
Abstract
Fusaric acid (FA), a food-borne mycotoxin, is a potent divalent metal chelator. The human immune system is complex and susceptible to environmental insult however, the immunotoxity of FA remains unknown. We investigated the immunotoxicity of FA on human peripheral blood mononuclear cells (PBMCs) and Thp-1 cells. FA was cytotoxic to PBMCs (IC50-240.8 μg/ml) and Thp-1 (IC50-107.7 μg/ml) cells at 24 h. FA induced early apoptosis but significantly decreased caspase activity in PBMCs, a characteristic of paraptosis. In Thp-1 cells, FA induced apoptosis and increased caspase −9 and −3/7 activities. In PBMCs, FA maintained mitochondrial membrane potential and decreased protein expression of Bax whilst increasing expression of p-Bcl-2; FA induced oxidative stress and depleted ATP levels in both cell types. In Thp-1 cells, FA increased mitochondrial membrane depolarization and decreased p-Bcl-2 expression. In PBMCs, FA significantly up-regulated the MAPK protein expression of p-ERK and p-JNK but down-regulated p-p38 expression. In Thp-1 cells, FA up-regulated MAPK protein expression of p-ERK whilst p-JNK and p-p38 expression were down-regulated. In conclusion FA induced programmed cell death and altered MAPK signaling in healthy PBMCs and Thp-1 cells strongly suggesting a possible mechanism of FA induced immunotoxicity in vitro.
Similar content being viewed by others

Introduction
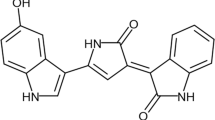
Endophytic fungi produce mycotoxins that are toxic to animals and humans1. Fusaric acid (FA, 5-butylpicolinic acid), is a picolinic acid derivative produced by several strains of Fusarium species2, 3. These fungal strains are ubiquitous in soil and are known to parasitize maize and many other cereal grains4, 5.
FA contains a pyridine ring with a butyl side chain that allows it to easily permeate cell membranes6. The toxicity of FA is also due to its ability to chelate divalent ions such as magnesium, calcium, zinc and iron2, 7. The nitrogen in the pyridine ring and the deprotonated, negatively charged oxygen on the carboxylic acid group are responsible for FA’s divalent metal chelating ability8, 9.
The human immune system functions in host defense against environmental exposure to bacteria, viruses, parasites, fungi and other perturbations, and in acquiring immunity against invading pathogens10, 11. In response to foreign particle or pathogen, several signaling pathways are activated in immune cells12. Foremost of these pathways, is the activation of mitogen-activated protein kinases (MAPKs)12. MAPK activity directs diverse immune responses ranging from stress, cell death/survival and immune defense12,13,14.
Optimal cellular mitochondrial function increases ATP synthesis and reactive oxygen species (ROS) that mediate cell signaling pathways8. The amount of intracellular ROS will significantly influence the MAPK pathway6. The MAPK family comprises of three universal serine/threonine protein kinases; these include the extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 kinase15, 16; each group of MAPK is activated via a series of phosphorylation events16. The first event involves the phosphorylation and activation of a MAPK kinase kinase (MAPKKK), which in turn, phosphorylates and activates a MAPK kinase (MAPKK). MAPKKs activate MAPKs through dual phosphorylation on both threonine and tyrosine residues located within the tri-peptide motif of the MAPK14, 15, 17, 18. Once activated, MAPKs phosphorylate several transcription factors, thereby regulating gene expression and cellular functions13, 14.
Apoptosis is executed by immune cells to maintain homeostasis of the immune system19,20,21. Apoptosis occurs via two main pathways, the intrinsic and extrinsic apoptotic pathways19, 22, 23. Both the intrinsic and extrinsic pathways are activated by caspases; the initiator caspases (−8 and −9) are involved in the intrinsic pathway, whilst the executioner caspases (−3/7) are integral to the extrinsic pathway19, 24. Paraptosis is distinct from necrotic and apoptotic cell death and its features are defined by the lack of apoptotic morphology and independent of caspase activation19, 22, 23, 25,26,27.
The phytotoxicity of FA is well documented and includes altered mitochondrial membrane potential and inhibition of ATP synthesis28, 29. In animals, FA inhibits the activity of dopamine-β-hydroxylase, synthesis of nucleic acids (zinc finger proteins involved in DNA repair) and impairs protein synthesis30. In young swine, FA showed moderate toxicity, induced vomiting and increased concentration levels of tryptophan and serotonin in the brain31. Elevated levels of serotonin results from its impaired regulation and consequently amplifies behaviors distinctive of the firing of serotonergic neurons such as loss of appetite and lethargy32. In zebrafish, FA induced teratogenic effects by inhibition of lysyl oxidase (a copper-dependent enzyme)33. FA also decreased norepinephrine levels in the brain, heart, spleen and adrenal gland of rats34.
To date, no study has investigated the effect of FA on the mammalian immune system. In this study, we assessed the immunotoxicity of FA associated with MAPK activity in healthy human peripheral blood mononuclear cells (PBMCs) and the acute monocytic leukemic (Thp-1) cell line. It was hypothesized that FA altered MAPK signaling was immunotoxic in both cell types. This study shows that FA, a common food borne mycotoxin, is toxic to the human immune system. This data may help develop a better understanding of the immune risks associated with FA consumption. This is of importance in South Africa, the epicenter of infectious diseases, where the majority population relies on maize as a food staple.
Results
Cell viability of PBMCs and Thp-1 cells
The WST-1 assay showed that FA induced a dose dependent decrease in PBMC and Thp-1 cell viability over 24 h (Supplementary Tables: S3-PBMC, S4-Thp-1). Thp-1 cells were more susceptible than PBMCs to FA toxicity. An IC50 of 240.8 μg/ml (Fig. 1A) and 107.7 μg/ml (Fig. 1B) determined for PBMCs and Thp-1 cells respectively; and was used in all subsequent assays.

