
Abstract
Pathogenic Vibrio spp., Aeromonas spp. and Plesiomonas shigelloides are associated with human gastroenteritis and wound infections, as well as fish diseases. The comprehensive and accurate identification of these pathogens is crucial for the current public health. The present study describes the development of a multiplex assay for the simultaneous identification of ten bacterial pathogens in a single reaction by using a multiplex ligation reaction based on probe melting curve analysis (MLMA). The specificity for target genes was 100%, as assessed with a panel of 67 bacterial pathogens, which indicated no cross-reactions. The detection limit of this assay ranged from 0.8 × 107 CFU/mL to 1.5 × 108 CFU/mL at the pure bacterial culture level and from 0.1 ng to 1.0 ng at the DNA level. The MLMA assay was used to detect ten species of pathogens in 269 clinical and seafood samples, and for further validation, the results were compared with the conventional culture method. The results indicated greater than 90% sensitivity and 100% specificity for each bacterial pathogen tested, and the kappa correlation for all the pathogens ranged from 0.95 to 1.00. Overall, this assay is well suited for public health laboratories for its high throughput, accuracy, and low cost.
Similar content being viewed by others


Introduction
Vibrio spp. are gram-negative bacteria that usually appear as motile rods and are abundant in estuarine and marine environments; there are approximately 72 species of vibrios (www.bacterio.cict.fr)1,2,3 that require sodium chloride for survival. Among the numerous Vibrio spp., V. vulnificus, V. parahaemolyticus, V. mimicus, V. fluvialis, V. alginolyticus, and V. cholerae are routinely isolated from human, clinical, and environmental samples; these pathogens are primarily involved in causing gastrointestinal illnesses, septicemia, wound infections and, in certain severe cases, death1, 4,5,6. Moreover, vibriosis, caused by a group of bacteria in the genus Vibrio, has resulted in considerable economic losses in marine aquaculture7. V. parahaemolyticus and V. vulnificus are the two most common causes of seafood-associated illnesses and deaths due to the consumption of raw or undercooked shellfish or other seafood8. V. cholerae is the causative agent of the disease cholera and is generally acknowledged to be an agent used in bioterrorism9. To date, only the strains of serogroups O1 and O139 have been identified as being responsible for human epidemic and pandemic cholera10; non-O1 and non-O139 V. cholerae strains are associated with a relatively mild gastroenteritis, thus occasionally resulting in a cholera-like illness in some serotypes11. It is crucial to know whether V. cholerae isolates carry the toxin genes to determine whether an isolate could cause pandemic cholera. V. mimicus, V. fluvialis, and V. alginolyticus have all been isolated in cases of diarrhea and wound infection in humans and diseases in fish6, 12, 13. Plesiomonas shigelloides has been implicated as an agent of human gastroenteritis and was formerly included in the family Vibrionaceae 14; however, most microbiologists currently agree that P. shigelloides is better classified as a member of the family Enterobacteriaceae. Aeromonas, a member of the Aeromonadaceae family, is ubiquitous in aquatic environments and can cause gastroenteritis as well as soft tissue infection15, 16, the diseases and illnesses caused by Vibrio spp. in humans have made the accurate identification of these organisms critical to the maintenance of public health.
To diagnose the above pathogens, the conventional method requires bacterial culture in alkaline peptone water, biochemical tests, and slide agglutination assays with specific antisera. These methods are often time consuming (2 to 7 days) and laborious. Furthermore, the variability in the biochemical characteristics of the conventional method may lead to a more complex interpretation of the results. Moreover, the accuracy level of six commercially available systems for identifying members of the family Vibrionaceae cannot ensure that the results obtained in every trial will be accurate (the accuracy rates range from 63.9% to 80.9%)17. Currently, molecular diagnostic methods, particularly multiplex nucleic acid-based assays, have been the most effective approaches, owing to their high specificity, sensitivity, and detection. Current molecular methods include the multiplex Polymerase Chain reaction (PCR) and multiplex real-time PCR assays, which can simultaneously detect two to seven targets in one closed tube4, 18, 19, and the Luminex assay can simultaneously detect six bacterial pathogens and seven enteric viruses20, 21. Jie Liu et al. have reported a TaqMan Array Card system that can detect 19 enteropathogens, including viruses, bacteria, and parasites, in one reaction20. Furthermore, multiplex ligase-dependent probe amplification (MLPA) has achieved a much higher throughput of up to 40 targets detected simultaneously22. However, there are drawbacks to the abovementioned methods, including their cost and vulnerability to contamination, and their need for specialized training that may not be available to all laboratories. Furthermore, to date, no assay system is able to identify all the bacterial pathogens in one reaction. In this regard, the development of an efficient, rapid, and accurate molecular method that can identify bacterial pathogens in one assay would be a useful tool for clinical laboratories and for the surveillance and diagnosis of human infections.
