
Abstract
Epitope-specific enzymes are powerful tools for site-specific protein modification but generally require genetic manipulation of the target protein. Here, we describe the laboratory evolution of the bacterial transpeptidase sortase A to recognize the LMVGG sequence in endogenous amyloid-β (Aβ) protein. Using a yeast display selection for covalent bond formation, we evolved a sortase variant that prefers LMVGG substrates from a starting enzyme that prefers LPESG substrates, resulting in a >1,400-fold change in substrate preference. We used this evolved sortase to label endogenous Aβ in human cerebrospinal fluid, enabling the detection of Aβ with sensitivities rivaling those of commercial assays. The evolved sortase can conjugate a hydrophilic peptide to Aβ42, greatly impeding the ability of the resulting protein to aggregate into higher-order structures. These results demonstrate laboratory evolution of epitope-specific enzymes toward endogenous targets as a strategy for site-specific protein modification without target gene manipulation and enable potential future applications of sortase-mediated labeling of Aβ peptides.

Similar content being viewed by others
Main
The ability to covalently modify proteins enables researchers to effectively interrogate and perturb their biological functions. Most purely chemical methods for protein labeling modify many proteins in a biological mixture and yield heterogeneous products that are difficult to characterize1. While technologies such as unnatural amino acid incorporation2,3, inteins4, small molecule-reactive peptides5 and epitope-specific enzymes6 enable chemo- and site-selective modification in biological systems, they typically require genetic manipulation of the protein of interest to introduce an amber stop codon or peptide tag, potentially altering its biological properties and limiting applicability to settings where target gene manipulation is possible. The ability to manipulate endogenous proteins in a site-specific manner would enable target labeling even in complex biological mixtures and would be especially useful when genetic manipulation is impractical. To explore this possibility, we sought to evolve a versatile epitope-specific enzyme to recognize and covalently modify a peptide sequence natively present in a pathogenic protein.
Sortase transpeptidases are a superfamily of enzymes widely distributed throughout Gram-positive bacteria7. Staphylococcus aureus sortase A (SrtA) is responsible for attaching proteins that contain a C-terminal LPXTG sorting sequence to the cell wall8. The enzyme cleaves between the threonine and glycine of the sorting sequence, forming an acyl-enzyme intermediate that subsequently acylates the primary amine of the pentaglycine of the peptidoglycan9. SrtA shows a strong preference for its LPXTG sorting sequence10, but studies have revealed that it will accept a variety of glycine-based (and some non-glycine) nucleophiles11. These properties make SrtA an attractive tool for site-specific protein modification. Indeed, SrtA has been successfully used for both C-terminal and N-terminal protein labeling, as well as protein circularization and semisynthesis of multi-domain proteins12,13,14,15,16,17.
Engineering of sortases for improved activity on both their cognate and new substrates has been an area of active research for almost a decade18,19. Our group previously used yeast display and fluorescence-activated cell sorting (FACS) to improve the kinetics of SrtA on LPETG20 and to evolve sortase variants that accept single amino acid substitutions at the second or fourth position of the recognition sequence21. In this study, we sought to reprogram the specificity of SrtA to covalently modify the Alzheimer’s disease (AD)-associated Aβ protein. The formation of Aβ plaques in the central nervous system is the hallmark of AD22. Despite the clinical importance of Aβ, its physiological functions and its role in AD pathogenesis are not clearly understood23,24,25. The ability to modify Aβ site-specifically might help illuminate its biological role, impede Aβ plaque formation or facilitate our understanding of AD pathogenesis. Since Aβ monomers are predominantly extracellular26, unstructured27,28 and contain a five-amino-acid sequence (LMVGG at residues 34–38) that shares features with sortase’s native recognition sequence, sortase-mediated conjugation is an attractive strategy to achieve site-specific modification of Aβ.
Over 16 rounds of evolution we generated a sortase variant, SrtAβ, which mediates the covalent modification of Aβ peptides. We used SrtAβ to biotinylate and detect endogenous Aβ in clinical cerebrospinal fluid (CSF) samples at concentrations of 2–19 ng ml−1. We also demonstrated that SrtAβ-mediated conjugation of a hydrophilic pentapeptide to Aβ42 greatly slows the initiation of detectable aggregation. This work establishes the evolution of sortase enzymes to site-specifically modify naturally occurring proteins without requiring modification of endogenous genes.
Results
Initial evolution of SrtA to recognize Aβ
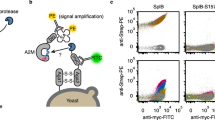
We sought to evolve SrtA variants that modify Aβ using yeast display20,29,30,31 and FACS (Fig. 1). Briefly, a yeast display library of sortase variants is conjugated to triglycine peptides with N termini that are free for sortase-catalyzed reactions. The library is then incubated with an N-terminally biotinylated target substrate and non-biotinylated off-target substrates. Sortase variants that catalyze transpeptidation between triglycine and the target substrate biotinylate the surfaces of the yeast cells that encode them. Activity on off-target substrates by promiscuous sortase variants leads to reduced biotinylation of the cells that encode them. After removal of cell surface-displayed sortases with Tobacco Etch Virus (TEV) protease (Supplementary Fig. 1), cells are stained with fluorophore-linked streptavidin and the biotinylated cells encoding active and selective sortase variants are isolated by FACS (Extended Data Fig. 1).

