
Abstract
Aberrant microRNA expression contributes to breast cancer progression and endocrine resistance. We reported that although tamoxifen stimulated miR-29b-1/a transcription in tamoxifen (TAM)-resistant breast cancer cells, ectopic expression of miR-29b-1/a did not drive TAM-resistance in MCF-7 breast cancer cells. However, miR-29b-1/a overexpression significantly repressed TAM-resistant LCC9 cell proliferation, suggesting that miR-29b-1/a is not mediating TAM resistance but acts as a tumor suppressor in TAM-resistant cells. The target genes mediating this tumor suppressor activity were unknown. Here, we identify miR-29b-1 and miR-29a target transcripts in both MCF-7 and LCC9 cells. We find that miR-29b-1 and miR-29a regulate common and unique transcripts in each cell line. The cell-specific and common downregulated genes were characterized using the MetaCore Gene Ontology (GO) enrichment analysis algorithm. LCC9-sepecific miR-29b-1/a-regulated GO processes include oxidative phosphorylation, ATP metabolism, and apoptosis. Extracellular flux analysis of cells transfected with anti- or pre- miR-29a confirmed that miR-29a inhibits mitochondrial bioenergetics in LCC9 cells. qPCR,luciferase reporter assays, and western blot also verified the ATP synthase subunit genes ATP5G1 and ATPIF1 as bone fide miR29b-1/a targets. Our results suggest that miR-29 repression of TAM-resistant breast cancer cell proliferation is mediated in part through repression of genes important in mitochondrial bioenergetics.
Similar content being viewed by others


Introduction
microRNAs (miRNA, miRs) are 22 nt non-coding RNAs that recognize and bind complementary seed sequences in the 3′-UTR region of a target messenger RNA (mRNA)1, 2. This results in translational repression and/or transcriptional degradation of the target gene. By targeting several mRNAs, miRNAs regulate several cellular and biological processes including cell cycle, cell proliferation and cell differentiation, apoptosis, cellular respiration and glycolysis (reviewed in refs 3,4,5,6). Aberrant miRNA expression mediates disease initiation and progression in breast and other cancers7.
Seventy percent of breast tumors express estrogen receptor alpha (ERα) implying eligibility for endocrine therapies including selective estrogen receptor modulators (SERMs), e.g., Tamoxifen (TAM), and aromatase inhibitors (AI), e.g., letrozole. The efficacy of endocrine therapy is limited by relapse in ~40% of patients8, 9. Endocrine resistance is mediated by multiple mechanisms depending on the type of therapy used (reviewed in refs 10,11,12,13,14). Alterations in miRNA expression are also implicated in endocrine-resistance (reviewed in refs 15 and 16).
Previously, we reported miR-29b-1 and miR-29a were downregulated by TAM in TAM- sensitive (TAM-S) MCF-7 BC cells and upregulated in TAM-resistant (TAM-R) LCC2, LCC9, and LY2 BC cells17. There are four miR-29 family members in the human genome: miR-29b-2 and miR-29c (chromosome 1q32.2) and miR-29b-1 and miR-29a (chromosome 7q32.3). miR-29b-1 and miR-29a are separated by ~652 bp on the same pri-miRNA transcript18,19,20,21. Upon processing, mature miR-29b-1 preferentially localizes to the nucleus, while mature miR-29a localizes to the cytoplasm22. miR-29 family members have been reported to exhibit both tumor suppressive and oncogenic roles in breast cancer17, 23,24,25.
We recently reported that ERα mediates 4-hydroxyTAM (4-OHT) repression of miR-29b-1/a in MCF-7 and upregulation in TAM-R LCC9 and LY2 BC cells. However, inhibition of miR-29b-1 and miR-29a did not sensitize LCC9 or LY2 cells to TAM17. Ectopic expression of miR-29b-1/a did not drive TAM resistance in MCF-7 cells, but it did significantly repress proliferation of TAM-R LCC9 and LY2 cells as compared to TAM-S MCF-7 cells. This suggests that miR-29b-1/a may regulate different transcripts, and thus pathways, in TAM-R cells and TAM-S cells. To identify and quantitate mRNA targets regulated by miR-29, we performed RNA-sequencing (RNA-seq) in MCF-7 and LCC9 BC cells transiently transfected with anti-miR-29a, pre-miR-29b-1 or pre-miR-29a. MetaCoreTM functional analysis of the RNA-seq data identified several metabolic processes including oxidative phosphorylation (OXPHOS) uniquely regulated by both miR-29b-1 and miR-29a in LCC9 cells. We observed that miR-29a overexpression inhibited mitochondrial bioenergetic function in LCC9 cells, thus correlating function with identified Gene Ontology (GO) processes. We used qPCR, a luciferase reporter assay, and western blot to validate that two subunits of ATP synthase (Complex V in the oxidative phosphorylation respiratory chain), ATP5G1 and ATPIF1, are bona fide miR-29b-1/a targets.
Results
Identification and characterization of the miR-29b-1 and miR-29a transcriptome in MCF-7 and LCC9 cells
To identify potential miR-29b-1 and miR-29a targets in breast cancer cells and their possible role in tumor suppression, TAM-S MCF-7 and TAM-R LCC9 cells were grown in hormonally depleted medium and transfected with anti-miR-29a (that inhibits both miR-29b-1 and miR-29a expression) and pre-miR-29b-1 or pre-miR-29a (Supplementary Fig. 1). The strategy of comparing the transcriptome of cells transfected with anti-miR-29a versus pre-miR-29b-1 and anti-miR-29a versus pre-miR-29a was selected to enrich for direct miR-29b-1 and miR-29a targets. As miRNAs historically repress target transcript translation and/or mRNA expression, we focused on genes downregulated with pre-miRNA treatment and upregulated with anti-miR treatment for transcriptome analysis.
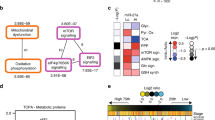
miR-29b-1 and miR-29a downregulated 447 and 139 genes in MCF-7 cells respectively (Fig. 1A) and 1,751 and 1,959 genes in LCC9 cells (Fig. 1B; p < 0.05), respectively. The identity of these genes with MetaCore pathway analysis is provided in Supplementary Tables 1 and 2. These data suggest that miR-29b-1 and miR-29a regulate more transcripts in TAM-R LCC9 cells compared to TAM-S MCF-7 cells.

