
Abstract
Hulless barley (Hordeum vulgare L. var. nudum) is a barley variety that has loose husk cover of the caryopses. Because of the ease in processing and edibility, hulless barley has been locally cultivated and used as human food. For example, in Tibetan Plateau, hulless barley is the staple food for human and essential livestock feed. Although the draft genome of hulless barley has been sequenced, the assembly remains fragmented. Here, we reported an improved high-quality assembly and annotation of the Tibetan hulless barley genome using more than 67X PacBio long-reads. The N50 contig length of the new assembly is at least more than 19 times larger than other available barley assemblies. The new genome assembly also showed high gene completeness and high collinearity of genome synteny with the previously reported barley genome. The new genome assembly and annotation will not only remove major hurdles in genetic analysis and breeding of hulless barley, but will also serve as a key resource for studying barley genomics and genetics.
Measurement(s) | RNA • transcriptome • DNA • genome • sequence_assembly • sequence feature annotation |
Technology Type(s) | RNA sequencing • DNA sequencing • sequence assembly process • sequence annotation |
Sample Characteristic - Organism | Hordeum vulgare |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.12162147
Similar content being viewed by others

Background & Summary
Hulless barley (Hordeum vulgare L. var. nudum) is a monophyletically originated variety of barley that has loose husk cover of the caryopses1 (Fig. 1). While many domesticated barley varieties are hulled and are mainly used for brewing malt and animal feed, hulless barley has been cultivated on a small scale and used as human food because of the ease in processing and edibility1. Although the cultivation of hulless barley is widely distributed, the frequency decreases from east to west2. The most frequently cultivated area is the Tibetan plateau, e.g. Nepal and Tibet, where hulless barley accounts for more than 95% of domesticated barley and is the staple food for people and an important livestock feed. Recently, hulless barley is also increasingly attracting attention as a potential crop for the development of value-added products and multiple food applications3.

Morphology of a Tibetan hulless barley. These pictures show (a) seedling; (b) heading stage; (c) mature stage; (d) filling stage spike; (e) grain of a hulless barley cultivated in Tibet.
Obtaining genomic sequences is critical for efficient molecular breeding and understanding of the evolutionary history of crops. Recently, studies, including one from our own group, have made significant progress in sequencing the genomes of hulless barley. Using the short-read sequencing approach, the genomes of two hulless barley strains that were grown in Tibet were sequenced and assembled4,5. The results suggest that many stress-related genes, which were expanded in hulless barley, might have facilitated the adaptation to the high-altitude environment4 and may provide a useful genetic resource for improving barley. Furthermore, by sequencing a population of 437 accessions, a study also showed that the current Tibetan hulless barley cultivars were derived from eastern domesticated barley and were introduced to southern Tibet between 4,500 and 3,500 years ago6. However, due to its large genome size and rich in transposable element sequences (80.8–84%)4,7,8, the genome assemblies of hulless barley using short-read sequencing approach remain incomplete and fragmented. This constrains the molecular breeding in hulless barley and the use of hulless barley for food applications.
Here, using a long-read sequencing technique (Pacific Biosciences), we sequenced a Tibetan hulless barley cultivar (Lasa Goumang) that has been previously sequenced using short-read, in high coverage (>67X). Using both Pacbio long reads and available Illumina reads, we assembled a significantly improved genome, of which the N50 contig size reached ~1.56 Mb (Table 1). Based on this improved assembly, we re-annotated the protein-coding genes in hulless barley and anchored the scaffolds to a linkage map of the barley cv. Morex9. The improved genome assembly and annotations will not only serve as a key resource for exploring the economic and genetic values of hulless barley varieties, but will also advance researches in barley genomics and genetics.
Methods
DNA isolation, libraries construction and sequencing
The seedlings were germinated from seeds of a Tibetan hulless barley (cultivar Lasa Goumang, NCBI BioSample ID SAMN09914874). Tissues were flash-frozen in liquid nitrogen and stored in the freezer until DNA extraction. DNA was extracted using the cetyltrimethyl ammonium bromide (CTAB) method10. The quality of the extracted genomic DNA was checked using electrophoresis on 1% agarose gel and the concentration was quantified using a Qubit fluorometer (Invitrogen, Carlsbad, CA, USA).