To increase the number of target genes and to improve the accuracy of detecting co-infection with two or more bacterial pathogens, we developed a multiplex ligation reaction based on a probe melting curve analysis (MLMA) assay for simultaneous identification of ten species of bacteria. After the multiplex ligation reaction, a linear-after-the-exponential (LATE)-PCR was carried out with a real-time PCR instrument, by using the fluorophore and melting temperature (Tm) value as dual labels, and a melting curve analysis (MCA) yielded a unique Tm code by which a certain pathogenic bacterium could be identified23. In this study, the analytical and clinical performances of the assay, compared with the conventional method, were determined by the evaluation of 67 reference isolates and 269 clinical and seafood samples. Early detection can help physicians correctly identify and treat patients affected by these bacterial pathogens and aid epidemiologists in tracking the source of the pathogens as quickly as possible, thereby limiting the economic loss associated with infected seafood.
Results
Genes Detected by the MLMA Assay
The MLMA assay is schematically illustrated in Fig. 1. During the detection of the ten bacterial pathogens, three probes that were fluorescently labeled with carboxyfluorescein (FAM), carboxy-X-rhodamine (ROX), and indodicarbocyanine-5 (Cy5) and 13 Tm tags (Supplementary Table S5) were used to hybridize the target DNA sequences to generate unique Tm values. In the FAM channel, Aeromonas, Plesiomonas shigelloides, the virulence gene thermostable direct hemolysin (tdh), and an internal control (IC) gene were detected. In the ROX channel, V. cholera, V. fluvialis, V. vulnificus, V. parahaemolyticus, V. mimicus, and V. alginolyticus were detected. In the CY5 channel, O1 V. cholera, O139 V. cholera, and the virulence gene CTX were detected. Thirteen specific genes (including an SUC2 gene) were developed for identification of the pathogenic vibrios, particularly V. cholera and V. parahaemolyticus. The MLMA assay not only simultaneously distinguished the O1 and O139 strains from non-O1 and non-O139 strains but also was capable of determining whether the detected bacteria had the toxin gene.

MLMA assay for the identification of ten bacterial pathogens in a single reaction. During the ligation process, 13 pairs of ligation oligonucleotides were hybridized onto the target genes of ten bacterial pathogens and one internal control (IC). In the LATE-PCR/MCA step, the ligated products were amplified by one pair of universal primers in the presence of three differently labeled fluorogenic probes; after PCR amplification, the unique Tm value was obtained via the MCA. FAM: IAC, P. shigelloides, Aeromonas, V. parahaemolyticus (tdh); ROX: V. cholerae, V. fluvialis, V. vulnificus, V. parahaemolyticus, V. mimicus, V. alginolyticus; Cy5: O1 V. cholerae, O139 V. cholerae, V. cholerae (CTX).
Evaluation of the Analytical Performance of the MLMA Assay
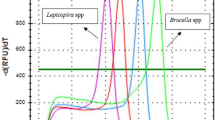
The newly developed MLMA assay exhibited high specificity (Fig. 2). No cross-reaction was observed when the reference strains were tested (Supplementary Table S1). The assay was able to detect all the target genes and yielded the expected Tm values (Supplementary Table S5), thus indicating that the developed assay has significant specificity.