Cytotoxicity of FA on PBMCs and Thp-1 cells. FA induced a dose dependent decrease in PBMC (A) and Thp-1 (B) cell viability.
FA activates caspase-independent cell death in PBMCs and intrinsic apoptosis in Thp-1 cells
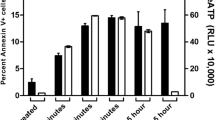
To confirm the toxicity of FA, we assessed the externalization of phosphatidylserine (PS) on the plasma membranes of PBMCs and Thp-1 cells. FA significantly increased the externalization of PS in PBMCs and Thp-1 cells by 1.42 (18.43 + 0.006% vs. 26.16 + 0.003%; p = 0.0003) and 2.27 (8.03 + 0.004% vs.18.19 + 0.002%; p < 0.0001) fold, respectively (Fig. 2). Propidium iodide (PI) staining showed a decreased percentage of necrotic cells by FA as compared to the controls in both PBMCs and Thp-1 cells (Supplementary Fig. S1). This was confirmed by quantifying the release of lactate dehydrogenase (LDH). FA did not induce membrane leakage in both PBMCs and Thp-1 cells (Supplementary Fig. S2), therefore, necrotic cell death was excluded.

Translocation of PS in PBMCs and Thp-1 cells. FA induced PS externalization in both PBMCs and Thp-1 cells. Data are expressed as mean ± SD. **p < 0.005 relative to respective control; ***p < 0.0001 relative to respective control.
Next, we measured caspase −8, −9 and −3/7 activities to determine the type of programmed cell death induced by FA in PBMCs and Thp-1 cells. Interestingly, despite the increase in PS externalization, FA significantly decreased caspase −8 activity by 0.81 fold (1.09 ± 0.001 × 104 RLU vs. 0.88 ± 0.042 × 104 RLU; p = 0.0022), caspase −9 by 0.73 fold (10.89 ± 0.609 × 104 RLU vs. 7.92 ± 0.241 × 104 RLU; p = 0.0070) and caspase −3/7 activities by 0.10 fold (1.19 ± 0.258 × 104 RLU vs. 0.12 ± 0.026 × 104 RLU; p = 0.0035) in PBMCs, relative to the control (Table 1). This result suggests that FA induced caspase-independent cell death in PBMCs. In Thp-1 cells, however, FA significantly decreased caspase −8 activity by 0.74 fold (4.52 ± 0.306 × 104 RLU vs. 3.3265 ± 0.021 × 104 RLU; p = 0.0211), but significantly increased both caspase −9 activity (1.43 fold; 62.67 ± 3.701 × 104 RLU vs. 89.37 ± 0.590 × 104 RLU; p = 0.0065) and caspase −3/7 activity (5.33 fold; 0.82 ± 0.482 × 104 RLU vs. 4.38 ± 0.604 × 104 RLU; p = 0.0041) when compared to the control (Table 2); an indicator of activation of intrinsic apoptosis.
FA induces oxidative stress in PBMCs and Thp-1 cells
Increased intracellular ROS is known to promote oxidative stress, that is not only injurious to cells but may also regulate cell signaling pathways8. Lipid peroxidation, a marker for oxidative stress, was measured by quantifying malondialdehyde (MDA) using the Thiobarbituric acid (TBARS) assay. FA significantly elevated MDA levels in PBMCs (7.59 fold; 0.02 ± 0.010 μM vs. 0.16 ± 0.016 μM; p = 0.0006) and Thp-1 cells (1.59 fold; 0.18 ± 0.020 μM vs. 0.28 ± 0.010 μM; p = 0.0039) (Fig. 3A). FA induced oxidative stress in both normal human PBMCs and Thp-1 cells.

Effect of FA on the oxidative status and mitochondrial function in PBMCs and Thp-1 cells. FA increased MDA levels in both PBMCs and Thp-1 cells (A) had no effect on the mitochondrial membrane potential in PBMCs but increased depolarization of the mitochondrial membrane in Thp-1 cells (B). FA depleted ATP levels in PBMCs and Thp-1 cells (C). Data are expressed as mean ± SD. **p < 0.005 relative to respective control; ***p < 0.0001 relative to respective control.
Mitochondria are important in maintaining cellular redox homeostasis and activation of the intrinsic apoptotic pathway. The evaluation of FA induced mitochondrial membrane integrity by flow cytometry in PBMCs showed that despite increased MDA levels, there was no effect on the mitochondrial membrane potential; however, in Thp-1 cells, mitochondrial membrane depolarization was significantly increased (1.95 fold; 33.58 ± 1.425% vs. 65.48 ± 0.329%; p = 0.0007) when compared to the controls (Fig. 3B).
Further, FA significantly depleted ATP levels in both PBMCs (0.16 fold; 38.97 ± 1.183 × 104 RLU vs. 6.11 ± 0.266 × 104 RLU; p = 0.0002) and in Thp-1 cells (0.52 fold; 225.21 ± 8.014 × 104 RLU vs. 117.26 ± 10.017 × 104 RLU; p = 0.0007) relative to controls (Fig. 3C).
Effect of FA on Bax and p-Bcl-2 protein expression in PBMCs and Thp-1 cells
To validate caspase-dependent and –independent cell death, protein expressions of pro-apoptotic Bax and anti-apoptotic p-Bcl-2 was determined. FA significantly decreased Bax protein expression (0.71 fold; 100 ± 10.33% vs. 70.90 ± 7.34%; p = 0.0201) and increased p-Bcl-2 protein expression (1.18 fold; 100 ± 6.40% vs. 118.30 ± 2.83%; p = 0.0455) in PBMCs compared to the control (Fig. 4A and B), whilst it only significantly decreased the protein expression of p-Bcl-2 (0.78 fold; 99.39 ± 0.89% vs. 77.40 ± 2.82%; p = 0.0007) in Thp-1 cells (Fig. 4C and D).