Single-molecule real-time (SMRT) long-reads sequencing was performed at NextOmics Technology Corporation (Wuhan, China) with a PacBio sequel sequencer (Pacific Biosciences, Menlo Park, CA, USA). The SMRT Bell library was prepared using a DNA Template Prep Kit (1.0). In total, six 20-kb SMRT Bell libraries were constructed. Genomic DNA (~10 μg) was mechanically sheared to fragments of approximately 20 kb using a Covaris g-TUBE. The fragment size distribution was assessed using a bioanalyzer 2100 12 K DNA Chip assay. A blunt-end ligation reaction followed by exonuclease treatment was conducted to generate the SMRT Bell template. The size-selection of SMRT Bell templates was performed using a BluePippin size-selection system (Sage Science) to enrich large fragments (>10 kb). The quality and quantity of size-selected libraries were assessed on a bioanalyzer12Kb DNA Chip (Agilent) and a Qubit fluorometer (Life Technologies), respectively. The SMRT bell libraries were prepared using the binding kit 2.0 (PacBio p/n 100-862-200) according to the manufacturer’s instructions. The libraries were sequenced using a PacBio Sequel instrument on PacBio SMRT cells v2.0 (Pacific Biosciences, acquiring one movie of 360 min per SMRT cell). The MagBead loading (Pacific Biosciences) method was used to improve the enrichment of the larger fragments. In total, ~300 Gb subreads sequences (average length: 9,358 bp) were generated on 64 SMRT cells.
RNA isolation and Iso-Seq sequencing
For RNA samples, plants were grown in a climate chamber in the laboratory (Lhasa). Roots, stems and leaves were sampled seven weeks after germination. To have sufficient materials for each RNA sample, we pooled plant tissue from 10 plants. Samples were placed on dry ice during sample collection and stored in −80 °C freezer until RNA isolation. In total, five pooled samples (one root, two stem and two leaf samples) were collected. Samples were ground with liquid nitrogen and total RNA was extracted using TRIzol reagent (Invitrogen) according to manufacturer’s protocol. RQ1 DNase (Promega) was used to remove DNA.
cDNA libraries were prepared using the ClontechSMARTer® cDNA synthesis kit according to the manufacturer’s recommendations. One μg total RNA was used for each of the five samples. Barcoded oligo dT was used to barcode samples. The cDNA products were purified with AMPure PB beads and quality control (QC) was performed on BioAnalyzer 2100 (Agilent). The purified cDNA libraries were pooled in an equal molar ratio. The pooled cDNA (~3.8 μg) was size fractionated using the Sage ELF system. Subsequent re-amplification was performed to yield four libraries (size of 1–2, 2–3, 3–6 and 5–10Kb) to minimize artifacts during large-scale amplification. The pooled PCR products were purified using AMPure PB beads. One to five μg of purified amplicons were subjected to Iso-Seq SMRT Bell library preparation (https://pacbio.secure.force.com/SamplePrep). A total of 17 SMRT cells were sequenced on the PacBio RS II platform using P6-C4 chemistry with 3–4 h movies. In total, 19.68 Gb sequence data (~1.5 million reads) were obtained (Table 2). The average subreads length was 4.0 kb and the average subreads quality was 0.9.
Genome assembly
In our assembly workflow, raw bam files from PacBio Sequel were first converted into subreads in fasta format using the PacBio software BAM2fastx. Then we used the falcon package (https://github.com/PacificBiosciences/falcon) to construct the primary assembly. Error correction was performed using an overlap-based strategy and the error-corrected reads were used to construct the contigs (parameters: length_cutoff = 5000; length_cutoff_pr = 10000). To correct errors in the primary assembly, we used the arrow pipeline from the SMRT link 4 toolkit to polish the genome (https://www.pacb.com/products-and-services/analytical-software/smrt-analysis/). The PacBio reads were aligned to the primary assembly using pbalign and variantCaller was used to call variants.
SSPACE11 was used to construct scaffolds from contigs. First, we aligned previously sequenced Illumina mate-pair libraries (20 kb and 40Kb fragment long)4 to the assembled contigs using bowtie v1.1.212 and constructed scaffolds with SSPACE-STANDARD-3.0. Second, we used PacBio long-reads to further improve the scaffolding using SSPACE-LongRead13. After scaffolding, the assembly contains 1,856 scaffolds, with a N50 contig size of 1.56 Mb and an N50 scaffold size of 4.0 Mb. The assembled genome size is 4.0 Gb (Table 3).