A population of yeast displays a library of approximately 107 SrtA variants: (1) triglycine is conjugated to the surface of each cell with Sfp; (2) cells are incubated with biotinylated target substrate and non-biotinylated off-target substrates; (3) after allowing the SrtA variants to catalyze transpeptidation between triglycine and the added substrates, cells are washed and the SrtA variants are removed from their surfaces using TEV protease. Cells are labeled with an anti-HA antibody (green) to quantify sortase expression and streptavidin-phycoerythrin (red) to quantify transpeptidation between triglycine and the positive selection substrate. Active sortase variants (blue) have higher on-target transpeptidation per unit expression than inactive variants (yellow) or promiscuous variants and can be isolated by FACS; (4) collected cells were grown, reinduced and further enriched for target recognition.
We started our evolution from a library of sortase variants previously evolved to recognize LPESG substrates (library 4S.6). Given that our target sequence, LMVGG, deviates from the wild-type (WT) sorting sequence, LPXTG, at the second and fourth positions, we reasoned that mutants already possessing altered substrate recognition at the fourth position were a more promising starting point than WT SrtA. We diversified this starting pool by error-prone PCR to create the round 1 library of 4.8 × 107 variants. To identify variants that preferred glycine over serine at the fourth position, we used biotinylated LPVGG as an initial positive selection substrate. The stringency of the screen was gradually increased by decreasing the biotinylated LPVGG concentration and increasing the off-target non-biotinylated LPESG substrate concentration (Supplementary Table 1). We isolated individual clones after five cycles of enrichment. Prominent mutations from round 1 included R94P, S118I, G134R and V189F (Table 1).
We rediversified the pool by error-prone PCR to create the round 2 library. This library showed sufficient activity on biotinylated LMVGG to permit FACS using this substrate. As in round 1, stringency was increased by reducing the amount of positive selection substrate while increasing the amounts of negative selection substrates, in this case LPVGG and LMVTG (Supplementary Table 1). After round 2, we observed that the S118I, G134R and V189F mutations from round 1 had persisted but that the identity of residue 94 was diverse (tyrosine, leucine, arginine, proline, histidine or glutamine) among sequenced clones. In addition, approximately 98% of sequenced clones had mutations at residue 124 (aspartic acid to glycine, leucine or tyrosine).
Rounds 3–7 consisted of iterative cycles of diversification by error-prone PCR and FACS screening for activity on biotinylated LMVGG with progressively higher stringencies (Supplementary Table 1). At the end of round 3, we observed a clone that represented 3.5% of the population and contained the new mutations K138I, V182A, T196S and R197S, in addition to the previously observed mutations R94Y, S118I, D124L, G134R and V189F. By the end of round 4, this clone represented 74% of the population (Table 1), suggesting a substantial fitness benefit from some combination of K138I, V182A, T196S and R197S.
The most common sequence emerging from round 5 (36% of the population) was the round 4 consensus sequence plus an I123L mutation. I123L was the most common new mutation emerging in round 5, present in 67% of sequenced clones. Notably, the V182A, T196S and R197S mutations that first appeared at the end of round 3 reached 100% prevalence in the population. Following two additional rounds of diversification and sorting, the consensus sequence of the round 7 pool (29% of the population) contained R94Y, S118I, I123L, D124L, G134R, K138I, K173E, V182A, V189F, T196S and R197S (Table 1). Analysis of previous sequencing data showed that this clone first appeared at the end of round 5, where it made up 9% of the population.
Of these 11 mutations, we were particularly interested in V182A, T196S and R197S because of their early prevalence. Additionally, mutations at residues 182 and 196 were previously observed in SrtA variants with improved activity or single-position altered substrate recognition20,21, while residue 197 is a crucial part of the active site in WT SrtA32,33,34,35. We generated the round 8 library using site saturation mutagenesis at these three positions followed by error-prone PCR. Mutations V182A, T196S and R197S remained fixed in sequences emerging from round 8, confirming the fitness advantage afforded by these three mutations. Additional well-represented mutations that appeared in round 8 include K62R (present in 84% of sequenced clones), I76L (60%), the reversion mutation Y94R (62%), N107D (20%), N127Y (15%), I138L (15%), K145T (15%), M155I (20%), R159C (15%), K173E (57%), K177R (24%) and F189I (35%).
SrtA evolution in human plasma
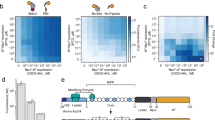
While the above screens for sortase activity on LMVGG were conducted in Tris-buffered saline (TBS) buffer, our goal of modifying Aβ in endogenous contexts requires that the evolved enzyme be active in biological fluids. Our previous work revealed that sortase enzymes evolved for LPESG recognition—including clone 4S.6, the starting point of this study—are capable of modifying fetuin-A in human plasma, presumably through its native LPPAG sequence21. Indeed, a fourfold molar excess of a round 8 clone also supported labeling of purified fetuin-A in Dulbecco’s PBS (Supplementary Fig. 2a) and overnight incubation of human plasma with 50 μM of evolved sortase and 1 mM of GGGK(biotin) also led to fetuin-A labeling (Supplementary Fig. 2b). To evolve decreased recognition of fetuin-A, we conducted additional rounds of evolution with negative selection against the LPPAG sequence of fetuin-A. This negative selection was achieved by including the LPPAG peptide in our sortase reaction mixtures (round 9) and by conducting the sortase reactions directly in human plasma (rounds 10–16). Over these eight rounds of evolution, we generated a sortase variant with greatly reduced activity on fetuin-A relative to the starting sortase 4S.6 (Supplementary Fig. 2c,d).