Detection results of the mixed infection simulation. V. vulnificus and V. mimicus were simultaneously detected and verified to co-exist in one sample in the same channel. Melting curves of artificial templates consisting of two vibrios with various percentage ratios (0:100, 1:99, 3:97, 5:95, 10:90, 20:80, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20, 90:10, 95:5, 97:3, 99:1 to 100:0) were tested. The overall template concentration was 108 copies per reaction. DEPC water was used because there were no template controls.
The reproducibility of this MLMA assay was evaluated by detecting two ten-fold serial dilutions (3 × 1010 and 3 × 108 copies/mL) in triplicate. The intra-assay and inter-assay standard deviations (SDs) and coefficient of variation (CV) values were calculated and are listed in Table 1. The largest SD value was no more than 1 °C, thus indicating that the assay was highly reproducible. The intra-assay CVs for each vibrio ranged from 0 to 2%, and the inter-assay CVs ranged from 0 to 1%. The limits of detection (LODs) were determined to be 0.1 ng to 1.0 ng at the DNA level and ranged from 0.8 × 107 CFU/mL to 1.5 × 108 CFU/mL at the bacterial level (Table 2).
Taking into account the co-infection in stool samples or seafood samples, two bacteria, either present simultaneously in the same fluorescence channel or present in two different fluorescence channels, were selected to simulate mixed infection by varying the relative ratios of the bacteria. The results (Fig. 3 and Supplementary S7) showed that the lowest level of one species of bacteria that could be detected was 3% in the presence of 97% of the other type of bacteria, thus indicating that the MLMA assay can detect mixed infections over a wide range.

Specificity of the MLMA assay in detecting reference strains from different sources.
Clinical Studies
A total of 269 clinical and seafood samples were tested with the MLMA assay, and the results were compared with those using the conventional method (Table 3). Additionally, four singleplex qPCR assays targeting the virulence gene for V. cholera and V. parahaemolyticus (Supplementary Table S4) were performed in parallel. All the samples that were positive for V. cholerae, V. fluvialis, V. vulnificus, V. parahaemolyticus, V. mimicus, V. alginolyticus, P. shigelloides, and Aeromonas according to the MLMA assay were also examined simultaneously through conventional methods, and the detection results for the virulence gene for V. cholera and V. parahaemolyticus were also consistent with the MLMA assay results. Among the 145 V. parahaemolyticus-positive samples, 91 tested positive for the tdh gene, and among the 62 V. cholera-positive samples, 41 tested positive for the CTX gene (Supplementary Table S8).
In addition, 54 samples were identified as being co-infected with V. parahaemolyticus and V. alginolyticus. However, with the conventional methods, only 48 samples were found to be positive for multiple pathogens. Most of the discrepancies (6/54) were MLMA-po-sitive/conventional-method-negative results, and the probable reason for the discrepant results (6/54) between the two methods was that the results that were MLMA positive but co-nventional method negative often had lower fluorescence intensities, because a lower bacterial concentration could not be isolated through the conventional method. The qPCR results appeared to support the MLMA results (Supplementary Table S6).
Overall, according to the detection results of the 269 clinical and seafood samples, the sensitivities of the MLMA assay versus the conventional method were 100% (V. cholerae), 93.3% (V. fluvialis), 100% (V. vulnificus), 95.8% (V. parahaemolyticus), 100% (V. mimicus), 100% (V. alginolyticus), 100% (P. shigelloides) and 100% (Aeromonas), and the specificities were 100%. The concordance between the MLMA assay and conventional method results for all the pathogens was >97%, and the kappa values ranged from 0.95 to 1.0 (Table 3).
Discussion
It has long been established that the pathogenic Vibrio spp., Aeromonas and Plesiomonas shigelloides not only are associated with human diarrhea, septicemia, and wound infections but also affect the seafood industry24,25,26. In developing countries, Vibrio infections occur more frequently, owing to the lack of safe drinking water and the consumption of raw seafood27. Therefore, the accurate identification of these high-risk organisms is essential.