Effect of FA on apoptotic regulator proteins Bax and p-Bcl-2 in PBMCs and Thp-1 cells. Protein expressions of Bax and p-Bcl-2 in healthy PBMCs (A) and Thp-1 cells (C). Percentage of Bax and p-Bcl-2 protein expression in healthy PBMCs (B) and Thp-1 cells (D). Percentage of protein expressions were represented as mean ± SD. *p < 0.05 relative to respective control; **p < 0.005 relative to respective control.
Effect of FA on MAPKs expression in PBMCs and Thp-1 cells
Finally, to evaluate the effect of FA on MAPK signaling, phosphorylation of MAPK protein expressions was determined using western blotting. In PBMCs, FA significantly increased expression of p-ERK (42 kDa fragment and 44 kDa fragment; 1.94 fold; 100 ± 5.73% vs. 194.19 ± 26.83%; p = 0.0271 and 1.36 fold; 99.80 ± 0.68% vs. 136.20 ± 5.29%; p = 0.0006, respectively) and p-JNK (46 kDa fragment and 54 kDa fragment; 1.46 fold; 97.10 ± 10.06% vs. 141.39 ± 0.92%; p = 0.0035 and 1.13 fold; 96.56 ± 11.92% vs. 108.77 ± 2.45%; p = 0.0454, respectively) (Fig. 5A and B). In Thp-1 cells, FA also significantly increased p-ERK (42 kDa and 44 kDa fragments; 1.35 fold; 100 ± 3.54% vs. 135.13 ± 15.06%; p < 0.0001 and 1.05 fold; 100 ± 5.15% vs. 104.68 ± 6.93%; p = 0.0006, respectively) (Fig. 5C and D), whilst significantly decreasing p-JNK (46 kDa and 54 kDa fragments; 1.02 fold; 105.85 ± 20.25% vs. 108.37 ± 0.75%; p = 0.0461 and 0.62 fold; 103.48 ± 12.05% vs. 63.98 ± 7.03%; p = 0.0055, respectively) (Fig. 5C and D). Furthermore, FA significantly decreased the expression of p-p38 in PBMCs and Thp-1 cells (0.75 fold; 100 ± 9.17% vs. 74.73 ± 11.08%; p = 0.0401 and 0.70 fold; 100 ± 17.40% vs. 69.86 ± 9.89%; p = 0.0033, respectively) (Fig. 5A–D).

Effect of FA on MAPK protein expression. MAPK protein expression in healthy PBMCs (A) and Thp-1 cells (C). Percentage of MAPK protein expression in healthy PBMCs (B) and Thp-1 cells (D). Percentage of protein expressions were represented as mean ± SD. *p < 0.05 relative to respective control; **p < 0.005 relative to respective control; ***p < 0.0001 relative to respective control.
Discussion
The promising role of divalent ion chelators in proliferative and virulent diseases has led to growing interest35,36,37. FA, a picolinic acid analogue and potent divalent metal chelator, has shown potential as an anti-cancer, anti-microbial and anti-viral agent35,36,37. Recently, the role of divalent ion chelators in proliferative and virulent diseases has been extensively studied. However, the toxicity of FA, a food-borne mycotoxin, on the immune system containing a diverse population of cells, has to date not been studied.
In this study, we showed the immunotoxic potential of FA to both healthy PBMCs (diverse population of immune cells) and the distinct immune Thp-1 cells. Interestingly, FA inhibited Thp-1 cell proliferation at an IC50 value less than half that of PBMCs (Fig. 1A and B). This result is in agreement with other studies that showed cytotoxic effects of FA on WI-38 cells (fibroblastic cells), LoVo cells (colorectal adenocarcinoma cells) and MDA-468 cells (human breast adenocarcinoma cells) in which FA had preferentially inhibited the proliferation of cancerous cells (LoVo and MDA-468) when compared to the normal cells (WI-38)35. Our data suggests that FA may exert selective toxicity to distinct immune cell types as evidenced by the Thp-1 response, albeit a leukemic cell line. Additionally, in comparison to the anti-neoplastic drug, ellipticine failed to inhibit PBMC and Thp-1 cell viability; which further exemplifies the potency of FA.
Contrary to the study by Fernandez-Pol (1998), FA significantly increased the externalization of PS in both PBMCs and Thp-1 cells by 26.16% and 18.19%, respectively (Fig. 2). Given that the externalization of PS occurs during both apoptosis and paraptosis, activities of caspases −8, −9 and −3/7 were assayed to determine type of cell death induced by FA in both cell lines; apoptosis requires caspase activation whilst paraptosis is independent of caspase activation. FA substantially decreased caspase activities in PBMCs (Table 1), strongly suggesting that paraptosis was the preferred mode of cell death. In Thp-1 cells, however, FA significantly increased caspase −9 and −3/7 activities were (Table 2), indicating the induction of intrinsic apoptosis in Thp-1 cells.