Enrichment analysis of RNA-seq data. Differentially expressed genes were identified in pairwise comparisons: Pre-miR-29b-1 vs. Anti-miR-29a and Anti-miR-29a vs. Pre-miR-29a using tophat and cufflink-cuff diff2. The Venn diagrams show the number of differentially and commonly regulated genes identified. GeneGo Pathways Software (MetaCoreTM) was then used to identify genes significantly downregulated by miR-29b-1 and miR-29a in (A) MCF-7 and (B) LCC9 cells. (C) Pathway analysis of the downregulated genes was performed using MetaCoreTM. The order of the pathways identified for each comparison are listed in the order provided by MetaCoreTM analysis.
In MCF-7 cells, 132 genes were identified as common targets of both miR-29b-1 and miR-29a (Fig. 1A). In LCC9 cells, 1,508 genes were common targets to both miR-29b-1 and miR-29a (Fig. 1B). MetaCoreTM pathway analysis of the common and uniquely regulated genes is provided in Supplementary Figs 2 and 3. Of the common targets, 60 were unique to MCF-7 and 1,436 were unique to LCC9 (Fig. 1A and Supplementary Tables 3). 72 targets were common to both MCF-7 and LCC9 cells (Fig. 1C). Together, these data confirm that miR-29b-1 and miR-29a regulate common and unique targets in MCF-7 and LCC9 cells. MetaCoreTM pathway enrichment analysis was performed for each dataset and the top pathways identified are listed in Fig. 1C.
Gene ontology (GO) analysis of the uniquely downregulated transcripts by both miR-29b-1 and miR-29a in LCC9 cells (Fig. 1C) identified organonitrogen compound metabolic and biosynthetic processes and cotranslational protein targeting to the membrane as the top three regulated processes (Supplementary Fig. 4A). The top five regulated process networks included translation initiation, elongation, termination, and cell cycle S phase (Supplementary Fig. 4B).
Pathway enrichment analysis of the uniquely downregulated miR-29b-1/a target genes in LCC9 cells identified pathways including oxidative phosphorylation, ATP metabolism, apoptosis, and cell survival. MetaCoreTM network analysis on mitochondrial respiratory complex genes downregulated by miR-29b-1/a is shown in Supplementary Fig. 4C. Several genes including components of respiratory Complex II and III, Cytochrome c oxidase, and ATP synthase (Complex IV) were uniquely downregulated by miR-29b-1/a in LCC9 cells (Supplementary Fig. 4C). We previously reported that six genes encoding subunits of ATP synthase that are not regulated by miR-29 were more highly expressed in LCC9 than MCF-7 cells26. The role of these genes in endocrine resistance will require further evaluation.
miR-29a regulates mitochondrial bioenergetics in tamoxifen-resistant LCC9 cells
Because OXPHOS was identified as the top enrichment pathway modulated by miR-29b-1/a in LCC9 cells, and not in MCF-7 cells (Fig. 1C), we postulated that miR-29a regulates mitochondrial bioenergetics activity in LCC9 cells. We focused on miR-29a because its basal expression is higher in LCC9 and other breast cancer cells compared to miR-29b-117. To examine the impact of miR-29a on mitochondrial respiration, MCF-7 and LCC9 cells were transfected with either anti-miR-29a or pre-miR-29a, versus control, and grown in hormonally depleted phenol red-free medium for 48 h prior to determining oxygen consumption rate (OCR) and extracellular acidification rate (ECAR; reflects glycolytic rate)27 using the Seahorse extracellular flux assay26, 27 (Supplementary Fig. 5).
In agreement with our previous work26, TAM-R LCC9 cells grown in hormonally depleted medium have increased basal OCR versus MCF-7 cells (Fig. 2A). Transfection of LCC9 cells with anti-miR-29b-1 increased while pre-miR-29a repressed basal OCR, ATP-linked OCR (which measures the rate of mitochondrial ATP synthesis) and mitochondrial reserve (also known as reserve respiratory capacity)28 (Fig. 2A). Similar results were seen in pre-miR-29a-transfected MCF-7 cells except that growth in hormonally depleted medium ablates reserve capacity, and thus, no effect of miR-29a on reserve capacity was detected (Fig. 2A). The changes in basal OCR suggest that miR-29a modulates metabolic rate in part through electron flow. The regulation of reserve capacity, which is a measurement of the ability to respond to increased energy demand after injection of the uncoupler FCCP, suggests that miR-29-regulated genes adversely impact LCC9’s mitochondrial ability to mitigate cellular stress. Inverse changes, albeit of lesser magnitude, were seen in LCC9 cells transfected with anti-miR-29a.

Upregulation of miR-29a represses mitochondrial function in MCF-7 and LCC9 cells. MCF7 and LCC9 cells were plated in XF-24 plates in hormonally depleted medium and transfected with control, anti-miR-29a, or pre-miR-29a for 28 h prior to running the Seahorse extracellular flux assay to determine mitochondrial activity. (A and B) OCR parameters; (C and D) ECAR parameters. Each bar is an average of 4 separate wells ± SEM in one experiment. *p < 0.05 versus LCC9 or MCF-7 control for the indicated parameter. For LCC9, data were analyzed by one-way ANOVA followed by Tukey’s test. For MCF-7, two-tailed Student’s t-test was performed (NS).
Compared to MCF-7 cells, LCC9 cells have higher basal proton leak. Proton leak is required for protecting against cellular oxidative damage through lowering ROS produced by the electron transport chain (ETC)29. Pre-miR-29a transfection repressed proton leak in LCC9 cells (Fig. 2A). Non-mitochondrial activity, which is attributed to cytoplasmic oxidases, was lower in LCC9 cells transfected with pre-miR-29a (Fig. 2A).
Transfection of LCC9 cells with anti-miR-29a increased while pre-miR-29a transfection decreased maximal mitochondrial (mt) capacity (Fig. 2B). The maximal mitochondrial capacity is the OCR measured after injection of the uncoupler FCCP and is indicative of the mt ability to take up substrates30.