While the genome size of barley cv. Morex is ~5.1 Gb9, our previous work has suggested that the genome size of Tibetan hulless barley is ~4.5 Gb using k-mer analysis4 (Table 1). However, it is well-known that genome size estimation from both k-mer approach and flow cytometry can have substantial standard deviations (e.g., 10%)14,15. To draw concrete conclusion on genome size differences between hulless barley and cv. Morex, additional experiments are required. However, this is beyond the scope of this study.
We further generated pseudochromosomes using the assembled scaffolds and linkage map of barley cv. Morex9. We used blastn to map the marker sequences of cv. Morex genome to the scaffolds. Only uniquely mapped markers with coverage greater than 0.8 and identity greater than 0.95 were considered. To anchor the scaffolds to pseudochromosomes, ALLMAPS16 was used (Fig. 2a). The synteny comparison between the newly assembled Tibetan hulless barley and barley cv. Morex (Fig. 3a) was performed using CoGe platform17 (https://genomevolution.org/coge/).

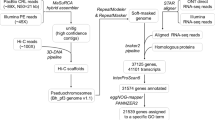
The workflows of genome assembly and annotation used in this study. (a) Genome assembly pipeline; (b) Protein-coding gene annotation pipeline. Software and tools were indicated at lines, data and database information were shown in rectangles.

Genome comparison between Tibetan hulless barley and barley cv. Morex. (a) The plot shows the LAST alignments of predicted protein-coding genes in barley cv. Morex assembly and Tibetan hulless barley assembly. (b–d) Dot plots show the MUMMER alignments of Tibetan hulless barley scaffolds and assembled barley cv. Morex bacterial artificial chromosome sequences. Different coverages were shown in B (65%), C (85%) and D (99.9%). The differences can be due to either true sequence divergences or assembly errors.
Repetitive sequences annotation
Repetitive DNA sequences are highly abundant in many organisms and their variations in abundancy resulted in remarkable genome size variations in plant18. In many Gramineae crop plants, repetitive elements represent more than 80% of their genome8,19,20. Repetitive elements can be classified as simple repeats and transposable elements (TE). Using tandem repeats finder21, we annotated ~155 Mb (3.89%) sequences as simple repeats. To annotate TE, we used both homology-based and de novo TE annotation tools: RepeatMasker22, RepeatProteinMask22, RepeatModeler (http://www.repeatmasker.org/RepeatModeler/) and LTR_FINDER23. For RepeatMasker, Repbase 21.0124 was used. In total, ~87.5% of the assembled genome were identified as TE (Table 4).
Protein-coding gene prediction
For the annotation of protein-coding genes, we used a previously established gene annotation pipeline (Fig. 2b) with minor modifications4. For de novo gene prediction, we first extracted the full-length transcripts from the Iso-Seq data using SMRTLINK. In total, 39,442 full-length transcripts were obtained and were subsequently aligned to the assembled genome using GMAP25. Among the 38,013 aligned transcripts, we removed all transcripts that had coverage less than 0.9 or sequence identity less than 0.85. This resulted in 14,099 high-quality full-length transcripts, which were used for open reading frame (ORF) prediction by TransDecoder (https://github.com/TransDecoder). In total, 13,936 (98.8%) transcripts contained at least one open reading frame (ORF) that is larger than 50 amino acids. These ORF containing transcripts were then assigned to 9,360 genes, which were considered as authentic genes. The authentic genes were then used for training the gene prediction models using AUGUSTUS v3.2.326. Based on the trained models, AUGUSTUS predicted 128,400 putative genes.
For homology based gene prediction, we used the protein sequences of seven monocot species (Triticum urartu (progenitor of wheat A genome)20, Triticum tauscii (progenitor of wheat D genome)27, Brachy podiumdistachyon28, Hordeum vulgare8, Oryza sativa29, Sorghum bicolor30 and Zea mays31) from public databases. All protein sequences were aligned to the hulless barley genome using tblastn32. The gene structure was predicted using GeneWise33 with the input protein sequence as reference.
To provide further evidence for evaluating the predicted gene models, we assembled the transcriptome using available RNA-seq Illumina short-reads from different libraries34. The transcriptome was assembled using both reference-guided approach (mapping: hisat235, assembly: stringtie36,37) and de novo approach (Trinity pipeline38). The reference-guided approach resulted in 47,490 transcripts and the de novo approach resulted in 722,803 transcripts.