Immune cells respond to stimuli by activating MAPK signaling to amplify other signals to elicit an appropriate physiological response for programmed cell death14, 38. Previous studies showed that prolonged activation of MAPK signaling induced cell death via ROS-activation of MAPK signaling pathways16. ROS are continuously generated by cellular processes, with the mitochondrion being the major source6. Excessive ROS generated during oxidative phosphorylation can cause oxidative damage to proteins, DNA and phospholipids16; oxidative degradation of lipids results in the formation of lipid peroxides such as MDA39. FA significantly increased MDA levels in both PBMCs and Thp-1 cells (Fig. 3A), indicative of a prolonged oxidative stress environment. Furthermore, FA disrupted mitochondrial membrane potential by increasing mitochondrial depolarization in Thp-1 cells (Fig. 3B). This could be due to the weak acidic nature of the carboxylic acid group of FA40. Weak acids act as proton carriers across lipid membranes, thereby disrupting the proton gradient along the electron transport chain (ETC)41, 42. Interestingly, FA did not disrupt the mitochondrial membrane potential in normal PBMCs (Fig. 3B) despite the significant depletion of ATP levels in both normal PBMCs and Thp-1 cells (Fig. 3C). In Thp-1 cells, this is substantiated by the decreased cell viability (decreased redox potential) and increased mitochondrial membrane depolarization. Also, the activation of ATP dependent caspases −9 and −3/7 may further deplete ATP levels43. In PBMCs, however, the decreased ATP levels may be due to increased activation and prolonged activation of ATP dependent protein kinases.
Intracellular ROS not only alters cellular integrity but is also important to MAPK signaling cascades6; FA induced increased ROS production and up-regulated protein expression of ERK in Thp-1 cells (Fig. 5C and D). Although ERK signaling pathways are well known for their role in promoting cell survival, recent studies have demonstrated their ability to potentiate apoptosis44. Prolonged activation of ERK may be due to the inhibition of tyrosine phosphatases, a group of enzymes responsible for the removal of phosphate groups on phosphorylated tyrosine residues, hence inactivating the protein3. However, tyrosine phosphatases are sensitive to increased ROS and become oxidized, thereby inhibiting their activity and prolonging ERK activation3, 45.
JNK and p38 MAPK signaling pathways are generally directed towards initiating cell death upon activation by stress signals. Recently, however, these signaling pathways have been associated in both cell death and survival38. In Thp-1 cells, FA significantly decreased p-JNK activation and p-p38 protein expressions (Fig. 5C and D). A study by Pedram et al. (1998) documented the cross-talk between the ERK and JNK MAPKs where the activation of JNK by ERK MAPK was followed by the activation of ERK by vascular endothelial growth factor (VEGF) whilst JNK stimulated ERKs proliferative signaling. Therefore, it can be inferred that a decrease in JNK activity hinders the cross-talk between JNK and ERK MAPKs, preventing survival signaling by ERK46.
Additionally, JNK and ERK MAPKs regulate the expression of Bcl-2 family proteins that are central in regulating the mitochondrial apoptotic death pathway3, 47. Bcl-2 inhibits apoptosis by forming a complex with pro-apoptotic proteins such as Bax47, 48. Phosphorylation of Bcl-2 compromises it’s protein stability and affects dimerization with Bax49. Thus, dissociation from the complex at the mitochondrial membrane leads to the formation of mitochondrial permeability transition pore (mPTP) and subsequent caspase activation50. In support of the increased caspase −9 and −3/7 activities in Thp-1 cells, FA decreased p-Bcl-2 expression resulting in apoptotic cell death (Fig. 4C and D). JNK signaling regulates the expression of Bcl-2 and is up-regulated in response to JNK activation. FA activated ERK death signaling, decreased p-Bcl-2 expression and induced apoptosis in Thp-1 cells, but had no significant effect on Bax expression (Fig. 4C and D). This may be due to the deletion of the p53 gene in the Thp-1 cell line as p53 acts as a transcription factor for Bax expression and recruitment to the mitochondrial membrane24, 51,52,53,54,55. Additionally, increased ERK activity regulates mitochondrial membrane potential56 and corresponds with the increased caspase −9 and −3/7 activities, and the subsequent activation of cell death in Thp-1 cells by FA. In PBMCs, FA increased p-Bcl-2 expression (Fig. 4A and B) and decreased Bax expression (Fig. 4A and B), with a corresponding decrease in caspase −8, −9 and −3/7 activities. Increased p-Bcl-2 expression helps maintain the mitochondrial membrane integrity and subsequent mitochondrial membrane potential by preventing the release of cytochrome c, activation of caspase −9 and the initiation of intrinsic apoptosis, further validating the induction of paraptosis in normal PBMCs by FA.
Although the molecular activation of paraptosis remains unknown, studies have suggested the involvement of MAPK signaling in the induction of cell death. In PBMCs, FA significantly increased the expression of ERK and JNK whilst decreasing p38 expression (Fig. 5A and B). Sperandio et al., (2000) reported that ERK and JNK activity mediated paraptosis stimulation by insulin-like growth factor 1 receptor, and that inhibition of these MAPKs prevented the induction of paraptosis in 293 T cells57. Another study by Yumnam et al., (2014) showed the involvement of ERK MAPK in hesperidin-induced paraptosis of human hepatocellular carcinoma (HepG2) cells58. Sugimori and colleagues (2015) recently showed that activated JNK induced paraptosis induction in HL-60 and NB4 human promyelocytic leukemic cell lines and in bone marrow blasts treated with benfotiamine59. Contrary to the studies by Sperandio et al., (2000) and Yumnam et al., (2014), benfotiamine inhibited the activity of ERK in bone marrow blasts and had no effect on ERK activity in HL-60 and NB4 cell lines57, 58. This suggests that the involvement of MAPK in the induction of paraptosis may be dependent on the cell line and type of activation. Additionally, caspase −9 was reported to be a direct target of ERK MAPK, and that phosphorylation at threonine 125 on caspase −9 inhibits its pro-apoptotic activity60. These findings support the activation of MAPK signaling pathways in the induction of paraptosis in PBMCs treated by FA.