We previously reported that ECAR measurements agree with conversion of radio-labelled glucose to H2O in MCF-7 and T47D breast cancer cells, demonstrating that ECAR is a reliable measure of glycolysis27. LCC9 cells alone or upon transfection with anti-miR-29a had increased basal ECAR than MCF-7 cells (Fig. 2C). The glycolytic response to oligomycin, considered as the glycolytic reserve31, was increased by miR-29a repression and reduced by miR-29a overexpression in LCC9 cells (Fig. 2D). These data suggest that miR-29a regulates genes with roles in glycolysis in LCC9 cells. Examination of the RNA-seq data identified TPI1, PGAM1, ENO1, and LDHA as possible targets of miR-29b-1/a (Supplementary Fig. 6A). qPCR analysis confirmed that overexpression of miR-29b-1/a inhibited ENO1 and LDHA transcript expression in MCF-7 and LCC9 cells (Supplementary Fig. 6B). Future studies will further evaluate these observations.
Validation of ATP5G1 and ATPIF1 as bona fide targets of miR-29b-1 and miR-29a
miR-29a regulation of mitochondrial activity is suggestive of direct regulation of mitochondrial complex genes. Analysis of our RNA-seq data revealed miR-29 regulation of mitochondrial genes including ATP5G1, ATP5C1, ATPIF1, ATP5G3, NDUFS6, and NDUFC2 (Supplementary Fig. 7). To confirm miR-29 regulation of these putative targets, qPCR was performed in MCF-7 and LCC9 cells transfected with either anti-miR-29a, pre-miR-29b-1, pre-miR-29a or their negative controls (Fig. 3). Anti-miR-29a transfection increased ATP5G1 and ATPIF1 mRNA expression in MCF-7 and LCC9 cells (Fig. 3A and B). Conversely, pre-miR-29b-1 and pre-miR-29a transfection repressed ATP5G1 in LCC9 cells alone and ATPIF1 mRNA expression in both MCF-7 and LCC9 cells (Fig. 3A and B). These data suggest regulation of ATP5G1 and ATPIF1 by miR-29b-1/a.

miR-29b-1/a regulates mRNA expression of putative targets in MCF-7 and LCC9 cells. MCF-7 and LCC9 cells were grown in ‘hormone-depleted’ media and transfected with anti-miR-control, anti-miR-29a (3p), pre-miR control or pre-miR-29b-1/a (3p), as indicated. Twenty-four h post transfection, RNA was isolated and qPCR performed. Values were normalized to 18S. Values are the mean ± SEM of 3 independent experiments. Within each experiment, each sample was run in triplicates. Statistical evaluation was performed using one-way ANOVA followed by Tukey’s test. *p < 0.05 versus control transfected cells.
In LCC9 cells, anti-miR-29a transfection increased ATP5C1 and NDUFC2 while pre-miR-29b-1 and pre-miR-29a transfection repressed ATP5C1, ATP5G3, NDUFS6 and NDUFC2 transcript expression (Fig. 3C–F). In MCF-7 cells anti-miR-29a transfection increased NDUFC2 expression while pre-miR-29b-1 and pre-miR-29a transfection repressed expression of ATP5G3 and NDUFC2 transcripts (Fig. 3D and F). Taken together, these data suggest cell line specific differential regulation of miR-29 targets between MCF-7 and LCC9 cells. Notably, ATP5G1 and ATP1F1 were downregulated by miR-29b-1/a in both LCC9 and MCF-7 cells (Fig. 3A and B). Further, basal mRNA expression of ATP5G1, ATP5IF1 and ATP5G3 were higher (lower CT) relative to ATP5IC1, NDUFS6, and NDUFC2 in both MCF-7 and LCC9 cells (Supplementary Fig. 8). Hence, we selected ATP5G1 and ATPIF1 for further study.
To determine whether ATP5G1 and ATPIF1 are bona fide miR-29 targets, we tested the ability of miR-29 to repress the luciferase activity from reporter vectors containing wild type (WT) or mutant (mut) 3′ UTRs of these genes. The miR-29 binding region at the 3′ UTR region of ATP5G1 and ATPIF1 are conserved across species (Fig. 4A). We created mutations in the miR-29 recognition sequence in ATP5G1 (Fig. 4A). Cotransfection of miR-29b or miR-29a mimics with the luciferase reporter containing ATP5G1 3′ UTR WT sequence repressed luciferase activity (Fig. 4B). Mutation of the ATP5G1 3′ UTR (Mut) abrogated repression by the miR-29b and miR-29a mimics (Fig. 4B). These data demonstrate that ATP5G1 is a bona fide direct target of miR-29b and miR-29a. Additionally, miR-29b and miR-29a mimics repressed luciferase activity from the ATPIF1 3′ UTR WT luciferase vector and repression was abrogated when cells were cotransfected with anti-miR-29a (Fig. 4C). These data suggest that ATPIF1 is also a bona fide target of miR-29b and miR-29a.

miR-29b-1/a regulate ATP5G1 and ATPIF1 3′-UTRs luciferase reporter activity. (A) Alignment of miR-29b-1 and miR-29a, their seed elements (in bold) and the 3′-UTRs of ATP5G1 and ATPIF1. The mutated seed element in ATP5G1 used in luciferase assay is indicated in red. miR-29 family members have the same seed sequence and their binding sites in the 3′ UTRs of ATP5G1 and ATPIF1 are conserved across several species. (B and C) HEK-293 cells grown in hormonally depleted medium were transiently transfected with pEZ-MT10 dual luciferase reporter containing the 3′ UTR of ATP5G1 wildtype (WT), ATPIF1 WT, miR-29 seed element mutated (Mut) constructs and an RNA mimic negative control or miR-29b or miR-29a mimics +/− Anti-miR-29a for 24 h prior to dual luciferase assay. Data are the avg. of three replicate wells ± SEM. *p < 0.05 versus negative control. Statistical analysis used one-way ANOVA followed by Newman-Keuls Multiple Comparison Test.
To further confirm ATP5G1 and ATPIF1 as miR-29 bona fide targets, we examined whether miR-29b-1 and miR-29a decreased protein expression of these genes. We transfected LCC9 cells with miR-29b and miR-29a mimics and evaluated ATP5G1 and ATPIF1 protein expression in whole cell extracts (Fig. 5). Compared to the negative control transfected cells, mimics of miR-29b-1 and miR-29a decreased protein expression of ATP5G1 and ATPIF1. These data further confirm ATP5G1 and ATPIF1 as validated targets of miR-29.

