invertebr Neurosci 10, 5–10 (2010).
Clark, J. M., Scott, J. G. & Campos, F. Resistance to avermectins: extent, mechanisms, and management Implications. Annu Rev Entomol. 40, 1–30 (1994).
Kwon, D. H., Kang, T. J., Kim, Y. H. & Lee, S. H. Phenotypic- and genotypic-resistance detection for adaptive resistance management in Tetranychus urticae Koch. Plos One 10, doi:10.1371/journal.pone.0139934 (2015).
Riga, M. et al. Abamectin is metabolized by CYP392A16, a cytochrome P450 associated with high levels of acaricide resistance in Tetranychus urticae. Insect Biochem Molec. 46, 43–53 (2014).
Pavlidi, N. et al. Functional characterization of glutathione S-transferases associated with insecticide resistance in Tetranychus urticae. Pestic Biochem Phys 121, 53–60, doi:10.1016/j.pestbp.2015.01.009 (2015).
Campos, F., Krupa, D. A. & Dybas, R. A. Susceptibility of populations of twospotted spider mites (Acari: Tetranychidae) from Florida, Holland, and the Canary Islands to abamectin and characterization of abamectin resistance. J Econ Entomol. 89, 594–601 (1996).
Stumpf, N. & Nauen, R. Biochemical markers linked to abamectin resistance in Tetranychus urticae (Acari: Tetranychidae). Pestic Biochem Phys 72, 111–121 (2002).
Herron, G. A., Rophail, J. & Wilson, L. J. The development of bifenthrin resistance in two-spotted spider mite (Acari: Tetranychidae) from Australian cotton. Exp Appl Acarol 25, 301–310 (2001).
Van Leeuwen, T., Van Pottelberge, S. & Tirry, L. Comparative acaricide susceptibility and detoxifying enzyme activities in field-collected resistant and susceptible strains of Tetranychus urticae. Pest Manag Sci 61, 499–507 (2005).
Ding, T. B. et al. Molecular characterisation of a sodium channel gene and identification of a Phe1538 to Ile mutation in citrus red mite. Panonychus citri. Pest Manag Sci 71, 266–277 (2015).
Nyoni, B. N. et al. Pyrethroid resistance in the tomato red spider mite, Tetranychus evansi, is associated with mutation of the para-type sodium channel. Pest Manag Sci 67, 891–897 (2011).
Tan, J. G. et al. Identification of amino acid residues in the insect sodium channel critical for pyrethroid binding. Mol Pharmacol 67, 513–522 (2005).
Dekeyser, M. A. in Proceedings of the BCP Conference, Pests and Diseases. 487–492.
Grosscurt, A. & Avella, L. in Proceedings of the BCP Conference, Crop Science and Technology. 49–56.
Van Nieuwenhuyse, P., Van Leeuwen, T., Khajehali, J., Vanholme, B. & Tirry, L. Mutations in the mitochondrial cytochrome b of Tetranychus urticae Koch (Acari: Tetranychidae) confer cross-resistance between bifenazate and acequinocyl. Pest Manag Sci 65, 404–412 (2009).
Herron, G., Edge, V. & Rophail, J. Clofentezine and hexythiazox resistance in Tetranychus urticae Koch in Australia. Exp Appl Acarol 17, 433–440 (1993).
Uesugi, R., Goka, K. & Osakabe, M. H. Genetic basis of resistances to chlorfenapyr and etoxazole in the two-spotted spider mite (Acari: Tetranychidae). J Econ Entomol. 95, 1267–1274 (2002).
Kobayashi, M., Kobayashi, S. & Nishimori, T. Occurrence of etoxazole resistance individuals of the two-spotted spider mite, Tetranychus urticae Koch from a limited region. Jpn J Appl Entomol Z. 45, 83–88 (2001).
Asahara, M., Uesugi, R. & Osakabe, M. Linkage between one of the polygenic hexythiazox resistance genes and an etoxazole resistance gene in the twospotted spider mite (Acari: Tetranychidae). J. Econ Entomol. 101, 1704–1710 (2008).
Ishida, T., Suzuki, J., Tsukidate, Y. & Mori, Y. in BCP Crop protection conference pests and pests diseases Vol. 1 37–44 (Brighton, UK, 1994).
Yamada, T., Kaeriyama, M., Matsui, N. & Yoneda, H. Development of a new miticide, hexythiazox. J Pestic Sci. 12, 327–335 (1987).
Aveyard, C. S., Peregrine, D. J. & Bryan, K. M. G. Biological activity of clofentezine against egg and motile stages of tetranychid mites. Exp Appl Acarol 2, 223–229 (1986).
Acknowledgements
This project was supported by the Research Foundation Flanders (FWO) (grant G009312N to L.T. and T.V.L. and grant G053815N to L.T., T.V.L. and W.D.). W.D is a postdoctoral fellow of the Research Foundation Flanders. Part of this research has been co-financed by Greek national funds through the Operational Program “Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF) Research Funding Program: THALES (projects 377301 and 380264). Authors thank Nicky Wybouw for help in performing toxicity test and manuscript proofreading and Naciye Sena Cagatay for her contribution in toxicity assays. Authors also thank Prof dr. Richard M. Clark for kindly providing T. urticae strains Wasatch and GH and dr. Juan M. Alba Cano for providing the KOP8 strain.
Author information
Authors and Affiliations
Contributions
T.V.L., M.R., S.B. designed research. M.R. and S.B. performed the experiments with contribution of S.P., C.T. and M.P. M.R. and S.B. analyzed data. S.B., M.R. and T.V.L. wrote the manuscript, with input from W.D. and J.V. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Riga, M., Bajda, S., Themistokleous, C. et al. The relative contribution of target-site mutations in complex acaricide resistant phenotypes as assessed by marker assisted backcrossing in Tetranychus urticae . Sci Rep 7, 9202 (2017). https://doi.org/10.1038/s41598-017-09054-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09054-y
This article is cited by
-
A glutamate-gated chloride channel as the mite-specific target-site of dicofol and other diphenylcarbinol acaricides
Communications Biology (2023)
-
Development of abamectin resistance in Tetranychus urticae in Australian cotton and the establishment of discriminating doses for T. lambi
Experimental and Applied Acarology (2021)
-
The G126S substitution in mitochondrially encoded cytochrome b does not confer bifenazate resistance in the spider mite Tetranychus urticae
Experimental and Applied Acarology (2021)
-
Mechanisms and management of acaricide resistance for Tetranychus urticae in agroecosystems
Journal of Pest Science (2021)
-
Resistance risk assessment of the novel complex II inhibitor pyflubumide in the polyphagous pest Tetranychus urticae
Journal of Pest Science (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