Between the end of round 8 and the beginning of round 9, we generated and analyzed the activity of a series of single reversion mutants from the round 7 consensus sequence. These data (Supplementary Fig. 3) revealed the importance of mutations at residues 94, 123 and 124. As such, we conducted site saturation mutagenesis at these three residues and adjacent residue 122, followed by error-prone PCR to generate the round 9 library. Increased off-target LPPAG concentration and decreased reaction times were used to increase selection stringency over the course of round 9 screening (Supplementary Table 1). Sequencing the pool at the end of round 9 revealed enrichment of many mutations that we first observed in round 8. This included K62R (up to 100% from 84% of sequenced clones), I76L (up to 97% from 60%), the reversion mutation Y94R (up to 91% from 62%), N107D (up to 61% from 20%), I138L (up to 42% from 15%), K145T (up to 55% from 15%), R159C or H (up to 21% and 53% from 15% and 4%, respectively), K173E (up to 85% from 57%), K177R (up to 72% from 24%) and F189I (up to 72% from 35%) (Table 1).
To maintain selection against fetuin-A recognition while introducing selection against other motifs that exist in human plasma, we conducted the sortase reactions for our screens directly in human plasma from round 10 onward; 100-fold-higher concentrations of biotinylated LMVGG were initially needed to observe sortase conjugation in human plasma than were needed to observe conjugation in TBS, suggesting that specific labeling of the desired target is more difficult in plasma (Supplementary Table 1). Analysis of the round 10 sequencing results showed further enrichment of N107D (present in 95% of sequenced clones), N127Y (56%), M155I (35%) and K145T (98%). The Y94R reversion, I138L, R159C and K173E all reached 100% abundance by the end of round 10. New mutations included N127H (27%) and Q172H (65%). Round 11 resulted in further enrichment of N127H (present in 75% of sequenced clones), M155I (70%), Q172H (100%) and appearance of the G139D (28%). N172H and M155I were further enriched in round 12 (both to 87% abundance), where an E105D mutation appeared in 32% of clones. By the end of round 13, E105D was found in 88% of sequenced clones.
Our inability to use more stringent conditions in round 14 than in round 13 (Supplementary Table 1), coupled with the low convergence of the resulting pools, prompted us to use DNA shuffling in an attempt to escape a potential fitness plateau. We shuffled the round 14 pool with the evolved SrtA pentamutant in a 1:1 ratio and subjected the products to error-prone PCR to create the round 15 library. In round 15, we used Aβ40 conjugated to biotin at its N terminus through an aminohexanoic acid linker (biotinylated LC-Aβ40) as the target substrate instead of biotinylated LMVGG to ensure activity on the full target peptide and not only the recognition motif. In round 16, we used a 1:1 mixture of biotinylated LC-Aβ40 and biotinylated LC-Aβ42 to select for activity on two different Aβ alloforms. The most notable mutation to emerge in round 16 was S102C, which was present in all 4 of the most active individual clones. Other mutations of note include M141I, K152R and K206R. Given the level of activity already observed and the lack of additional strongly enriching mutations after round 13, we ended the evolution campaign and characterized the evolved sortase enzymes.
Flow cytometry analysis of the pools at the end of each screening round revealed an upward trend in activity on the LMVGG substrate (from round 1 through to round 9; Extended Data Fig. 2). An initial downward trend in pool activity was observed on switching to human plasma as the reaction buffer. This downward trend was reversed in round 12, but activity dropped and plateaued again in rounds 13 and 14. The noticeable increase in activity from round 14 to round 15 both in TBS and plasma (43% increase in TBS, 54% in plasma) suggested that DNA shuffling was a successful strategy to escape an apparent fitness plateau; the overall trend in strongly increased activity between round 1 and round 16 confirmed a successful evolutionary campaign for a SrtA variant with activity on Aβ.
Characterization of key mutants and mutational analysis
At the end of round 8, individual variants were isolated by sorting single cells into a 96-well plate. The activity of 32 evolved sortase clones towards LMVGG and LPVGG was assessed by a flow cytometry assay (Extended Data Fig. 3). Clone 8.5-H3 demonstrated the best combination of activity on LMVGG and selectivity over LPVGG. When expressed and purified, this variant was active on LMVGG in an established HPLC assay for SrtA activity36, converting 10% of 10 μM of substrate to product in 2 h, an improvement from previous rounds (Extended Data Fig. 4). Kinetic parameters for 8.5-H3 were determined to be Kcat = 0.012 s−1 (95% confidence interval (CI) = 0.009–0.017 s−1) to with KM = 52 μM (95% CI = 29–103 μM) using an established fluorescence assay36,37. Western blot analysis revealed that variant 8.5-H3 conjugated a variety of Aβ isoforms to GGGK(biotin), demonstrating that sortases evolved to process LMVGG also show activity on Aβ (Extended Data Fig. 5).