We successfully developed an MLMA assay for the simultaneous identification of ten pathogenic bacteria in clinical samples and seafood in a single reaction tube. To our knowledge, the MLMA assay is the first to identify these bacterial pathogens in a single reaction, and thus this method has extensive application prospects. All the target genes were accurately identified, and no cross-reaction was observed, even in the presence of genetically related bacterial species. The LOD for each gene was 0.8 × 107 CFU/mL to 1.5 × 108 CFU/mL at the level of pure bacterial cultures and 0.1 ng to 1 ng at the genomic DNA level, and the reproducibility was 100% in all aspects. Moreover, this method accurately detected multiple pathogens in one sample over a broad range. The MLMA assay displayed greater than 90% sensitivity and 100% specificity for each pathogen, and the kappa correlation values for the detection of all the pathogens ranged from 0.95 to 1.00.
An increasing number of recently published studies have focused on the development of molecular methods to solve multiple detection in one reaction24, 28,29,30. Recently, MCA has been used to achieve a higher throughput assay for multiplex detection31, 32. The MLMA assay developed and described in this study combined MLPA, LATE-PCR, and MCA techniques to achieve multiplex nucleic acid detection. MLMA and MLPA differ in their respective detection methods. MLPA detects target genes through a single capillary electrophoresis, in which each probe gives rise to an amplification product of a unique size between 130 and 480 bp22, whereas MLMA uses MCA to detect the target DNA sequence with a real-time PCR instrument, thus yielding unique Tm values23. A universal primer amplifies all the ligated products after the ligation reaction. Then, three detection probes hybridize with the artificial sequences contained in the ligated product, thus generating a unique Tm value through MCA in the FAM, ROX, and Cy5 fluorogenic channels. The MLMA assay, by allowing these bacterial pathogens to be identified in a single reaction, has broad application prospects.
The MLMA assay has three advantages, as compared with the existing methods. First, a detection probe is used to hybridize all the target sequences in one channel. A real-time PCR instrument with four fluorescence channels can distinguish at least 24 target genes through MCA. Second, compared with currently available methods, such as the conventional method or a multiplex PCR assay, which may lead to ambiguous results, the results of the MLMA assay can be automatically and accurately obtained within 3 h. Third, the MLMA assay is extremely cost-effective; the cost of a fluorescence detection method primarily depends on the consumption of the fluorescence probe. For example, if 15 target genes require detection, three fluorescence probes can be used in the MLMA assay, whereas at least 18 fluorescence probes are required to complete the same task by using multiplex real-time PCR. Therefore, the MLMA assay is very cost-effective.
Another distinct advantage of our MLMA assay is that this method can detect a mixed infection in one sample. Many studies have reported the occurrence of mixed infections in clinical and seafood samples with a small proportions33, 34. Understanding the significance of a mixed infection may help direct the development of treatment strategies. However, the conventional method often depends on the expertise of the operator when single colonies are extracted from plates, and therefore, only the dominant pathogen may be identified, while the minor and other suspicious colonies are frequently overlooked. In this study, the reason of the discrepant results (6/54) between the two methods may have been that minor single colonies in portions of the samples were ignored or not cultured. In our previous study35, we have used the multicolor combinational probe coding (MCPC) strategy to develop a multiplex assay for the simultaneous detection of five enteric viruses. Although this approach can achieve rapid detection, it is still difficult to determine whether a result indicates a mixed infection if both the FAM and HEX fluorescence signals from dual labeling appear with a high Ct value. Other current methods, such as DNA sequencing or restriction fragment length polymorphism, also focus on the dominant pathogen of the sample. However, our MLMA assay was able to identify a pathogen within a mixed infection with an abundance as low as 3% in the presence of the other pathogen. The consistency between the results of this method and those of singleplex rt-PCR fully illustrates the ability of the MLMA assay to distinguish mixed infections.
Notably, there are several limitations of the MLMA assay that require improvement. First, this method cannot be used for direct detection because the low LOD requires the sample to be cultured overnight before detection; therefore, the MLMA assay is more suitable for identification than for rapid detection. Second, compared with real-time PCR detection, the MLMA assay involves more complicated procedures in the ligation step. Therefore, further studies should address combining the ligation reaction with PCR amplification and MCA. Third, an insufficient number of V. mimicus strains were used to conclude significant evaluations regarding sensitivity and specificity; therefore, more samples will be collected for further validation in the future.