The full-length transcripts from Iso-Seq, assembled transcripts from short-reads were used as evidence to evaluate the predicted gene models using EVidenceModeler39. For the data integration, evidence from different sources was assigned to different weight parameters: 20 for Iso-Seq assembly, 8 for short-reads assembly, 5 for homology-based prediction, 2 for AUGUSTUS gene prediction. In total, 129,269 transcripts were obtained, and the structural optimization was performed using PASA. We removed transcripts that either do not show any homology to sequences in nr or uniport database (blast results with identity > = 50% and coverage > = 50%) or have no protein sequences containing any Pfam40 domain (hmmer results with e-value < = 1e-5). For each gene, only the transcript with the longest protein sequences was kept. The tandem duplicated genes were identified using MCScanX. Genes (4,530) that contain large TE sequences (90% coverage) were discarded. The pipeline generated 61,303 genes.
We further classified these genes into high-confidence (HC) genes (40,457), which are likely true protein-coding genes, and less reliable low-confidence (LC) genes (20,846), which potentially are fragmented genes, pseudogenes and/or non-coding genes. This was done in a two-step procedure as described previously8.
We annotated putative functions of the 61,303 protein sequences using public databases, including nr, KEGG41, SwissProt42, Trembl42, GO43, PFAM40, and InterPro44. Blastp was used to compare the predicted protein sequences with the protein databases(e-value < = 1e-5). Blast2GO45 was used to annotate the GO terms using nr database (downloaded in December 2017) with default parameters. The protein domains were annotated using PfamScan46 and InterProScan47 based on InterPro protein databases, including CATHGene3D48, HAMAP49, PANTHER50, PIRSF51, PRINTS52, ProDom53, PROSITE54, SMART55, SUPERFAMILY56 and TIGRFAMs57.
Non-coding gene prediction
tRNAs were annotated using tRNAscan-SE v1.3.158 and rRNAs were annotated using blastn with the rRNA sequences from Arabidopsis thaliana and Oryza sativa (5S rRNA: AJ307354, 5.8S rRNA: AJ232900, 18S rRNA: X16077, 28S rRNA: AH001750). In addition, we also used INFERNAL to predict the miRNA and snRNA.
Data Records
The genomic Pacbio sequencing data (SRS3725794) and Iso-Seq sequencing data (SRS4809149) are available in NCBI Sequence Read Archive under SRP15912959. The available Illumina genome sequencing data that was deposited under SRP0550424,60 was used in our genome assembly and validation processes. The final genome assembly and annotation was deposited at NCBI GenBank under SDOW0000000061 and NCBI Assembly under GCA_004114815.162. The previously generated RNA sequencing data (deposited in NCBI’s Sequence Read Archive under SRP07487063) was used in our genome annotation steps. All the files in this project, such as the assembled scaffolds, repeat annotation, gene predictions and gene function annotations were uploaded to figshare64.
Technical Validation
We evaluated the quality of the new assembly using three independent approaches. First, we mapped 682.57 Gb previously generated genomic Illumina paired-end reads4,60 to the assembled genome. Overall, ~99.9% of paired-end reads were mapped to the genome concordantly. Second, using the mapped short reads, we estimated the quality value (QV) of the assembly using a previously described method65 in which erroneous bases in the genome assembly were identified based on the variant calling software Genome Analysis Toolkit (GATK)66. The estimated base pair error rate is 6.3E-06, suggesting high accuracy of the assembly at base-pair level. Third, we mapped available RNA-seq Illumina reads to the new assembly34 using bowtie2 (parameters:--sensitive --score-min L,0,-0.1 -k 200 --no-discordant --gbar 99999999 --dpad 0 -p 24 --no-mixed -X 1000 --mp 1,1 -I 1 --np 1). The mapping rate with the new assembly (more than 73.6%) is more than 10% higher than the previous assembly. Fourth, we also mapped the PacBio long reads to the new assembly using blasr (parameters: -m 4 --minMatch 8 --minPctIdentity 70 --bestn 1 --nCandidates 20 --maxScore -500). Overall, 91.2% PacBio long-reads can be mapped back to the assembly. Fifth, we downloaded the assembled bacterial artificial chromosome (BAC) sequences from barley cv. Morex67 and mapped them back to the new hulless barley assembly using mummer68 (Fig. 3b–d). Among 299 BACs that were larger than 200 kb and can be mapped back to barley cv. Morex, 74.6% showed high collinearity with the new hulless barley assembly (coverage greater than 60%). Sixth, we also used BUSCO69 to assess the genome completeness. Among 1,440 conserved eukaryotic core genes, 1,378 (95.7%) were complete, 15 were fragmented and 47 were missing in the hulless barley genome assembly. Together, the results suggested that the newly assembled hulless barley genome is of high quality and will serve as a key resource for future research in barley genetics and genomics.