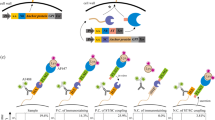
Individual variants from round 16 were sorted and reassayed for LMVGG activity at the end of the round. The top variant from round 16 (SrtAβ) was assayed by flow cytometry on a panel of substrates, which revealed a greatly altered activity profile from the starting enzyme 4S.6 (Fig. 2a). The results were consistent with positive selection for activity on LPVGG in round 1 and LMVGG in subsequent rounds with negative selection against LPESG in round 1 and against LPPAG in rounds 9–16. Compared to the starting enzyme 4S.6, SrtAβ had a 53-fold reduced activity on LPESG, 11-fold reduced activity on LPPAG and 28-fold increased activity on LMVGG (Fig. 2a). SrtAβ had a 30-fold preference for LMVGG over LPESG, whereas 4S.6 had a 49-fold preference for LPESG over LMVGG. Overall, SrtAβ evolved a 1,470-fold change in preference to favor LMVGG over LPESG.

a, The evolved SrtAβ and starting enzyme 4S.6 were displayed on yeast and assayed for their ability to catalyze transpeptidation on different substrates. b, SrtAβ, 4S.6 and all 25 single reversion mutants were displayed on yeast and assayed for their ability to catalyze transpeptidation between triglycine and biotinylated LMVGG. Reversion mutants with activity less than half that of SrtAβ are highlighted in pink. c, The predicted locations of the 6 reversion mutations that reduce SrtAβ activity by >50% are shown in pink on the NMR solution structure of WT S. aureus SrtA (Protein Data Bank: 2KID). An LPXTG substrate analog is shown in blue and the calcium ion required for activity is shown in orange. Residues 118, 182 and 197 are part of the substrate binding pocket, while other residues are further from the active site. d, The activity of 4S.6, a minimal mutant (4S.6 with S118I, G134R, R159C, K177R, V182A and R197S mutations) and SrtAβ on biotinylated LMVGG were compared by flow cytometry. Addition of these six mutations to 4S.6 improved activity on the LMVGG substrate but was insufficient to confer the level of activity displayed by SrtAβ, highlighting the importance of other mutations. All graphs represent the mean of three replicates ± s.d. Activity is defined as the ratio of cell surface biotinylation (PE) to sortase expression level (FITC).
SrtAβ contains 25 mutations relative to the starting sortase enzyme 4S.6. To determine the relative importance of individual mutations to activity on LMVGG, we reverted each mutation back to its corresponding residue in the starting enzyme. These 25 single-mutant variants were assayed alongside SrtAβ and 4S.6 by flow cytometry (Fig. 2b). Eleven of the reversions reduced enzyme activity by less than 25%, 8 reduced activity between 25 and 50% and 6 reduced enzyme activity by at least twofold. Notably, reversion mutations at residues 118 and 197 reduced activity on the LMVGG substrate greater than 90%, near the low level of activity demonstrated by the starting enzyme 4S.6. Two of these six mutations are at residues that were identified as modulators of sortase substrate specificity in our previous evolution campaigns (residues 118 and 182) (ref. 21), but the remaining four were at new residues. Notably, three of these four new residues were outside of the substrate binding pocket and the fourth, R197, was highly conserved across the sortase superfamily. It has been suggested that R197 in WT S. aureus SrtA stabilizes the binding of the LPXTG sorting signal or the oxyanion intermediates generated during catalysis35. That a nonconservative mutation at this residue is not only tolerated, but required, is surprising. These results highlight the challenge of a priori prediction of mutations that alter SrtA specificity and the importance of including random mutagenesis as a diversification strategy (Fig. 2c)35. A minimal mutant containing these six mutations in the 4S.6 background showed a fourfold improvement in LMVGG activity relative to 4S.6, but 23-fold lower activity than SrtAβ (Fig. 2d). This result confirms that other mutations, although less important individually, collectively contribute to substantially improved target activity. Four of these other mutations, in addition to R177K, are at residues located near the calcium binding site in the WT enzyme. Assaying SrtAβ activity at various calcium concentrations revealed compatibility with a broad range of concentrations (0.1–10 mM) that include physiological calcium concentrations, but confirmed that calcium is still required for activity (Supplementary Fig. 4).
To confirm that the shift in substrate specificity observed in the flow cytometry assay translated to purified enzymes, we determined the kinetic parameters of 4S.6 and SrtAβ on LPESG and LMVGG using an HPLC assay (Extended Data Fig. 6). Sortase 4S.6 showed Kcat = 0.36 s−1 (95% CI = 0.22–0.96 s−1) and KM = 610 μM (95% CI = 90–5,550 μM) on LPESG, whereas SrtAβ activity on LPESG was too low to establish accurate kinetic parameters. SrtAβ had Kcat = 0.018 s−1 (95% CI = 0.015–0.023 s−1) and KM = 128 μM (95% CI = 87–198 μM) on LMVGG, whereas 4S.6 activity on LMVGG was not detectable. These findings confirm that the evolution resulted in a large change in substrate preference, which is consistent with the >1,400-fold change observed in the flow cytometry assays (Fig. 2a).