miR-29-b-1/a downregulate ATP5G1 and ATPIF1 protein expression in LCC9 breast cancer cells. LCC9 cells were grown in hormonally depleted medium and transfected with mimic negative control (Neg Ctrl), miR-29b mimic, or miR-29a mimic for 48 h. ATP5G1 and ATPIF1 and β-actin (normalizer) was examined in WCE (10 µg protein/lane). These images were cropped from full-length blots (Supplementary Figs 13–15). The same blot was stripped and reprobed for each protein. Values are ratios of ATP5G1/β-actin or ATPIF1/ β-actin with the Neg Ctrl set to one. This blot if from one experiment.
Discussion
Dysregulation of the miR-29 family members plays a role in breast cancer progression, metastasis, and endocrine resistance17, 32. Using miRpower33, we observed that lower expression of miR-29b-1 and miR-29a is associated with decreased relapse-free survival (RFS) in overall breast cancer and in patients with ER + primary tumors (Supplementary Fig. 9). However, the precise mechanism(s) and targets of miR-29 in breast cancer are not fully elucidated. Here we identified the miR-29b-1 and miR-29a transcriptomes in MCF-7 TAM-S and LCC9 TAM-R breast cancer cells. Although miR-29b-1 and miR-29a have commonly regulated transcripts, they also have uniquely regulated transcripts. We observed that miR-29 regulates more transcripts in LCC9 than MCF-7 cells. While the biological reason for the higher number of targets of miR-29 identified in LCC9 compared to MCF-7 cells is unknown, we speculate that miR-29 may downregulate a suppressor(s) of transcription in LCC9 cells. One example is DNMT3B, a validated target of miR-29b34, miR-29a, and miR-29c35. DNMT3 family members are gene-specific, de novo DNA methyl transferases36. We observed that DNMT3B is a target of miR-29b-1/a in LCC9, but not MCF-7 cells (Supplementary Fig. 12). The identity of additional suppressors remains to be examined. Alternatively, the expression of components of the complexes involved in miRNA processing and activity, e.g., DICER1 and RISC complexes, may be higher in LCC9 than MCF-7 cells, cellular components that stabilize miR-29b-1/a may be more abundant in LCC9 cells, or the expression of lncRNAs acting as miR-29 sponges may be higher in MCF-7 cells. Further experiments are needed to address these speculation MetaCore enrichment analysis of unique miR-29 target transcripts in LCC9 cells identified GO pathways involved in oxidative phosphorylation, ATP metabolism, apoptosis and survival. In agreement with this literature-based association, ectopic miR-29a expression resulted in an inhibition of mitochondrial bioenergetics in LCC9 cells. Further, we demonstrate for the first time that ATP5G1 and ATPIF1 are validated, bona fide miR-29a targets.
ATP5G1 encodes a subunit (C1) of the inner mitochondrial membrane-bound Fo domain of mitochondrial ATP synthase (complex V) that is involved in H+ transport necessary for the proton-motive force for ATP synthesis37. Inhibition of ATP synthase blocks the proliferation of breast cancer cells, but not normal MCF-10A breast epithelial cells38. Therapeutic targeting ATP synthase is of interest in blocking proliferation of breast stem-like cancer cells (CSCs) to inhibit disease recurrence and metastasis39. ATPIF1 encodes ATPase Inhibitory Factor I that is in a 1:1 ratio with F1 domain of ATP synthase and functions to limit ATP depletion when the mitochondrial membrane potential falls below a threshold needed for ATP synthesis40. ATPIF1 is required to avoid the consumption of cellular ATP when the ATP synthase enzyme acts in reverse to hydrolyze ATP41.
Dysregulation of mitochondrial bioenergetics is common in cancer cells29. Thus, identifying impaired mitochondrial pathways is of pathological and therapeutic relevance42. Mitochondria dysregulation results from alterations in mitochondrial function and ultrastructure43, overproduction of reactive oxygen species (ROS)44, and mitochondrial DNA (mtDNA) mutations and deletions44, 45. miRNA regulation of mitochondria has previously been reported (reviewed in refs 46 and 47). However, this is the first report that miR-29b-1/a regulate genes in ATP synthase (Complex V).
We examined the expression of ATP5G1 and ATPIF1 in relation to overall survival and disease free survival of ERα+ breast cancer patients using KM plotter48 (Supplementary Fig. 10). We focused on ERα+ patients because they would be likely to receive TAM and/or AI therapy. We observed that high ATP1F1 was associated with increased OS in ERα+ breast cancer patients (Supplementary Fig. 10B) while higher ATP5G1 correlated with reduced RFS in ERα+ breast cancer patients (Supplementary Fig. 10C). Another breast tumor database of gene expression from microarrays also showed that higher ATP5G1 expression was associated with reduced disease free survival (DFS, which is the same as RFS) in ERα+ breast cancer patients in Ireland (http://glados.ucd.ie/BreastMark/mRNA_analysis.html)49. We note that all data in both databases are from primary tumors. Based on our LCC9 and LY2 cell experiments, we would expect that breast cancer patients with endocrine-resistant disease (metastasis) on TAM therapy will have increased miR-29b-1/a which would result in lower ATP5G1 and ATPIF1 transcript levels. The lower ATPIF1 associated with lower OS (Supplementary Fig. 10B) agrees with this speculation; however, the apparent protective effect of lower ATP5G1 (Supplementary Figs 10C and 11) does not. Because the gene expression data are from primary tumors, the impact of the expression of these genes in metastatic, endocrine-resistant tumors is unknown.
In agreement with the MetaCore pathway enrichment identification of OXPHOS as the top enriched pathway among miR-29b-1/a regulated genes in LCC9 cells, we observed that pre-miR-29a decreased basal OCR, ATP-linked OCR, and mt reserve. Concordantly, anti-miR-29a increased OCR and mt reserve, suggesting that repression of miR-29a may serve as an adaptive response to meet the increased energy demand of cancer cells and promote survival. Basal-like human breast tumors have lower levels of miR-29a, miR-29b, and miR-29c compared with Luminal A and B tumors50. Repression of miR-29a increased basal ECAR (glycolysis) in LCC9 cells (Fig. 2C), a finding in agreement with the Warburg effect of increased glycolysis seen in basal-like tumors51, 52. Overall changes in OCR and ECAR suggest overexpression of miR-29a has a greater impact on OXPHOS while repression of miR-29a increases both glycolytic activity and mitochondrial activity (OXPHOS) in LCC9 cells. Pre-miR-29a transfection repressed proton leak in LCC9 cells to levels below those seen in MCF-7 cells with hormone deprivation, suggesting miR-29a overexpression depletes mitochondrial OXPHOS, which could lead to activation of apoptosis or other cell death pathways.