Code availability
The software mentioned in methods section are described blow.
1) falcon: version 1.2.4, parameters: (length_cutoff=5000, length_cutoff_pr=10000);
2) pbalign: contained in SMRT Link 4 toolkit, parameters:(--algorithm=blasr);
3) variantCaller: contained in SMRT Link 4 toolkit, parameters:(--algorithm=arrow);
4) bowtie: verion 1.1.2, default parameters;
5) SSPACE-STANDARD: version 3.0, parameters:(-p 1 -g 2);
6) SSPACE-LongRead: version 1-1, default parameters;
7) blasr: version 1.3.1.121193, default parameters;
8) GATK: version 4.0.0.0, default parameters;
9) nucmer: contained in mummer version 4.0.0beta2, default parameters;
10) BUSCO: version 3, parameters:(-l embryophyta_odb9);
11) Tandem repeats finder: version 409, default parameters;
12) RepeatMasker: version open-4.0.7, parameters: (-nolow -no_is -norna -engine ncbi -parallel 1);
13) RepeatProteinMask: version open-4.0.7, parameters:(-engine ncbi -noLowSimple -pvalue 0.0001);
14) RepeatModeler: version open-1.0.11, parameters:(-engine ncbi -pa 9);
15) ltr_finder: version 1.06, parameters:(-w 2);
16) GMAP: version 2017-05-08, parameters:(-z sense_force -f samse -n 0);
17) TransDecoder: version 4.0.1, default parameters;
18) augustus: version 3.2.3, default parameters;
19) tblastn: version 2.6.0+, parameters:(-evalue 1e-5 -seg no);
20) GeneWise: version 2.4.1, default parameters;
21) hisat2: version 2.1.0, parameters:(--dta --no-discordant --no-mixed);
22) StringTie: version 1.2.4, default parameters;
23) EVidenceModeler: version 1.1.1, parameters:(--weights:PROTEIN GeneWise:5, TRANSCRIPT StringTie:8, ABINITIO_PREDICTION AUGUSTUS:2, OTHER_PREDICTION transdecoder:20);
24) Trinity: version 2.4.0, parameters: (--group_pairs_distance 500 --path_reinforcement_distance 80 --min_glue 3 --min_kmer_cov 3 --min_contig_length 100 --KMER_SIZE 25 --bflyHeapSpaceInit 1G --bflyHeapSpaceMax 4G --bfly_opts “-V 5 --edge-thr=0.1 --stderr”);
25) PASA: version 2.1.0, default parameters;
26) MCScanX: latest version, default parameters;
27) tRNAscan-SE: version 1.3.1, default parameters; 28) infernal: version 1.1.1, default parameters.
28) tRNAscan-SE: version 1.3.1, default parameters; 28) infernal: version 1.1.1, default parameters.
References
Taketa, S. et al. Monophyletic origin of naked barley inferred from molecular analyses of a marker closely linked to the naked caryopsis gene (nud). Theoretical and Applied Genetics 108, 1236–1242 (2004).
Zeng, X. et al. Origin and evolution of qingke barley in Tibet. Nature Communications 9, 5433 (2018).
Izydorczyk, M. S., Chornick, T. L., Paulley, F. G., Edwards, N. M. & Dexter, J. E. Physicochemical properties of hull-less barley fibre-rich fractions varying in particle size and their potential as functional ingredients in two-layer flat bread. Journal of food science and technology 108, 561–570 (2008).
Zeng, X. et al. The draft genome of Tibetan hulless barley reveals adaptive patterns to the high stressful Tibetan Plateau. Proceedings of the National Academy of Sciences of the United States of America 112, 1095–1100 (2015).