To obtain a more quantitative understanding of our evolved enzyme’s activity on Aβ40 in plasma, we developed an ELISA to measure biotinylated Aβ. Streptavidin was used to capture biotinylated peptide and detection was accomplished using 4G8, a monoclonal antibody that recognizes Aβ residues 17–24. Aβ40 labeled with GGGK(biotin) was used as the calibrant. Employing this assay, we confirmed SrtAβ activity on Aβ40 spiked into human plasma, with 1.5 μM of SrtAβ generating 2.3 μM of biotinylated product from 5 μM of Aβ40 in 2 h (Extended Data Fig. 7a). As we expected, increasing the amount of GGGK(biotin) nucleophile greatly improved reaction yields (Extended Data Fig. 7b).
Concentrations of Aβ peptides are important biomarkers of AD. This is especially true of Aβ42 in CSF, where a decrease to roughly 50% of baseline Aβ42 levels is typically observed in patients with AD38. To enable labeling and detection of physiologically relevant amounts of Aβ, we changed the format of the ELISA to capture the product with monoclonal antibody m266 (the epitope of which spans Aβ residues 13–26) and detect with horseradish peroxidase (HRP)-conjugated streptavidin. After optimizing the concentrations of various assay components, we consistently detected and quantified Aβ-biotin conjugates at concentrations comparable to commercial Aβ ELISA kits (Supplementary Table 2). The lower limit of quantitation is defined as the lowest standard with a signal higher than the average signal of the blank samples plus 9 s.d. and allows a percentage recovery of 80–120%. In six runs over 6 d, the lower limit of quantitation for our assay was 39–78 pg ml−1 or roughly 10–20 pM. Using this SrtAβ-mediated assay, we observed that labeling of Aβ40 spiked into human plasma at concentrations as low as 5 nM (Extended Data Fig. 7c). Given that typical Aβ concentrations in human CSF are on a similar order of magnitude, these observations suggest the possibility of using SrtAβ to label endogenous Aβ in CSF, where the generation, clearance and aggregation of Aβ are all intimately connected with AD etiology24,39.
SrtAβ labels endogenous Aβ in CSF
The ability to site-specifically modify endogenous Aβ in CSF would provide researchers with new ways to interrogate or influence these dynamic processes. Thus, we sought to demonstrate labeling of endogenous Aβ in CSF. First, we measured Aβ levels in CSF samples using immunoassays specific for Aβ terminating at Val40 or Ala42 (ref. 40). Because sortase-mediated conjugation of Aβ40 and Aβ42 destroys the C-terminal epitopes used for immunodetection, we reasoned that this reaction would cause a loss of ELISA-measured signal. Indeed, after treating the samples with SrtAβ and GGG, we observed losses in signal ranging from 47 to 77%, confirming that the enzyme was active in the CSF (Fig. 3a).

a, Transpeptidation of Aβ40 or Aβ42 with GGG should yield Aβ37-GGG, which is not detected by Aβ40- and Aβ42-specific ELISAs. Treatment of CSF specimens with SrtAβ and GGG caused a substantial reduction in ELISA-measured levels of both Aβ40 and Aβ42. b, Transpeptidation of Aβ40 or Aβ42 with GGGK(biotin) yields Aβ37-GGGK(biotin), which can be detected through its affinity handle. Detectable levels of transpeptidation product are observed in all ten CSF samples. Importantly, no product was observed (the asterisks indicate below the limit of detection) in the absence of SrtAβ or GGGK(biotin). Product yield is defined as the amount of product detected divided by the amount of Aβ40 + Aβ42 measured in each sample. For each labeling experiment, all reactions were set up in triplicate. The bars represent the mean of three replicates ± s.d. The GGG labeling experiment was performed once. The GGGK(biotin) labeling experiment was performed twice. The data presented are representative of both attempts.
While this loss of signal is consistent with transpeptidation, it might also be explained by hydrolysis or interference of the sortase enzyme with the binding of the detection antibody. Loss of signal due to aggregation is unlikely since we have previously shown that incubation of biological samples at room temperature for up to 24 h does not alter detection of Aβ42 (ref. 41). Besides the enzyme and GGG, the only difference between treated and untreated samples was the addition of calcium, a cation known to influence in vitro aggregation of Aβ42. However, CSF already contains micromolar levels of calcium and it is unlikely that a modest increase in calcium would induce aggregation of Aβ present at nanomolar concentrations.
To obtain a more direct readout of transpeptidation activity, we generated AβM1–37-GGGK(biotin) semisynthetically (Supplementary Fig. 5a) and used it as our standard to detect the reaction product generated by SrtAβ-catalyzed conjugation with GGGK(biotin). As before, Aβ peptides were captured using the m266 antibody and detected via HRP-conjugated streptavidin. We observed Aβ labeling efficiencies of 13–56% (Fig. 3b). Given that only transpeptidation can lead to a gain of signal in this assay, these efficiencies are, as expected, lower than those observed with GGG labeling. The lower efficiency is not likely due to SrtAβ preferring GGG over GGGK(biotin) since reactions of chemically synthesized Aβ with equimolar amounts of different triglycine nucleophiles yielded similar amounts of transpeptidation products (Supplementary Fig. 5b). The variable labeling efficiencies across samples suggests that this method requires further optimization for use in the absolute quantification of Aβ. Nonetheless, these data demonstrate the ability of the evolved SrtAβ enzyme to modify endogenous Aβ in human CSF.