LCC9 cells alone or when transfected with anti-miR-29a had a higher maximum mitochondrial capacity than the hormonally depleted MCF-7 cells, suggesting increased bioenergetics reserve. Indeed repression of miR-29a in LCC9 cells increased the glycolytic reserve. This may be attributable in part to the higher non-mitochondrial activity in LCC9 cells. Further metabolomics studies will be required to further assess changes in cellular metabolism with changes in miR-29b/a expression.
We identify several ETC proteins including ATP synthase genes ATP5G1 and ATPIF1 as targets of miR-29b-1/a. We experimentally validated that ATP5G1 and ATPIF1 are bona fide targets of miR-29b and miR-29a. Inhibition of ATP synthase in MCF7, T47D and MDA-MB-231 BC cells induced cell cycle arrest, decreased colony formation, and inhibited cell proliferation53. The targeting of ATP synthase complex subunits may account for miR-29’s repression of OCR and of BC cell proliferation17. Additional studies are warranted to confirm this suggestion.
Conclusions
Our RNA-seq transcriptome analysis revealed that miR-29b-1 and miR-29a regulate common and unique gene targets in TAM-S MCF-7 and TAM-R LCC9 BC cells. We focused on common miR-29b-1 and miR-29a targets downregulated uniquely or in common in MCF-7 and LCC9 cells. We report that miR-29b-1 and miR-29a regulate more transcripts in LCC9 than MCF-7 cells. MetaCore pathway enrichment analysis identified OXPHOS as the top GO pathway regulated by miR-29b-1/a target transcripts in LCC9 cells and cell cycle: chromosome condensation in prometaphase as the top GO pathway in MCF-7 cells. Seahorse extracellular flux analyses of LCC9 and MCF-7 cells confirmed regulation of cellular bioenergetics by miR-29a including repression of OXPHOS by ectopic miR-29a expression. We experimentally validated ATP5G1 and ATPIF1 as new miR-29 bona fide targets suggesting that these targets play a role in mediating miR-29’s anti-proliferative effects in LCC9 and MCF-7 breast cancer cells.
Materials and Methods
Cell lines and reagents
The following cells and reagents were used: HEK-293 (ATCC® CRL-1573™) and MCF-7 cells were purchased from ATCC. LCC9 cells are ERα+/progesterone receptor (PR) + cells resistant to antiestrogens (tamoxifen and fulvestrant) and were kindly provided by Dr. Robert Clarke, Georgetown University54. Transfection reagents included Anti-miR-29a (Anti-miRTMs, Ambion), pre-miR-29b-1–3p or pre-miR-29a-3p precursor (Pre-miRTMs, Ambion), miR-29a mimic or miR-29b-1 mimic (miR-VanaTM, Ambion), Lipofectamine RNAiMAX (Invitrogen), FuGENE® HD (Promega) Pre-miR™ negative control #1 (Ambion), Anti-miR™ negative control #1(Ambion), miR-VanaTM negative control (Ambion), pEZX-MT06_ATP5G1 wild type (WT), pEZX-MT06_ATPIF1 (WT), pEZX-MT06_negative control (miTargetTM miRNA 3′ UTR, GeneCopoeia).
Transient transfection of miRNAs and anti-miRNAs
Cells were grown as previously described in ref. 17. Briefly, MCF-7 and LCC9 cells were grown in ‘hormone-depleted’ medium: phenol-red free IMEM (Gibco, ThermoFisher) supplemented with 5% dextran-coated charcoal-stripped fetal bovine serum (DCC-FBS, Atlanta Biologicals)17 and concomitantly transfected with either anti-miR-29a, pre-miR-29b-1-3p, pre-miR-29a-3p, pre-miR™ negative control #1 or anti-miR™ negative control #1 for a 48 h, as indicated.
RNA-sequencing
RNA-sequencing was previously described in ref. 26. In brief, MCF-7 and LCC9 breast cancer cells were plated in 100 mm plates, ‘hormone-depleted’ and transfected in triplicate with either pre-miR-29b-1, pre-mir-29a and anti-miR-29a using Lipofectamine (Ambion) and OPTI-MEM® I (Gibco: Life technologies). 24 h post transfection, RNA was isolated using miRCURY RNA isolation kit (Exiqon) according to manufacturer’s instructions and RNA concentration and quality assessed with a NanoDrop spectrophotometer. Using TruSeq Stranded mRNA kit (Illumina, San Diego, CA, USA), mRNA libraries were made using 2 μg of RNA and validated with an Agilent 2100 Bioanalyzer (Santa Clara (CA). After further library quantification with the Illumina Library Quantification Kit, RT-PCR was performed using the ABI Prism qPCR Mix (Kapa Biosystems) on an ABI17900HT real-time PCR instrument. Single read sequencing (75–76 cycles) was then performed using the 500 High-output v2 (75cycle) sequencing kit on an Illumina NextSeq500 instrument. Obtained read sequences were mapped to the human reference genome version GRCh37.1 using the mapping algorithm tophat55 version 2.0.2. Using cufflinks version 2.2.1 and annotations found at ENSEMBL, Homo_sapiens GRCh37.73.gtf expression levels at loci were quantified. Raw sequencing data files obtained from our analysis are available at Gene Expression Omnibus (GEO) database: accession number GSE81620. Entrez gene identifiers for significantly expressed genes had a q-value cutoff of 0.05. Samples were then divided into fastq single end sequencing files representing comparisons of pre-miR-29b-1 vs anti-miR-29a and anti-miR-29a vs pre-miR-29a in MCF-7 and LCC9 cells.
In silico network analysis
Data from RNA-seq was analyzed such that transcripts selected had a log2 fold-change greater than 0.34 (or −0.34 for repressed transcripts) and a statistical significant threshold q value less than 0.05. Pathway and network analysis of differentially expressed genes was determined using the web-based software MetaCoreTM version 6.27 (GeneGO, Thomson Reuters, New York, N.Y). MetaCoreTM is a manually curated data base of over 6 million experimental findings interactions including protein-protein, protein-DNA, protein-RNA, and protein-compounds; metabolic and signaling pathways; and other additional information56.