In conclusion, the MLMA assay developed for the accurate identification of these ten bacterial pathogens differs from established molecular diagnostic methods in that it is high throughput, less costly, contamination-free, and easier to use. Furthermore, this strategy can be modified and extended to detect more pathogens associated with human diseases in clinical settings. This assay is a sensitive and specific tool for the simultaneous identification of the major bacterial pathogens causing gastroenteritis and wound infections in humans and diseases in fish.
Materials and Methods
Bacterial Strains and Genomic DNA Preparation
The present study used 67 bacterial isolates for the initial development and accuracy evaluation of the MLMA assay, including 51 reference strains from the American Type Culture Collection (ATCC) and the National Center for Medical Culture Collections of China (CMCC), and 16 positive clinical isolates (see Table S1 in the supplemental material). These isolates were obtained from Shenzhen Center for Disease Control and Prevention (Shenzhen CDC, Shenzhen, Guangdong Province, China) and Chinese Center for Disease Control and Prevention (China CDC, Changping, Beijing, China). Genomic DNA was isolate-d using a QIAamp DNA Mini Kit (QIA-GEN; Hilden, Germany), according to the manufacturer’s protocol. The isolated DNA was quantified using a Nanodrop spectrophotometer (N-anodrop Products, Wilmington, Delaware, USA). Prepared genomic DNA was stored at −2-0 °C for use.
Preparation of Clinical Isolate DNA
A total of 269 clinical specimens and seafood samples were collected from the Shenzhen Center for Disease Control and Prevention (Shenzhen CDC, Shenzhen, Guangdong Province, China). All the samples were first incubated with alkaline peptone water (APW [pH 8.6]; 3% NaCl) for 12 h at 37 °C. The previously described simple heating lysis method was used for DNA extraction from 1 mL of sample culture medium19. The DNA samples were analyzed using the MLMA assay, which is described in the subsequent section.
Procedures for Conducting the MLMA
In general, the MLMA assay system includes two steps: (1) Hybridization and ligation; and (2) PCR amplification and melt curve analysis. During the hybridization-ligation process, each probe consisted of two oligonucleotides that hybridized at adjacent sites of the target sequence in the DNA sample, and these oligonucleotides were then ligated with DNA ligase after hybridization. A universal PCR primer pair was designed to amplify the ligated product via LATE-PCR by using a standard real-time PCR instrument. During the detection process, an MCA was performed for each sample after the PCR amplification to detect the target DNA sequence, thus resulting in unique Tm values displayed by the hybrids formed between the Tm tags and their corresponding fluorescent probes in the respective fluorophore channels. Finally, the identification results for each pathogen were determined on the basis of the Tm23.
Step 1: Hybridization and Ligation
The hybridization-ligation reaction was performed with a T3 Thermocycler (Biometra, Germany) and 1.5 μL of ligation probe mix (1–4 fmol of each specific ligation oligonucleotide, including an IC, Supplementary Table S2). A 5-μl volume of the previously isolated genomic DNA was denatured at 98 °C for 5 min and allowed to reach 75 °C, and finally, a 3.5-μL reaction mixture containing 1 μL of DNA ligase buffer, 1 unit of Taq DNA ligase (New England Biolabs, Beijing, China), and 1.5 μL of diethylpyrocarbonate (DEPC) water was added to the reaction tube. The samples were then incubated at 60 °C for 80 min, 95 °C for 5 min, and finally cooled to 4 °C.
Step 2: PCR Amplification and MCA
The reactions for amplification and MCA were performed on a Bio-Rad CFX 96 real-time PCR system (Bio-Rad Inc., Hercules, CA). The reaction mixture (50 μL) contained 1 × PCR buffer, 3 mM MgCl2, 2 μL of deoxynucleoside triphosphate (2.5 mM), 1 unit of Taq polymerase, 0.015 μM limiting primer, 0.4 μM excess primer, 0.08 μM to 0.16 μM fluorogenic probes (Supplementary Table 3), and 5 μL of ligation product from step 1. The amplification procedure consisted of an initial denaturation at 95 °C for 3 min, followed by 40 cycles of 95 °C for 5 s, 56 °C for 15 s, and then 74 °C for 15 s. Then, the MCA began with denaturation for 2 min at 95 °C, hybridization for 2 min at 40 °C, and a stepwise temperature increase (1 °C per 5 s) from 40 °C to 85 °C. The levels of FAM, ROX, and Cy5 fluorescence were collected and recorded during the MCA procedure.