Conclusion
FA is immunotoxic to both healthy PBMCs and Thp-1 cells, albeit at a higher concentration in PBMCs. The cancerous Thp-1 cells are highly susceptible to FA toxicity. Collectively, the results show that the host response to FA exposure augmented MAPK signaling and induction of apoptosis in Thp-1 cells (via the mitochondrial apoptotic pathway) and paraptosis in PBMCs. This study shows that FA, a common food borne mycotoxin, is toxic to the human immune system. This data may help develop a better understanding of the immune risks associated with FA consumption. This has great importance in socio-economically challenged countries where the majority population relies on corn as a food staple.
Material and Methods
Materials
Cell culture reagents for PBMC maintenance and FA (Gibberella fujikuroi) were purchased from Sigma Aldrich (Johannesburg, SA). The Thp-1 cells and media were purchased from ATCC (University Boulevard Manassas, USA) and Scientific group (Johannesburg, SA), respectively. Luminometry reagents were obtained from Promega (Madison, USA).
Cell culture
PBMCs were isolated from whole blood using Histopaque 1077 (Sigma Aldrich) and gradient centrifugation from young healthy males following institutional (University of KwaZulu-Natal) ethical approval (BE057/15) and written informed consent. Collection and use of blood was in compliance with relevant institutional guidelines and procedures. Isolated PBMCs were maintained at 37 °C with 5% CO2 in RPMI 1640 medium (supplemented with 10% FCS, 1% L-glutamine and 1% pencillin-streptomycin). Thp-1 cells were cultured in RPMI 1640 medium supplemented with 10% FCS, 1% L-glutamine, 1% pencillin-streptomycin, 1 mM sodium pyruvate and 0.05 mM β-mecaptoethanol. Thp-1 cells were maintained at 3 × 105 cells/ml in 75 cm3 ventilated flasks at (37 °C, 5% CO2) and were split at a cell count of 8 × 105 cells/ml. Viability of cells was assessed using trypan blue exclusion.
Cell viability
The cytotoxicity of FA on PBMCs and Thp-1 cells was analyzed using the WST-1 reagent. Briefly, PBMC and Thp-1 cells (20,000 cells/well) were seeded into a 96-well microtitre plate. The cells were incubated with varying FA concentrations (30–300 µg/ml) in triplicate (200 µl/well) for 24 h (37 °C, 5% CO2). Ellipticine, anti-neoplastic agent, was used as a positive control to FA toxicity (Data shown in Supplementary Tables: S1 PBMC; S2 Thp-1). A positive control of cells with RPMI only and a negative control with RPMI/WST-1 reagent solution was also seeded. Following incubation, the plate was centrifuged at 24 °C, 400xg for 10 min. The supernatant was then aspirated and 110 µl/well of a RPMI/WST-1 reagent solution (1:10) was added and incubated for 3 h (37 °C, 5% CO2). The optical density of the colorimetric reaction was measured at a wavelength of 450 nm and reference wavelength of 620 nm using a spectrophotometer (Bio-Tek uQuant, Winooski, VT, USA). The percentage cell viability was calculated by standardizing untreated (control) cells to 100% and then comparing FA treated cells to the control cells (Detailed calculation shown as Supplementary Information). The concentration of half maximum inhibition (IC50) was determined using GraphPad Prism v5.0 software. All assays were performed in triplicate, twice independently.
Cell death parameters
PS Externalization
Flow cytometry was performed to determine the externalization of PS. Following treatment, 100 μl of an Annexin V-FITC Fluos solution (1:1:50; annexin V-FITC: PI: staining buffer) was added to each sample (200,000 cells in 100 μl PBS) and incubated in the dark at room temperature (RT, 15 min). Thereafter, the samples (20,000 events) were analyzed for apoptosis on the AccuriTM C6 flow cytometer. The cells were gated to exclude cellular debris using the Fl-1 channel (525 nm) (BD Biosciences, Johannesburg, SA). The results were expressed as a percentage.
LDH activity
The LDH cytotoxicity detection kit (Roche, Mannheim, Germany) was used to confirm damaged/necrotic cells. In brief, cell homogenates (100 μl) were seeded into a 96-well opaque polystyrene microtitre plate in triplicate. Subsequently, 100 μl of a substrate mixture containing a catalyst (diaphorase/NAD+) and dye solution (INT/sodium lactate) was added to each homogenate and incubated in the dark for 25 min (RT). The optical density was measured (500 nm) using a spectrophotometer (Bio-Tek uQuant, Winooski, VT, USA). The results were reported as mean optical density.
Caspase activity
Caspase activities of −8, −9 and −3/7 were determined using luminometry. Cells (20,000 cells/well) were seeded into a 96-well opaque polystyrene microtitre plate in triplicate. 20 µl/well of the reagent (Caspase-Glo® 3/7, Caspase-Glo® 8 and Caspase-Glo® 9 Assays) was added to each sample and incubated in the dark for 30 min (RT). Thereafter, the luminescence was measured on a Modulus™ microplate luminometer (Turner Biosystems, Sunnyvale, USA). All data was expressed as relative light units (RLU).
Evaluation of oxidative stress
Lipid peroxidation
The TBARS was used to determine FA generation of ROS61. Briefly, cell homogenates (400 μl) were added to a 7% phosphoric acid (400 μl) and a thiobarbituric acid (1%W w/v)/butylated hydroxytoluene (0.1 mM) (TBA/BHT) solution. A positive control containing MDA (1 μl) and a negative control containing 3 mM hydrogen chloride were prepared. All samples were heated in a water bath (100 °C, 15 min) and allowed to cool (RT). Thereafter, lipids were extracted with butanol (1.5 ml) and were measured on a spectrophotometer (Bio-Tek uQuant, Winooski, VT, USA) at 532 nm with reference wavelength of 600 nm. The mean optical density for each sample was calculated and divided by the absorption coefficient (156 mM−1). The results were expressed in μM.