Metabolic analysis with the Seahorse XF24 extracellular flux analyzer
Metabolic profiles were performed as previously described in refs 26 and 27. Briefly, LCC9 cells were plated at 25,000 cells/well in XF24 plates. Twenty-four h post plating, cells were transfected with either anti-miR-29a or pre-miR-29a for 48 h. One hour prior to running the Seahorse Mito Stress Test assay, the media was replaced with DMEM assay media containing 1 mM sodium pyruvate, 25 mM glucose, and 1.85 g/L NaCl (all from Sigma), 2 mM Glutamax (ThermoFisher), pH 7.4, and maintained at 37 °C in an non-CO2 incubator. Sensor cartridges were incubated in XF24 calibrant solution (Agilent Technologies) at least 6 h prior to running assay. Initial measurement of basal ECAR (mpH/min) and OCR (pmol O2/min) were taken prior to determining mitochondrial function using sequential injections of oligomycin A (1.5 μM), FCCP (0.5 μM) and combination of antimycin A (10 μM) and rotenone (2 μM)26. The protein concentration in each well was determined using the BioRad DC™ Protein Assay (BioRad, Hercules, CA, USA). OCR and ECAR values were normalized to protein concentration/well. Each experimental condition was run in quadruplicate within one experiment and separate experiments were run 3–4 times for statistical evaluation.
Quantitative real-time PCR (qRT-PCR)
RNA isolation, RT-PCR and qPCR were performed as previously described in ref. 17. TaqMan (Thermo Fisher Scientific) master mix was used for miR-29b-1/a primers (Thermo Fisher Scientific) and SYBR green (QIAGEN) master mix was used for LDHA 57, ENO1 58, CYCS 59, ATP5G1 60, ATP5C1 61, ATPIF1 62, ATP5G3 60, NDUFS6 63, NDUFC2 64. Normalizers included RNU6B or U48 (Thermo Fisher Scientific) for miRNA and GAPDH (SYBR green)65 and 18S (TaqMan; Thermo Fisher) for mRNA.
Site-directed mutagenesis, transient transfection of HEK-293 cells, and Dual luciferase assay
Mutation of the predicted 3′ UTR binding site of miR-29b-1/a on pEZX-MT06 miRNA 3′ UTR target dual luciferase expression vectors was performed using the GeneArt® Site-Directed Mutagenesis PLUS Kit (ThermoFisher Scientific) according to manufacturer’s instructions. Where indicated, HEK-293 cells were plated in 24 well plates and co-transfected with 200 ng of wild type (wt) or mutant (mut) vectors plus a 20 nM final concentration of miR-29b-1/a mimic, anti-miR-29a or negative control mimic using FuGENE HD (Promega) according to manufacturer’s protocol. Twenty-four h post transfection, a dual luciferase assay (Promega) was performed. Luciferase expression was determined relative to negative control mimic and statistical evaluation performed using GraphPad Prism Software (Graph Pad Software, San Diego, CA).
Western blot
LCC9 cells were transfected with miRNA MIMIC 2.0 (NEG #1, Life Technologies), miR-29b mimic, or miR-29a mimic (Life Technologies) for 48 h. Whole cell extracts (WCE) were prepared RIPA buffer (Sigma) in the presence of complete protease and phosphatase inhibitors. 10 μg of protein, determined by Bio-Rad DC protein quantification assay, were electrophoresed on a 10% SDS-PAGE gel. Following separation, the proteins were transferred to a nitrocellulose membrane and immunoblotted for ATP5G1 (GeneTex, Cat # GTX104671), ATPIF1 (Cell Signaling, Cat #8528) and β-actin (Sigma; loading control). Bands were visualized on a Bio-Rad ChemiDoc™MP imaging system with Image Lab version 5.2.1 software and quantified by UN-SCAN-IT Graph Digitizer version 7.1 software.
Statistics
Data are represented as mean ± standard error of the mean (SEM) of at least three independent experiments. Statistical analyses were performed using GraphPad Prism 5 (Graph Pad Software, Inc.). One-way analysis of variance (ANOVA) was followed by Tukey’s or Newman-Keuls multiple comparison post hoc tests where indicated17.
Data availability statement
Raw sequencing data files obtained from our analysis are available at GEO: accession number GSE81620. All data analysed during this study are included in this published article (and its Supplementary Information files).
References
Iorio, M. V. & Croce, C. M. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol 27, 5848–5856, doi:10.1200/jco.2009.24.0317 (2009).
Iorio, M. V. & Croce, C. M. microRNA involvement in human cancer. Carcinogenesis 33, 1126–1133, doi:10.1093/carcin/bgs140 (2012).
McGuire, A., Brown, J. A. & Kerin, M. J. Metastatic breast cancer: the potential of miRNA for diagnosis and treatment monitoring. Cancer Metastasis Rev. 34, 145–155, doi:10.1007/s10555-015-9551-7 (2015).
Negrini, M. & Calin, G. A. Breast cancer metastasis: a microRNA story. Breast cancer research: BCR 10, 203, doi:10.1186/bcr1867 (2008).
O’Day, E. & Lal, A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res 12, 201, doi:10.1186/bcr2484 (2010).
Singh, R. & Mo, Y.-Y. Role of microRNAs in breast cancer. Cancer Biology & Therapy 14, 201–212, doi:10.4161/cbt.23296 (2013).
Wiemer, E. A. The role of microRNAs in cancer: no small matter. European journal of cancer 43, 1529–1544, doi:10.1016/j.ejca.2007.04.002 (2007).
Ring, A. & Dowsett, M. Mechanisms of tamoxifen resistance. Endocrine-related cancer 11, 643–658, doi:10.1677/erc.1.00776 (2004).
Guest, S. K. et al. Src Is a Potential Therapeutic Target in Endocrine-Resistant Breast Cancer Exhibiting Low Estrogen Receptor-Mediated Transactivation. PLOS ONE 11, e0157397, doi:10.1371/journal.pone.0157397 (2016).
Anzick, S. L. et al. AIB1, a Steroid Receptor Coactivator Amplified in Breast and Ovarian. Cancer. Science (New York, N.Y.) 277, 965–968, doi:10.1126/science.277.5328.965 (1997).
Osborne, C. K. et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J. Natl. Cancer Inst. 95, 353–361, doi:10.1093/jnci/95.5.353 (2003).