Dai, F. et al. Assembly and analysis of a qingke reference genome demonstrate its close genetic relation to modern cultivated barley. Plant Biotechnology Journal 16, 760–770 (2018).
Fu, D. X., Ruan, R. W., Dai, X. M. & Liu, Y. M. A study on ancient barley, wheat and millet discovered at Changguo in Tibet. Acta Agronomica sinica 26, 392–398 (2000).
Mayer, K. et al. A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716 (2012).
Mascher, M. et al. A chromosome conformation capture ordered sequence of the barley genome. Nature 544, 427–433 (2017).
Mascher, M. et al. Anchoring and ordering NGS contig assemblies by population sequencing (POPSEQ). The Plant Journal 76, 718–727 (2013).
Rogers, S. O. & Bendich, A. J. In Plant Molecular Biology Manual (eds. Stanton B. Gelvin, Robbert A. Schilperoort & Desh Pal S. Verma) 73–83 (Springer Netherlands, 1989).
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D. & Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579 (2011).
Langmead, B. Aligning short sequencing reads with Bowtie. Current protocols in bioinformatics, Chapter 11, Unit 11.7 (2010).
Boetzer, M. & Pirovano, W. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics 15, 211 (2014).
He, K., Lin, K., Wang, G. & Li, F. Genome sizes of nine insect species determined by flow cytometry and k-mer analysis. Frontiers in physiology 7, 569 (2016).
Guo, L. T. et al. Flow cytometry and K-mer analysis estimates of the genome sizes of Bemisia tabaci B and Q (Hemiptera: Aleyrodidae). Frontiers in physiology 6, 144 (2015).
Tang, H. et al. ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biology 16, 3 (2015).
Lyons, E. & Freeling, M. How to usefully compare homologous plant genes and chromosomes as DNA sequences. The Plant Journal 53, 661–673 (2008).
Paterson, A. H. Leafing through the genomes of our major crop plants: strategies for capturing unique information. Nature Reviews Genetics 7, 174–184 (2006).
Schnable, P. S. et al. The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115 (2009).
Ling, H. Q. et al. Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 557, 424–428 (2018).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 25, 4.10.1–14.10.14 (2009).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 (2005).
Stanke, M. & Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, ii215–ii225 (2003).
Luo, M. C. et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551, 498–502 (2017).
Initiative, I. B. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768 (2010).
Du, H. et al. Sequencing and de novo assembly of a near complete indica rice genome. Nature Communications 8, 15324 (2017).
McCormick, R. F. et al. The Sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. The Plant Journal 93, 338–354 (2018).
Jiao, Y. et al. Improved maize reference genome with single-molecule technologies. Nature 546, 524–527 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of molecular biology 215, 403–410 (1990).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Research 14, 988–995 (2004).
Wei, Z. et al. Comparative transcriptome analysis revealed genes commonly responsive to varied nitrate stress in leaves of Tibetan hulless barley. Frontiers in Plant Science 7, 1067 (2016).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33, 290–295 (2015).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature Protocols 11, 1650–1667 (2016).
Grabherr, M. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29, 644–652 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7 (2008).
Bateman, A. et al. The Pfam protein families database. Nucleic Acids Research 32, D138–D141 (2004).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Research 40, D109–D114 (2012).
Boeckmann, B. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research 31, 365–370 (2003).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nature Genetics 25, 25–29 (2000).
Mitchell, A. et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Research 43, D213–D221 (2015).
Conesa, A. & Gotz, S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. International Journal of Plant Genomics 2008, 619832, 12 (2008).
Mistry, J., Bateman, A. & Finn, R. D. Predicting active site residue annotations in the Pfam database. BMC Bioinformatics 8, 298 (2007).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Lees, J. G. et al. Gene3D: multi-domain annotations for protein sequence and comparative genome analysis. Nucleic Acids Research 42, D240–D245 (2014).
Pedruzzi, I. et al. HAMAP in 2013, new developments in the protein family classification and annotation system. Nucleic Acids Research 41, D584–D589 (2013).
Mi, H. et al. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Research 38, D204–D210 (2010).
Nikolskaya, A. N., Arighi, C. N., Huang, H., Barker, W. C. & Wu, C. H. PIRSF family classification system for protein functional and evolutionary analysis. Evolutionary Bioinformatics Online 2, 197–209 (2006).