Mitochondrial membrane potential
Mitochondrial membrane potential was measured using the JC-1 Mitoscreen kit (BD Biosciences, Johannesburg, SA) and flow cytometry. Briefly, 100 µl of a JC-1 working solution was added to each sample (200,000 cells in 100 µl PBS) and incubated in the dark for 30 min (RT). Following incubation, 100 µl flow cytometry sheath fluid was added to each sample and were analyzed on the AccuriTM C6 flow cytometer. A total of 20 000 events were gated using AccuriTM C6 flow cytometer Fl-1 channel (525 nm) (BD Biosciences, Johannesburg, SA). The results were expressed as a percentage.
ATP levels
Intracellular ATP levels were measured using the ATP CellTitre Glo reagent (Promega, Madison, USA). Following treatment, 20,000 cells/well were seeded into a 96-well opaque polystyrene microtitre plate in triplicate. The reagent (20 μl/well) was added to each sample and incubated in the dark for 30 min (RT). Thereafter, the luminescence was measured on a ModulusTM microplate luminometer (Turner Biosystems, Sunnyvale, USA). The data was expressed as relative light units (RLU).
Western blotting
Western blots were performed to analyze the protein expressions of p-ERK, p-JNK, p-p38, Bax and p-Bcl-2. Briefly, total protein was isolated using CytobusterTM reagent (Novagen, San Diego, CA, USA). Cells were re-suspended in 200 µl Cytobuster and incubated on ice for 30 min. Following incubation, the cells were centrifuged for 10 min (180xg, 4 °C). Protein samples were quantified by the bicinchoninic acid (BCA) assay and standardized to 0.2 mg/ml (PBMCs) and 1.0 mg/ml (Thp-1). The samples were then boiled in Laemmli Sample buffer (dH20, 0.5 M Tris-HCl (pH 6.8), glycerol, 10% SDS, β-mercaptoethanol, 1% bromophenol blue) for 5 min (100 °C). Thereafter, the samples (25 μl- Thp-1; 40 μl- PBMC) were electrophoresed in sodium dodecyl sulphate polyacrylamide (SDS-PAGE) gels (4% stacking gel, 7.5% resolving gel) for 1.5 h at 150 V (Bio-Rad compact power supply). The separated proteins were electro-transferred onto nitrocellulose membranes for 30 min (400 mA) using the Trans-Blot Turbo Transfer system (Bio-Rad). The membranes were then blocked for 1 h (RT) with 5% bovine serum albumin (BSA) in Tris Buffer Saline with tween 20 (TTBS- NaCl, KCl, Tris, dH2O, 0.5% tween 20, pH 7.5) or 5% Non-Fat Dry Milk (NFDM) in TTBS for phospho- and non-phospho- antibodies, respectively. Thereafter, the membranes were incubated with primary antibody [mouse anti-p-ERK (9106), mouse anti-p-JNK (9255), rabbit anti-Bax (5023), rabbit anti-p-Bcl-2 (2827), Cell Signalling, 1: 1000; mouse anti-p-p38 (M8177), β-actin (A3854), Sigma Aldrich, 1: 5000] for 1 h (RT) and then overnight at 4 °C. The membranes were washed 5 times with TTBS (10 min intervals) and incubated (RT) with horseradish peroxidase (HRP)- conjugated secondary antibody [goat anti-rabbit (ab6112), anti-mouse (ab97046), Abcam, 1: 5000] for 1 h. Once more, the membranes were washed 5 times with TTBS (10 min intervals). Protein band images were detected using Clarity Western ECL Substrate (Bio-Rad) and captured using Alliance 2.7 Image documentation system (UViTech, Cambridge, UK). The expression of protein bands was analyzed using UViBand Advanced Image Analysis software v12.14 (UViTech, Cambridge, UK). All proteins were normalized to β-actin before comparison (i.e. control vs. FA treatment. The data was expressed as relative band intensity (RBI).
Statistical analysis
Statistical analysis was performed using GraphPad Prism v5.0 software (GraphPad Software Inc., La Jolla, USA). GraphPad Prism Software was used for the unpaired t-test with Welch’s correction to assess the differences between samples. Level of significance (p) was established at a p < 0.05.
References
Murphy, P. A., Hendrich, S., Landgren, C. & Bryant, C. M. Food mycotoxins: an update. Journal of food science 71, R51–R65, doi:10.1111/jfds.2006.71.issue-5 (2006).
Amin, A. N., Douglas, M. G. & Fernandez-Pol, J. A. (Google Patents WO 2001060349 A2., 2001).
Harper, M. E., Bevilacqua, L., Hagopian, K., Weindruch, R. & Ramsey, J. J. Ageing, oxidative stress, and mitochondrial uncoupling. Acta Physiologica Scandinavica 182, 321–331, doi:10.1111/j.1365-201X.2004.01370.x (2004).
Bacon, C., Porter, J., Norred, W. & Leslie, J. Production of fusaric acid by Fusarium species. Applied and Environmental Microbiology 62, 4039–4043 (1996).
Wang, H. & Ng, T. Pharmacological activities of fusaric acid (5-butylpicolinic acid). Life sciences 65, 849–856, doi:10.1016/S0024-3205(99)00083-1 (1999).
Kim, D., Park, G. & Hur, D. Apoptotic signaling through reactive oxygen species in cancer cells. World J Immunol 4, 158–173 (2014).
Kuznetsov, A. V., Margreiter, R., Amberger, A., Saks, V. & Grimm, M. Changes in mitochondrial redox state, membrane potential and calcium precede mitochondrial dysfunction in doxorubicin-induced cell death. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1813, 1144–1152, doi:10.1016/j.bbamcr.2011.03.002 (2011).