Jeselsohn, R., Buchwalter, G., De Angelis, C., Brown, M. & Schiff, R. ESR1 mutations[mdash]a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol 12, 573–583, doi:10.1038/nrclinonc.2015.117 (2015).
Fanning, S. W. et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 5, doi:10.7554/eLife.12792 (2016).
Harrod, A. et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene, doi:10.1038/onc.2016.382 (2016).
Muluhngwi, P. & Klinge, C. M. Roles for miRNAs in endocrine resistance in breast cancer. Endocrine-related cancer 22, R279–300, doi:10.1530/erc-15-0355 (2015).
Muluhngwi, P. & Klinge, C. M. Identification of miRNAs as biomarkers for acquired endocrine resistance in breast cancer. Mol Cell Endocrinol, doi:10.1016/j.mce.2017.02.004 (2017).
Muluhngwi, P. et al. Tamoxifen differentially regulates miR-29b-1 and miR-29a expression depending on endocrine-sensitivity in breast cancer cells. Cancer Lett. 388, 230–238, doi:10.1016/j.canlet.2016.12.007 (2017).
Mott, J. L. et al. Transcriptional Suppression of mir-29b-1/mir-29a Promoter by c-Myc, Hedgehog, and NF-kappaB. Journal of cellular biochemistry 110, 1155–1164, doi:10.1002/jcb.22630 (2010).
Kriegel, A. J., Liu, Y., Fang, Y., Ding, X. & Liang, M. The miR-29 family: genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol Genomics 44, 237–244, doi:10.1152/physiolgenomics.00141.2011 (2012).
Mishmar, D. et al. Molecular characterization of a common fragile site (FRA7H) on human chromosome 7 by the cloning of a simian virus 40 integration site. Proc Natl Acad Sci USA 95, 8141–8146 (1998).
Eyholzer, M., Schmid, S., Wilkens, L., Mueller, B. U. & Pabst, T. The tumour-suppressive miR-29a/b1 cluster is regulated by CEBPA and blocked in human AML. British journal of cancer 103, 275–284, doi:10.1038/sj.bjc.6605751 (2010).
Hwang, H. W., Wentzel, E. A. & Mendell, J. T. A hexanucleotide element directs microRNA nuclear import. Science (New York, N.Y.) 315, 97–100, doi:10.1126/science.1136235 (2007).
Wang, Y., Zhang, X. W., Li, H., Yu, J. P. & Ren, X. B. The role of miRNA-29 family in cancer. European Journal of Cell Biology 92, 123–128, doi:10.1016/j.ejcb.2012.11.004 (2013).
Schmitt, M. J., Margue, C., Behrmann, I. & Kreis, S. MiRNA-29: a microRNA family with tumor-suppressing and immune-modulating properties. Curr Mol Med 13, 572–585, doi:10.2174/1566524011313040009 (2013).
Jiang, H., Zhang, G., Wu, J. H. & Jiang, C. P. Diverse roles of miR-29 in cancer (review). Oncol. Rep. 31, 1509–1516, doi:10.3892/or.2014.3036 (2014).
Radde, B. N. et al. Nuclear respiratory factor-1 and bioenergetics in tamoxifen-resistant breast cancer cells. Experimental cell research 347, 222–231, doi:10.1016/j.yexcr.2016.08.006 (2016).
Radde, B. N. et al. Bioenergetic differences between MCF-7 and T47D breast cancer cells and their regulation by oestradiol and tamoxifen. The Biochemical journal 465, 49–61, doi:10.1042/bj20131608 (2015).
Hill, B. G. et al. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biological chemistry 393, 1485–1512, doi:10.1515/hsz-2012-0198 (2012).
Pike Winer, L. S. & Wu, M. Rapid Analysis of Glycolytic and Oxidative Substrate Flux of Cancer Cells in a Microplate. PLoS ONE 9, e109916, doi:10.1371/journal.pone.0109916 (2014).
Dranka, B. P. et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 51, 1621–1635, doi:10.1016/j.freeradbiomed.2011.08.005 (2011).
Mookerjee, S. A., Nicholls, D. G. & Brand, M. D. Determining Maximum Glycolytic Capacity Using Extracellular Flux Measurements. PLoS One 11, e0152016, doi:10.1371/journal.pone.0152016 (2016).
Jiang, H. L. et al. MicroRNA-195 regulates steroid receptor coactivator-3 protein expression in hepatocellular carcinoma cells. Tumour Biol 35, 6955–6960, doi:10.1007/s13277-014-1933-x (2014).
Lanczky, A. et al. miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast cancer research and treatment 160, 439–446, doi:10.1007/s10549-016-4013-7 (2016).
Yan, B. et al. Micro-ribonucleic acid 29b inhibits cell proliferation and invasion and enhances cell apoptosis and chemotherapy effects of cisplatin via targeting of DNMT3b and AKT3 in prostate cancer. OncoTargets and therapy 8, 557–565, doi:10.2147/ott.s76484 (2015).
Hysolli, E. et al. Regulation of the DNA Methylation Landscape in Human Somatic Cell Reprogramming by the miR-29 Family. Stem Cell Reports 7, 43–54, doi:10.1016/j.stemcr.2016.05.014 (2016).
Nguyen, T. et al. Downregulation of microRNA-29c is associated with hypermethylation of tumor-related genes and disease outcome in cutaneous melanoma. Epigenetics 6, 388–394, doi:10.4161/epi.6.3.14056 (2011).
Jonckheere, A. I., Smeitink, J. A. M. & Rodenburg, R. J. T. Mitochondrial ATP synthase: architecture, function and pathology. Journal of Inherited Metabolic Disease 35, 211–225, doi:10.1007/s10545-011-9382-9 (2012).
Huang, T. C. et al. Targeting therapy for breast carcinoma by ATP synthase inhibitor aurovertin B. J Proteome Res 7, 1433–1444, doi:10.1021/pr700742h (2008).
Fiorillo, M. et al. Bedaquiline, an FDA-approved antibiotic, inhibits mitochondrial function and potently blocks the proliferative expansion of stem-like cancer cells (CSCs). Aging (Albany NY) 8, 1593–1606, doi:10.18632/aging.100983 (2016).
van Raaij, M. J. et al. The ATPase inhibitor protein from bovine heart mitochondria: the minimal inhibitory sequence. Biochemistry (Mosc). 35, 15618–15625, doi:10.1021/bi960628f (1996).