Attwood, T.K. et al. The PRINTS database: a fine-grained protein sequence annotation and analysis resource–its status in 2012. Database: The Journal of Biological Databases and Curation 2012, bas019 (2012).
Bru, C. et al. The ProDom database of protein domain families: more emphasis on 3D. Nucleic Acids Research 33, D212–D215 (2005).
Sigrist, C. J. A. et al. New and continuing developments at PROSITE. Nucleic Acids Research 41, D344–D347 (2013).
Letunic, I., Doerks, T. & Bork, P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Research 40, D302–D305 (2011).
de Lima Morais, D. A. et al. SUPERFAMILY 1.75 including a domain-centric gene ontology method. Nucleic Acids Research 39, D427–D434 (2011).
Haft, D. H. et al. TIGRFAMs and genome properties in 2013. Nucleic Acids Research 41, D387–D395 (2013).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research 25, 955–964 (1997).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP159129 (2018).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP055042 (2015).
Zeng, X. et al. Hordeum vulgare cultivar Lasa Goumang, whole genome shotgun sequencing project. Genbank, https://identifiers.org/ncbi/insdc:SDOW00000000 (2019).
NCBI Assembly, https://identifiers.org/ncbi/insdc.gca:GCA_004114815.1 (2019).
NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP074870 (2016).
Xu, T. et al. An improved high-quality genome assembly and annotation of Tibetan hulless barley. Figshare, https://doi.org/10.6084/m9.figshare.7582181.v5 (2019).
Xia, M. et al. Improved de novo genome assembly and analysis of the Chinese cucurbit Siraitia grosvenorii, also known as monk fruit or luo-han-guo. Gigascience 7, 6 (2018).
Heldenbrand, J. R. et al. Performance benchmarking of GATK3.8 and GATK4. Preprint at, https://doi.org/10.1101/348565v1 (2018).
IBSC. (ed IPK Gatersleben) (Plant Genomics and Phenomics Research Data Repository (PGP), IPK Gatersleben, 2016).
Kurtz, S. et al. Versatile and open software for comparing large genomes. Genome Biology 5, R12 (2004).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This research was supported by the following funding sources: the Tibet Autonomous Region Financial Special Fund 2017CZZX001, 2017CZZX002, and the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2019QZKK0502). We acknowledge Zuoyi Jian for helping on the genome assembly, Hao Tang for developing genome annotation pipeline and Zhen Zeng for annotating repetitive sequences. We are grateful to the help from Zhenhua Zhuang and Jie Li during project development, and Martin Schäfer for the proof reading. We thank NextOmics and BGI-Shenzhen for sequencing supports.
Author information
Authors and Affiliations
Contributions
Project coordination: X.Z., T.X., Y.W., Z.L., S.X. and N.T.; Sampling and experimentation: X.Z., Y.W., H.Y., Q.X., S.Z., W.Q., Y.B., J.D. and M.Y.; Supervision on genome assembly, genome annotation: T.X.; Data analysis: T.X., S.X., X.Z., Y.W., H.Y., Q.X., S.Z., W.Q., Y.B., J.D. and M.Y.; Data submission: T.X.; Writing: T.X., H.Y. and S.X. All authors read and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Zeng, X., Xu, T., Ling, Z. et al. An improved high-quality genome assembly and annotation of Tibetan hulless barley. Sci Data 7, 139 (2020). https://doi.org/10.1038/s41597-020-0480-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-020-0480-0
This article is cited by
-
Identification of transcriptionally active transposons in Barley
BMC Genomic Data (2023)
-
Inferring regulatory element landscapes and gene regulatory networks from integrated analysis in eight hulless barley varieties under abiotic stress
BMC Genomics (2022)
-
Accumulation and regulation of anthocyanins in white and purple Tibetan Hulless Barley (Hordeum vulgare L. var. nudum Hook. f.) revealed by combined de novo transcriptomics and metabolomics
BMC Plant Biology (2022)
-
Pathogenesis-related protein-4 (PR-4) gene family in Qingke (Hordeum vulgare L. var. nudum): genome-wide identification, structural analysis and expression profile under stresses
Molecular Biology Reports (2022)
-
Genome-wide identification and expression profile under abiotic stress of the barley non-specific lipid transfer protein gene family and its Qingke Orthologues
BMC Genomics (2021)