Gutterman, D. D. Mitochondria and reactive oxygen species an evolution in function. Circulation research 97, 302–304, doi:10.1161/01.RES.0000179773.18195.12 (2005).
Kwiatkowski, M., Sund, C., Ylikoski, J., Mukkala, V. M. & Hemmilä, I. (Google Patents EP 0298939 A1., 1989).
Calder, P. C. & Kew, S. The immune system: a target for functional foods? British Journal of Nutrition 88, S165–S176, doi:10.1079/BJN2002682 (2002).
MacGillivray, D. M. & Kollmann, T. R. The role of environmental factors in modulating immune responses in early life. Frontiers in immunology 5, 434, doi:10.3389/fimmu.2014.00434 (2014).
He, W. et al. Naturally activated Vγ4 γδ T cells play a protective role in tumor immunity through expression of eomesodermin. The Journal of Immunology 185, 126–133, doi:10.4049/jimmunol.0903767 (2010).
Morrison, D. K. MAP kinase pathways. Cold Spring Harbor perspectives in biology 4, a011254, doi:10.1101/cshperspect.a011254 (2012).
Roux, P. P. & Blenis, J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiology and molecular biology reviews 68, 320–344, doi:10.1128/MMBR.68.2.320-344.2004 (2004).
Dhillon, A. S., Hagan, S., Rath, O. & Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290, doi:10.1038/sj.onc.1210421 (2007).
Son, Y. et al. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? Journal of signal transduction 2011 (2011).
Boldt, S., Weidle, U. H. & Kolch, W. The role of MAPK pathways in the action of chemotherapeutic drugs. Carcinogenesis 23, 1831–1838, doi:10.1093/carcin/23.11.1831 (2002).
Cuenda, A. & Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1773, 1358–1375, doi:10.1016/j.bbamcr.2007.03.010 (2007).
Auffray, C., Sieweke, M. H. & Geissmann, F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annual review of immunology 27, 669–692, doi:10.1146/annurev.immunol.021908.132557 (2009).
Devitt, A. & Marshall, L. J. The innate immune system and the clearance of apoptotic cells. Journal of leukocyte biology 90, 447–457, doi:10.1189/jlb.0211095 (2011).
Kaushik, S., Pandav, S. & Ram, J. Neuroprotection in glaucoma. Journal of postgraduate medicine 49, 90–5, doi:10.4103/0022-3859.917 (2003).
Marchi, S. et al. Mitochondria-ros crosstalk in the control of cell death and aging. Journal of signal transduction 2012 (2011).
Sperandio, S. et al. Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death & Differentiation 11, 1066–1075, doi:10.1038/sj.cdd.4401465 (2004).
Akashi, M., Osawa, Y., Koeffler, H. P. & Hachiya, M. p21WAF1 expression by an activator of protein kinase C is regulated mainly at the post-transcriptional level in cells lacking p53: important role of RNA stabilization. Biochemical Journal 337, 607–616, doi:10.1042/bj3370607 (1999).
Zhang, Y. et al. Kinase AKT controls innate immune cell development and function. Immunology 140, 143–152, doi:10.1111/imm.2013.140.issue-2 (2013).
Bury, M. et al. Ophiobolin A induces paraptosis-like cell death in human glioblastoma cells by decreasing BKCa channel activity. Cell death & disease 4, e561, doi:10.1038/cddis.2013.85 (2013).
Wang, Y. et al. An alternative form of paraptosis-like cell death, triggered by TAJ/TROY and enhanced by PDCD5 overexpression. Journal of cell science 117, 1525–1532, doi:10.1242/jcs.00994 (2004).
D’Alton, A. & Etherton, B. Effects of fusaric acid on tomato root hair membrane potentials and ATP levels. Plant Physiology 74, 39–42, doi:10.1104/pp.74.1.39 (1984).
Pavlovkin, J., Mistrik, I. & Prokop, M. Some aspects of the phytotoxic action of fusaric acid on primary Ricinus roots. Plant Soil and Environment 50, 397–401 (2004).
Bharathiraja, B., Jayakumar, M., Ramaprabu, C. & Jayamuthunagai, J. Production Of Fusaric Acid From Cassava Using Fusarium oxysporum MTCC 1755. Production of Fusaric Acid From Cassava using Fusarium oxysporum MTCC 1755 9, 26–31 (2010).
Smith, T. & MacDonald, E. Effect of fusaric acid on brain regional neurochemistry and vomiting behavior in swine. Journal of animal science 69, 2044–2049, doi:10.2527/1991.6952044x (1991).
Smith, T. Recent advances in the understanding of Fusarium trichothecene mycotoxicoses. Journal of animal science 70, 3989–3993, doi:10.2527/1992.70123989x (1992).
Yin, E. S. et al. Fusaric acid induces a notochord malformation in zebrafish via copper chelation. BioMetals 28, 783–789, doi:10.1007/s10534-015-9855-7 (2015).
Terasawa, F. & Kameyama, M. The clinical trial of a new hypotensive agent, “fusaric acid (5-butylpicolinic acid)”: the preliminary report. Japanese circulation journal 35, 339–357, doi:10.1253/jcj.35.339 (1971).
Fernandez-Pol, J. A. (Google Patents US 5767135 A., 1998).
Stack, B. C. et al. Fusaric acid: a novel agent and mechanism to treat HNSCC. Otolaryngology–Head and Neck Surgery 131, 54–60, doi:10.1016/j.otohns.2004.02.038 (2004).
Pan, J.-H. et al. Antimycobacterial activity of fusaric acid from a mangrove endophyte and its metal complexes. Archives of pharmacal research 34, 1177–1181, doi:10.1007/s12272-011-0716-9 (2011).