Faccenda, D. & Campanella, M. Molecular Regulation of the Mitochondrial F(1)F(o)-ATPsynthase: Physiological and Pathological Significance of the Inhibitory Factor 1 (IF(1)). International Journal of Cell Biology 2012, 367934, doi:10.1155/2012/367934 (2012).
Tombol, Z. et al. Integrative molecular bioinformatics study of human adrenocortical tumors: microRNA, tissue-specific target prediction, and pathway analysis. Endocrine-related cancer 16, 895–906, doi:10.1677/ERC-09-0096 (2009).
Gong, Y., Chai, Y., Ding, J. H., Sun, X. L. & Hu, G. Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neuroscience letters 488, 76–80, doi:10.1016/j.neulet.2010.11.006 (2011).
Woo, Y. M. et al. Inhibition of Aerobic Glycolysis Represses Akt/mTOR/HIF-1α Axis and Restores Tamoxifen Sensitivity in Antiestrogen-Resistant Breast Cancer Cells. PLoS ONE 10, e0132285, doi:10.1371/journal.pone.0132285 (2015).
Kujoth, G. C. et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science (New York, N.Y.) 309, 481–484, doi:10.1126/science.1112125 (2005).
Li, P., Jiao, J., Gao, G. & Prabhakar, B. S. Control of mitochondrial activity by miRNAs. Journal of cellular biochemistry 113, 1104–1110, doi:10.1002/jcb.24004 (2012).
Beltrán-Anaya, F. O., Cedro-Tanda, A., Hidalgo-Miranda, A. & Romero-Cordoba, S. L. Insights into the Regulatory Role of Non-coding RNAs in Cancer Metabolism. Frontiers in physiology 7, doi:10.3389/fphys.2016.00342 (2016).
Gyorffy, B. et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 123, 725–731, doi:10.1007/s10549-009-0674-9 (2010).
Madden, S. F. et al. BreastMark: An Integrated Approach to Mining Publicly Available Transcriptomic Datasets Relating to Breast Cancer Outcome. Breast cancer research: BCR 15, R52–R52, doi:10.1186/bcr3444 (2013).
Chou, J. et al. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol 15, 201–213, doi:10.1038/ncb2672 (2013).
Lu, J., Tan, M. & Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 356, 156–164, doi:10.1016/j.canlet.2014.04.001 (2015).
Dong, C. et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer cell 23, 316–331, doi:10.1016/j.ccr.2013.01.022 (2013).
Chang, H. Y., Huang, T. C., Chen, N. N., Huang, H. C. & Juan, H. F. Combination therapy targeting ectopic ATP synthase and 26S proteasome induces ER stress in breast cancer cells. Cell Death Dis 5, e1540, doi:10.1038/cddis.2014.504 (2014).
Brunner, N. et al. MCF7/LCC9: an antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer research 57, 3486–3493 (1997).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protocols 7, 562–578, doi:10.1038/nprot.2012.016 (2012).
Bolser, D. M. et al. MetaBase–the wiki-database of biological databases. Nucleic Acids Res 40, D1250–1254, doi:10.1093/nar/gkr1099 (2012).
Xiao, X. et al. The miR-34a-LDHA axis regulates glucose metabolism and tumor growth in breast cancer. Scientific reports 6, 21735, doi:10.1038/srep21735 (2016).
Fu, Q.-F. et al. Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. Journal of Hematology & Oncology 8, 22, doi:10.1186/s13045-015-0117-5 (2015).
Bertini, I., Grassi, E., Luchinat, C., Quattrone, A. & Saccenti, E. Monomorphism of human cytochrome c. Genomics 88, 669–672, doi:10.1016/j.ygeno.2006.07.010 (2006).
De Marchi, E., Bonora, M., Giorgi, C. & Pinton, P. The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium 56, 1–13, doi:10.1016/j.ceca.2014.03.004 (2014).
Pihlajamaki, J. et al. Thyroid hormone-related regulation of gene expression in human fatty liver. J Clin Endocrinol Metab 94, 3521–3529, doi:10.1210/jc.2009-0212 (2009).
Chen, W. W. et al. Inhibition of ATPIF1 ameliorates severe mitochondrial respiratory chain dysfunction in mammalian cells. Cell reports 7, 27–34, doi:10.1016/j.celrep.2014.02.046 (2014).
Loeffen, J. et al. cDNA Sequence and Chromosomal Localization of the Remaining Three Human Nuclear Encoded Iron Sulphur Protein (IP) Subunits of Complex I: The Human IP Fraction Is Completed. Biochemical and Biophysical Research Communications 247, 751–758, doi:10.1006/bbrc.1998.8882 (1998).
Rubattu, S. et al. Ndufc2 Gene Inhibition Is Associated With Mitochondrial Dysfunction and Increased Stroke Susceptibility in an Animal Model of Complex Human Disease. Journal of the American Heart Association: Cardiovascular and Cerebrovascular Disease 5, e002701, doi:10.1161/JAHA.115.002701 (2016).
More, E. et al. Activation of the MAP kinase pathway induces chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) expression in human breast cancer cell lines. J. Endocrinol. 176, 83–94, doi:10.1677/joe.0.1760083 (2003).
Acknowledgements
We thank Dr. Christine Schaner-Tooley for reading the manuscript and suggesting corrections. This research was funded by a grant from the University of Louisville School of Medicine to C.M.K. and by a University of Louisville EVPRI internal grant to C.M.K.
Author information
Authors and Affiliations
Contributions
P.M., N.A.-R., and S.L.V. performed experiments; P.M. and C.M.K. designed experiments; P.M. and C.M.K. performed statistical analyses; P.M. and C.M.K. wrote the manuscript with editing by T.S.K.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muluhngwi, P., Alizadeh-Rad, N., Vittitow, S.L. et al. The miR-29 transcriptome in endocrine-sensitive and resistant breast cancer cells. Sci Rep 7, 5205 (2017). https://doi.org/10.1038/s41598-017-05727-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05727-w
This article is cited by
-
Crosstalk Between miRNA and Protein Expression Profiles in Nitrate-Exposed Brain Cells
Molecular Neurobiology (2023)
-
Regulation of breast cancer metastasis signaling by miRNAs
Cancer and Metastasis Reviews (2020)
-
HNRNPA2/B1 is upregulated in endocrine-resistant LCC9 breast cancer cells and alters the miRNA transcriptome when overexpressed in MCF-7 cells
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