Sortagging Aβ42 alters aggregation kinetics
Next, we sought to conjugate Aβ to a molecule that would impede its aggregation. Previous studies showed that the hydrophobic C terminus of Aβ42 is well-resolved in the NMR solution structure of Aβ42 fibrils43. We hypothesized that the replacement of hydrophobic C-terminal residues with more hydrophilic residues would alter the aggregation propensity of the resulting peptides.
To test this possibility, we expressed and purified Aβ42 as reported previously44. Immediately after batch purification, we treated a portion of the recombinant Aβ42 (20 ml of approximately 40 μM) overnight with 20 μM of SrtAβ and 200 μM of GGGRR. Transpeptidation should replace the last five residues of AβM1–42, GVVIA, with GGGRR yielding a more hydrophilic 43-mer. As expected, AβM1–37-GGGRR, the identity of which was confirmed by MS, eluted from reverse-phase HPLC before AβM1–42 (Supplementary Fig. 5c). We then directly compared the aggregation propensity of the HPLC-isolated AβM1–37-GGGRR to that of recombinant AβM1–42 from the same initial batch purification.
Using a continuous thioflavin T (ThT) binding assay45, the SrtAβ-modified peptides were found to take much longer to nucleate into aggregates. The lag time to initiation of detectable aggregation for 20 μM of AβM1–42 occurred within 5 min, whereas the lag time for 20 μM of AβM1–37GGGRR was 8.2 h. The modified peptides also took approximately 40-fold longer to reach half maximal aggregation (0.70 or 0.56 h for 10 or 20 μM of AβM1–42 versus 28 or 14.6 h for 10 or 20 μM of AβM1–37GGGRR) (Fig. 4). The impaired aggregation of SrtAβ-modified AβM1–42 was replicated with recombinant AβM1–37GGGRR (Extended Data Fig. 8). In contrast to the delayed kinetics of aggregation, the maximum ThT signals of C-terminally modified fibrils were higher (46,000 versus 14,000 relative fluorescence units (RFU) at 20 μM, 30,000 versus 5,000 RFU at 10 μM). Thus, while the lag time for AβM1–37GGGRR was much longer than for AβM1–42, the rate of aggregation and the extent of ThT binding was greater for AβM1–37GGGRR. These results indicate that modification of the Aβ C terminus delays nucleation; once nuclei are formed, elongation is rapid and the structure formed binds ThT in a manner distinct from Aβ42. Indeed, EM analysis of aggregation end products revealed substantial ultrastructural differences in the fibrils formed by AβM1–37GGGRR and AβM1–42 (Extended Data Fig. 9). Collectively, these results establish the modification of a disease-associated form of Aβ to a form less prone to aggregation by transpeptidation using a laboratory-evolved sortase enzyme.

ThT binding was used to monitor the aggregation of AβM1–-42 and AβM1–37GGGRR. Data points from the time course are shown for each replicate (n = 3 for AβM1–42, n = 4 for 10 μM of AβM1–37GGGRR and n = 6 for 20 μM of AβM1–37GGGRR) and curves fitted to each replicate by Boltzmann equation are indicated. The initiation of aggregation of the AβM1–37GGGRR monomer was greatly delayed compared to AβM1–42, with an average t1/2 = 14.6 h at 20 μM and 28.3 h at 10 μM (compared to 0.6 and 0.7 h for AβM1–42 at 20 μM and 10 μM, respectively).
Discussion
We used a yeast display selection strategy over many rounds of evolution to generate a sortase enzyme capable of site-specifically modifying Aβ peptides. We leveraged the ability to tailor reaction conditions during selection by lowering the concentration of target substrate, altering the kinetic requirements to survive selection and introducing various decoy off-target substrates, thereby tuning selection stringency for activity and specificity. After 16 total rounds of evolution, we generated a sortase variant that prefers LMVGG 30-fold over LPESG, a large change in specificity from a starting enzyme that prefers LPESG 49-fold over LMVGG. To our knowledge, this work represents the first example of a sortase enzyme evolved for activity on a substrate with mismatches at multiple amino acids in its recognition sequence.
Previous efforts to reprogram sortase activity yielded orthogonal variants that were highly active on singly mutated LAXTG and LPXSG substrates21. The present results demonstrate that it is possible to evolve an epitope-specific enzyme capable of recognizing an endogenous peptide tag in a disease-associated protein (LMVGG, residues 34–38 of Aβ), although the catalytic efficiency of the evolved enzyme (143 M−1s−1) on this new substrate is lower than evolved to recognize the singly mutated targets (approximately 103–104 M−1s−1) (ref. 21). This difference in efficiency largely arises from a lower Kcat since the KM of SrtAβ for LMVGG (128 μM) is improved relative to the KM of 4S.6 for LPESG (610 μM) and falls within the range of KM values reported for other sortase variants evolved by yeast display20,21. In our current scheme, each copy of a given library member is only allowed a single turnover with which to generate signal. Development of a multiple turnover variant of this selection for bond-forming enzymes, perhaps by displaying two sets of Aga2p-fusions on the cell surface46, could facilitate further improvements in turnover number and catalytic efficiency. The identification of new peptide ligases as evolutionary starting points could also broaden access to targets that otherwise would be inaccessible to sortase A47,48,49.