Evaluation of the Analytical Performance of the MLMA Assay
Assessment of the Specificity of the MLMA Assay
Evaluation of the analytical specificity was performed by assessing DNA lysates prepared from pure cultures of 67 reference strains obtained from different sources (Supplementary Table 2), according to the procedures of the MLMA assay and comparison of the results with traditional culture-based methods.
Measurement of the LOD and Reproducibility of the MLMA Assay
The LOD was determined by using both a series of 10-fold dilutions of the purified genomic DNA from 10 ng to 0.01 ng and lysates obtained from the reference strains starting from 1010 to 105 CFU/mL. A turbidity meter was used for estimation.
Two ten-fold serial dilutions (3 × 1010 and 3 × 108 copies/mL) were prepared to analyze reproducibility.
DNA extraction was conducted by using a QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany). Each concentration was detected in triplicate with the MLMA assay.
MLMA Assay Detection of a Mixed Infection
Both clinical samples and seafood samples often contain mixed infections. Therefore, to increase the relevance of the study results to real world situations, in the same fluorescence channel, V. mimicus and V. vulnificus, V. vulnificus and V. alginolyticus, V. parahaemolyticus and V. alginolyticus, V. fluvialis and V. vulnificus, as well as V. cholera and V. vulnificus were used as examples to simulate co-infection. In different fluorescence channels, V. cholera and P. shigelloides, as well as V. mimicus and Aeromonas, were used as examples to simulate co-infection. The MLMA assay was performed with various percentage ratios of the two species (100:0, 97:3, 95:5, 90:10, 80:20, 70:30, 60:40, 50:50, 40:60, 30:70, 20:80, 10:90, 5:95, 3:97, and 0:100), and DEPC water was used as a negative control.
Verification of Multiplex Results through Singleplex Real-Time PCR Reactions
To verify the multiplex results, four rt-PCR reactions were carried out to detect V. cholera, CTX, V. parahaemolyticus, and the tdh gene of V. parahaemolyticus (Supplementary Table S4) by using a CFX96 real-time system (Bio-Rad) in a 25-μl reaction mixture. The PCR components were 1 × PCR buffer, 3 mM MgCl2, 2 mM dNTP, 1 U/μL Taq polymerase, 10 pmol each of forward and reverse primers, 4 pmol of TaqMan probe, and 5 μL of DNA template under the following conditions: 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s, 55 °C for 30 s, and 72 °C for 20 s, and the fluorescence intensity in the FAM detection channel was recorded.
Clinical Specimens and Statistical Analysis
A double-blind test was performed for all samples to evaluate the performance of the MLMA assay compared with the conventional method. A total of 269 clinical specimens and seafood samples were enriched in parallel with pathogen-specific enrichment broth. Then, a loopful of bacteria from each enrichment culture was streaked onto appropriate agar plates, and a minimum of five typical colonies were selected and subjected to biochemical and serological tests for identification according to the Chinese National Food Safety Standard “Food microbiological examination” (GB4789-2010, China).
The results of the multiplex rt-PCR assay were compared with those of the traditional culture-based methods by using a 2 × 2 table to estimate indices of sensitivity and specificity. The kappa correlation was also calculated.
References
Tarr, C. L. et al. Identification of Vibrio isolates by a Multiplex PCR assay and rpoB sequence determination. Journal of Clinical Microbiology 45, 134–140 (2006).
Thompson, F. L. et al. Phylogeny and molecular identification of Vibrios on the basis of multilocus sequence analysis. Applied and Environmental Microbiology 71, 5107–5115 (2005).
Dickinson, G., Lim, K. Y. & Jiang, S. C. Quantitative microbial risk assessment of pathogenic Vibrios in marine recreational waters of southern California. Applied and Environmental Microbiology 79, 294–302 (2012).