Zhang, W. & Liu, H. T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell research 12, 9–18, doi:10.1038/sj.cr.7290105 (2002).
Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN oncology 2012 (2012).
Parks, S. K., Chiche, J. & Pouysségur, J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nature Reviews Cancer 13, 611–623, doi:10.1038/nrc3579 (2013).
Alberts, B. et al. Electron-transport chains and their proton pumps. (2002).
van Beilen, J., de Mattos, M. T., Hellingwerf, K. & Brul, S. Distinct Effects of Sorbic Acid and Acetic Acid on the Electrophysiology and Metabolism of Bacillus subtilis. Applied and environmental microbiology 80, 5918–5926, doi:10.1128/AEM.01391-14 (2014).
McIlwain, D. R., Berger, T. & Mak, T. W. Caspase functions in cell death and disease. Cold Spring Harbor perspectives in biology 5, a008656, doi:10.1101/cshperspect.a008656 (2013).
Park, J.-I. Growth arrest signaling of the Raf/MEK/ERK pathway in cancer. Frontiers in biology 9, 95–103, doi:10.1007/s11515-014-1299-x (2014).
Ray, P. D., Huang, B.-W. & Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular signalling 24, 981–990, doi:10.1016/j.cellsig.2012.01.008 (2012).
Pedram, A., Razandi, M. & Levin, E. R. Extracellular signal-regulated protein kinase/Jun kinase cross-talk underlies vascular endothelial cell growth factor-induced endothelial cell proliferation. Journal of Biological Chemistry 273, 26722–26728, doi:10.1074/jbc.273.41.26722 (1998).
Hemmati, P. G. et al. Adenovirus-mediated overexpression of p14^ A^ R^ F induces p53 and Bax-independent apoptosis. Oncogene 21, 3149–3161, doi:10.1038/sj.onc.1205458 (2002).
Wan, K. F., Chan, S.-L., Sukumaran, S. K., Lee, M.-C. & Victor, C. Y. Chelerythrine induces apoptosis through a Bax/Bak-independent mitochondrial mechanism. Journal of Biological Chemistry 283, 8423–8433, doi:10.1074/jbc.M707687200 (2008).
Huang, S.-T. J. & Cidlowski, J. A. Phosphorylation status modulates Bcl-2 function during glucocorticoid-induced apoptosis in T lymphocytes. The FASEB journal 16, 825–832, doi:10.1096/fj.01-0852com (2002).
Zhou, F., Yang, Y. & Xing, D. Bcl‐2 and Bcl‐xL play important roles in the crosstalk between autophagy and apoptosis. FEBS journal 278, 403–413, doi:10.1111/j.1742-4658.2010.07965.x (2011).
Chipuk, J. E., Maurer, U., Green, D. R. & Schuler, M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer cell 4, 371–381, doi:10.1016/S1535-6108(03)00272-1 (2003).
Chipuk, J. E. et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303, 1010–1014, doi:10.1126/science.1092734 (2004).
Sugimoto, K. et al. Frequent mutations in the p53 gene in human myeloid leukemia cell lines. Blood 79, 2378–2383 (1992).
Ng, K. W. et al. The role of the tumor suppressor p53 pathway in the cellular DNA damage response to zinc oxide nanoparticles. Biomaterials 32, 8218–8225, doi:10.1016/j.biomaterials.2011.07.036 (2011).
Zhao, Z. et al. p53 loss promotes acute myeloid leukemia by enabling aberrant self-renewal. Genes & Development 24, 1389–1402, doi:10.1101/gad.1940710 (2010).
Zhuang, S. & Schnellmann, R. G. A death-promoting role for extracellular signal-regulated kinase. Journal of Pharmacology and Experimental Therapeutics 319, 991–997, doi:10.1124/jpet.106.107367 (2006).
Sperandio, S., de Belle, I. & Bredesen, D. E. An alternative, nonapoptotic form of programmed cell death. Proceedings of the National Academy of Sciences 97, 14376–14381, doi:10.1073/pnas.97.26.14376 (2000).
Yumnam, S. et al. Hesperidin induces paraptosis like cell death in hepatoblatoma, HepG2 cells: Involvement of ERK1/2 MAPK. PloS one 9, e101321, doi:10.1371/journal.pone.0101321 (2014).
Sugimori, N. et al. Paraptosis cell death induction by the thiamine analog benfotiamine in leukemia cells. PloS one 10, e0120709, doi:10.1371/journal.pone.0120709 (2015).
Allan, L. A. et al. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nature cell biology 5, 647–654, doi:10.1038/ncb1005 (2003).
Bruce, A. J. & Baudry, M. Oxygen free radicals in rat limbic structures after kainate-induced seizures. Free Radical Biology and Medicine 18, 993–1002, doi:10.1016/0891-5849(94)00218-9 (1995).
Acknowledgements
This study was supported by grants from the National Research Foundation (Grant UID: 98378) and the College of Health Science (UKZN).
Author information
Authors and Affiliations
Contributions
S.D. performed all experiments and analyzed data. S.D., S.N. and A.A.C. wrote main manuscript text. A.A.C. designed the study. D.B.N. assisted in cell culture and analyzed data. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dhani, S., Nagiah, S., Naidoo, D.B. et al. Fusaric Acid immunotoxicity and MAPK activation in normal peripheral blood mononuclear cells and Thp-1 cells. Sci Rep 7, 3051 (2017). https://doi.org/10.1038/s41598-017-03183-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-03183-0
This article is cited by
-
Targeting paraptosis in cancer: opportunities and challenges
Cancer Gene Therapy (2024)
-
Interconnections among major forms of regulated cell death
Apoptosis (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