We demonstrated the ability of our evolved enzyme, SrtAβ, to generate conjugates with Aβ monomers, validating the evolution of epitope-specific enzymes as a strategy for site-specific labeling of endogenous peptides. An enzyme capable of site-specific Aβ modification enables a variety of applications. We successfully generated conjugates of purified Aβ with GGGK(biotin) and GGGRR, among other peptides. The ability of sortase enzymes to use a wide array of glycine-based nucleophiles means that the applications of our evolved enzyme with purified Aβ monomers are not limited to those described herein. For example, SrtAβ could be used to generate new Aβ-adjuvant conjugates for vaccine development efforts50.
SrtAβ can also conjugate peptides to endogenous Aβ in human CSF, raising the possibility of biomedical applications on endogenous Aβ. Attachment of fluorophores could enable imaging studies that further our understanding of AD etiology. Tagging monomers to inhibit their aggregation, as demonstrated above, or to mark them for degradation, could modify disease state. As such, SrtAβ or other variants could help illuminate the biological role of Aβ, increase our understanding of AD pathogenesis and potentially contribute to the development of new AD treatments.
Methods
Library diversification by error-prone PCR
Genes were isolated from collected yeast libraries by PCR using the primers pCTCon2CTEV.HR2.Fwd and pCTCon2CTEV.HR2.Rev, purified by gel electrophoresis and subsequently mutagenized by using the GeneMorph II Random Mutagenesis Kit (Agilent Technologies) for 25 cycles of PCR amplification using the primers pCTCon2CTEV.HR2.Fwd and pCTCon2CTEV.HR2.Rev. Reactions were purified by spin column and combined with NheI/BamHI-digested pCTCon2CTev vectors in a 5:1 insert:backbone mass ratio and electroporated into ICY200 as described below to yield yeast libraries: pCTCon2CTEV.HR2.Fwd: 5′-CCCATACGACGTTCCAGACTATGCAGGATCTGAG AACTTGTACTTTCAAGGTGCT-3′; pCTCon2CTEV.HR2.Rev: 5′-CTGTTGTTATCAGATCTCGAGCTATTACAAGTCCT CTTCAGAAATAAGCTTTTGTTCGGA-3′.
Library diversification by site saturation mutagenesis (rounds 8 and 9)
Genes were isolated from collected yeast libraries by PCR using the primers pCTCon2CTEV.HR2.Fwd and pCTCon2CTEV.HR2.Rev, purified by gel electrophoresis and subcloned into pET29 via restriction digest with NheI/BamHI. This plasmid was used as the template for site saturation mutagenesis with polynucleotide kinase-treated primers: 182-NNK-Fwd: 5′-NNKACCTGCGATGATTATAACTTTGAAACCG-3′, 182-NNK-Rev: 5′-CAGGGTCAGCTGTTTATCTTTGCC-3′, 196-197-NNK-Fwd: 5′-NNKNNKAAAATTTTTGTGGCGACCGAAGTG-3′ and 196-197-NNK-Rev: 5′-TTCCCACACGCCGGTTTC-3′ in round 8 and 94-NNK-Fwd-1: 5′-NNKGAACAGCTGGATCGTGGCGTGAGC-3′, 94-NNK-Fwd-2: 5′-NNKGAACAGCTTGATCGTGGCGTGAGC-3′, 94-NNK-Rev: 5′-GGTCGCCGGGCCCGG-3′, 122-124-NNK-Fwd-1: 5′-NNKNNKNNKCGTCCGAACTATCAGTTTACCAACCTG-3′, 122-124-NNK-Fwd-2: 5′-NNKNNKNNKCGTCCGTACTATCAGTTTACCAACCTG-3′ and 122-124-NNK-Rev: 5′-GGTATGGCCGATAATGCTAATGTTCTGATC-3′ in round 9. Site-saturated genes were then amplified out of the pET29c backbone using the primers pCTC-HR-pET29-Fwd and pCTC-HR-pET29-Rev and purified by gel electrophoresis: pCTC-HR-pET29-Fwd: 5′-CCCATACGACGTTCCAGACTATGCAGGATCTGAGAACTTGTACTTTCAAGGTGCTAGCCAGGCGAGACCGCAGATTCC-3′ and pCTC-HR-pET29-Rev: 5′-CTGTTGTTATCAGATCTCGAGCTATTACAAGTCCTCTTCAGAAATAAGCTTTTGTTCGGA TCCTTTCACTTCGGTCGC-3′.