Singh, D. V., Isac, S. R. & Colwell, R. R. Development of a hexaplex PCR assay for rapid detection of virulence and regulatory genes in Vibrio cholerae and Vibrio mimicus. Journal of Clinical Microbiology 40, 4321–4324 (2002).
Huehn, S. et al. Pathogenic vibrios in environmental, seafood and clinical sources in Germany. International Journal of Medical Microbiology 304, 843–850 (2014).
Austin, B. Vibrios as causal agents of zoonoses. Veterinary Microbiology 140, 310–317 (2010).
Gonzalez, S. F., Krug, M. J., Nielsen, M. E., Santos, Y. & Call, D. R. Simultaneous detection of marine fish pathogens by using Multiplex PCR and a DNA microarray. Journal of Clinical Microbiology 42, 1414–1419 (2004).
Johnson, C. N. et al. Relationships between environmental factors and pathogenic Vibrios in the northern Gulf of Mexico. Applied and Environmental Microbiology 76, 7076–7084 (2010).
Paauw, A. et al. OmpU as a biomarker for rapid discrimination between toxigenic and epidemic Vibrio cholerae O1/O139 and non-epidemic Vibrio cholerae in a modified MALDI-TOF MS assay. BMC Microbiol 14, 158 (2014).
Khuntia, H. K., Pal, B. B. & Chhotray, G. P. Quadruplex PCR for simultaneous detection of serotype, biotype, toxigenic potential, and central regulating factor of Vibrio cholerae. Journal of Clinical Microbiology 46, 2399–2401, doi:10.1128/jcm.00024-08 (2008).
Dutta, D. et al. Vibrio cholerae Non-O1, Non-O139 serogroups and cholera-like diarrhea, Kolkata, India. Emerging Infectious Diseases 19, 464–467 (2013).
Chowdhury, M. A., Aziz, K. M., Kay, B. A. & Rahim, Z. Toxin production by Vibrio mimicus strains isolated from human and environmental sources in Bangladesh. J Clin Microbiol 25, 2200–2203 (1987).
Chowdhury, G. et al. Vibrio fluvialisin patients with diarrhea, Kolkata, India. Emerging Infectious Diseases 18, 1868–1871 (2012).
Gonzalez-Rey, C. et al. Specific detection of Plesiomonas shigelloides isolated from aquatic environments, animals and human diarrhoeal cases by PCR based on 23S rRNA gene. FEMS Immunol Med Microbiol 29, 107–113 (2000).
Liu, J., Janaki, L. & Houpt, E. Precaution for using nucleic acid-based methods to detect Aeromonas. Journal of Clinical Microbiology 50, 1829–1829 (2012).
Nam, I. Y. & Joh, K. Rapid detection of virulence factors of Aeromonas isolated from a trout farm by hexaplex-PCR. J Microbiol 45, 297–304 (2007).
O’Hara, C. M., Sowers, E. G., Bopp, C. A., Duda, S. B. & Strockbine, N. A. Accuracy of six commercially available systems for identification of members of the family Vibrionaceae. Journal of Clinical Microbiology 41, 5654–5659 (2003).
Al-Talib, H., Latif, B., Mohd-Zain, Z. & Land, G. A. Pentaplex PCR assay for detection of hemorrhagic bacteria from stool samples. Journal of Clinical Microbiology 52, 3244–3249 (2014).
Huang, Q., Hu, Q. & Li, Q. Identification of 8 foodborne pathogens by multicolor combinational probe coding technology in a single real-time PCR. Clinical Chemistry 53, 1741–1748 (2007).
Liu, J. et al. A laboratory-developed TaqMan array card for simultaneous detection of 19 enteropathogens. Journal of Clinical Microbiology 51, 472–480 (2012).
Liu, Y. et al. Simultaneous detection of seven enteric viruses associated with acute gastroenteritis by a multiplexed Luminex-based assay. Journal of Clinical Microbiology 50, 2384–2389 (2012).
Schouten, J. P. et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 30, e57 (2002).
Liao, Y. et al. Combination of fluorescence color and melting temperature as a two-dimensional label for homogeneous multiplex PCR detection. Nucleic Acids Research 41, e76–e76 (2013).
Panicker, G., Call, D. R., Krug, M. J. & Bej, A. K. Detection of pathogenic Vibrio spp. in shellfish by using multiplex PCR and DNA microarrays. Applied and Environmental Microbiology 70, 7436–7444 (2004).
Jones, J. L. et al. Abundance of Vibrio cholerae, V. vulnificus, and V. parahaemolyticus in oysters (Crassostrea virginica) and clams (Mercenaria mercenaria) from long island sound. Applied and Environmental Microbiology 80, 7667–7672 (2014).
Dechet, A. M., Yu, P. A., Koram, N. & Painter, J. Nonfoodborne vibrio infections: an important cause of morbidity and mortality in the United States, 1997–2006. Clinical Infectious Diseases 46, 970–976 (2008).
Newton, A., Kendall, M., Vugia, D. J., Henao, O. L. & Mahon, B. E. Increasing rates of Vibriosis in the United States, 1996–2010: review of surveillance data from 2 systems. Clinical Infectious Diseases 54, S391–S395 (2012).
Kawatsu, K., Ishibashi, M. & Tsukamoto, T. Development and evaluation of a rapid, simple, and sensitive immunochromatographic assay to detect thermostable direct hemolysin produced by Vibrio parahaemolyticus in enrichment cultures of stool specimens. Journal of Clinical Microbiology 44, 1821–1827 (2006).
Wei, S., Zhao, H., Xian, Y., Hussain, M. A. & Wu, X. Multiplex PCR assays for the detection of Vibrio alginolyticus, Vibrio parahaemolyticus, Vibrio vulnificus, and Vibrio cholerae with an internal amplification control. Diagn Microbiol Infect Dis 79, 115–118 (2014).
Vinothkumar, K., Bhardwaj, A. K., Ramamurthy, T. & Niyogi, S. K. Triplex PCR assay for the rapid identification of 3 major Vibrio species, Vibrio cholerae, Vibrio parahaemolyticus, and Vibrio fluvialis. Diagn Microbiol Infect Dis 76, 526–528 (2013).
Souza, T. B. et al. Real-Time Multiplex PCR assay and melting curve analysis for identifying diarrheagenic Escherichia coli. Journal of Clinical Microbiology 51, 1031–1033 (2013).
Xu, Y. et al. Fluorescent probe-based lateral flow assay for multiplex nucleic acid detection. Analytical Chemistry 86, 5611–5614 (2014).
Lindsay, B. et al. Diarrheagenic pathogens in polymicrobial infections. Emerging Infectious Diseases 17, 606–611 (2011).
Zhou, D. et al. A Foodborne outbreak of gastroenteritis caused by Vibrio parahaemolyticus and Norovirus through non-seafood vehicle. Plos One 10, e0137848 (2015).
Jiang, Y. et al. Simultaneous detection of five enteric viruses associated with gastroenteritis by use of a PCR assay: a single real-time multiplex reaction and its clinical application. J Clin Microbiol 52, 1266–1268 (2014).
Acknowledgements
We thank Prof. Biao Kan from the Chinese Center for Disease Control and Prevention for providing the positive strains. This study was supported by the Shenzhen Municipal Science and Technology Program (CXZZ20140411105636301) and China National Science and Technology Major Project (No. 2017ZX10303406).
Author information
Authors and Affiliations
Contributions
Qinghua Hu planned the experiments. Yixiang Jiang, Lianhua He, Pingfang Wu, Xiaolu Shi, Min Jiang, Yaqun Qiu, Yiman Lin, Yinghui Li and Fang Bai performed the experiments. Yiqun Liao, Qingge Li, and Renli Zhang conducted the data analysis. Yixiang Jiang wrote the paper, and Qinghua Hu reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, Y., He, L., Wu, P. et al. Simultaneous Identification of Ten Bacterial Pathogens Using the Multiplex Ligation Reaction Based on the Probe Melting Curve Analysis. Sci Rep 7, 5902 (2017). https://doi.org/10.1038/s41598-017-06348-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06348-z
This article is cited by
-
Multiplex ligation reaction based on probe melting curve analysis: a pragmatic approach for the identification of 30 common Salmonella serovars
Annals of Clinical Microbiology and Antimicrobials (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