Library diversification by DNA shuffling (round 15)
The library collected from the end of round 14 and the evolved sortase A pentamutant (5 M) were each amplified with pCTCon2CTEV.HR2.Fwd and pCTCon2CTEV.HR2.Rev and purified by gel electrophoresis51. Then, 1 μg of each PCR product was added to 5 μl of 500 mM Tris-HCl pH 7.4, 100 mM of MnCl2 and brought to 50 μl total volume. This mixture was incubated at 15 °C for 5 min at which point 0.5 U of DNase I was added. After 90 s, 1 μl of 500 mM of EDTA was added to the reaction and the enzyme was heat-killed at 90 °C for 10 min. The digest was run on a 3% agarose gel and 25–150-base pair fragments were isolated. Then, 200 ng of DNA fragments were added to a 100-μl primerless reassembly reaction with 5 μl of 4 mM of deoxynucleoside triphosphate, 4 μl of 50 mM of MgSO4, 10 μl of 600 mM of Tris-SO4 (pH 8.9)/180 mM ammonium sulfate, 1 U of Taq polymerase and 1 U of Phusion polymerase. This reaction was cycled at 94 °C for 2 min, then 35 cycles of 94 °C for 15 s, 65 °C for 45 s, 62 °C for 45 s, 59 °C for 45 s, 56 °C for 45 s, 53 °C for 45 s, 50 °C for 45 s, 47 °C for 45 s, 44 °C for 45 s, 41 °C for 45 s, 68 °C for 45 s and then 68 °C 1 min. After PCR cleanup, a portion of the primerless reassembly product was amplified with the primers CJP66-Fwd and CJP66-Rev, digested with NheI/BamHI and ligated into pCTCon2CTev vector: CJP66-Fwd: 5′-GTACTTTCAAGGTGCTAGCC-3′; CJP66-Rev: 5′-CAGAAATAAGCTTTTGTTATC-3′.
Yeast library construction
Fresh plates of ICY200 Saccharomyces cerevisiae cells were streaked from long-term glycerol stocks and grown for 72 h at 30 °C before use. A single colony was picked and grown in 10 ml of yeast extract peptone dextrose (YPD) + 100 U ml−1 of penicillin, 100 μg ml−1 of streptomycin and 100 μg ml−1 of kanamycin overnight with shaking at 30 °C. This suspension culture was freshly diluted into 125 ml of YPD and electrocompetent cells were prepared as described by Chao et al.52. All library transformations were performed by gap repair homologous recombination into pCTCon2CTev vectors linearized by NheI and BamHI digestion. After transformation, 105 and 106 dilutions were plated and used to estimate library size.
Yeast library induction
Libraries were grown in synthetic complete dextrose (SCD)-tryptophan-uracil dropout medium + 100 U ml−1 of penicillin, 100 μg ml−1 of streptomycin and 100 μg ml−1 kanamycin at 30 °C. Library expression was induced by transfer to SGR-tryptophan-uracil medium at 20 °C overnight.
GGGK-coenzyme A synthesis
Fmoc-GGGK-CONH2 was dissolved in DMSO to a final concentration of 100 mM, then combined with 1.5 equivalents of LC-SMCC (Thermo Fisher Scientific) and 2 equivalents of DIPEA (Sigma-Aldrich) in DMSO. The reaction was incubated for 1 h at room temperature, then combined with 1.5 equivalents of CoA trilithium salt hydrate (Sigma-Aldrich) in DMSO to a final peptide concentration of 25 mM and mixed at room temperature overnight. The Fmoc protecting group was removed with 20% vol/vol piperidine and incubation for 20 min. The reaction was quenched by adding 1 equivalent of trifluoroacetic acid (TFA); the product was purified on a preparative Kromasil 100-5-C18 column (21.2 × 250 mm; Peeke Scientific) by reverse-phase HPLC (flow rate: 9.5 ml min−1; gradient: 10–70% acetonitrile with 0.1% TFA in 0.1% aqueous TFA gradient over 30 min; retention time: 17.1 min). Electrospray ionization MS: [M-H]− m/z = 1,300.1 (observed); calculated for C45H72N14O23P3S− = 1,301.4. The concentration of GGGK-CoA peptide was determined from the measured A259 using the known molar extinction coefficient of CoA53—15,000 M−1 cm−1.
Surfactin phosphopantetheinyl transferase expression and purification
Escherichia coli BL21(DE3) harboring the pET29 expression plasmid for surfactin phosphopantetheinyl transferase (Sfp) was cultured at 37 °C in lysogeny broth (LB) with 50 μg ml−1 of kanamycin until OD600 was approximately 0.6. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM and protein expression was induced at 37 °C for 3 h. Cells were collected by centrifugation and lysed by resuspension in B-PER (Thermo Fisher Scientific) containing 260 nM of aprotinin, 1.2 μM of leupeptin, 2 U ml−1 DNase I and 1 mM of phenylmethylsulfonyl fluoride. The clarified supernatant was purified on Ni-NTA agarose and fractions that were >95% pure were consolidated and dialyzed against 10 mM of Tris pH 7.5 + 1 mM of EDTA + 5% glycerol. Enzyme concentration was calculated from the measured A280 using the published extinction coefficient of 27,220 M−1cm−1(ref. 54).
TEV protease expression and purification
E. coli BL21(DE3) harboring the pRK793 plasmid for TEV S219V expression and the pRIL plasmid (Addgene) was cultured in LB with 50 μg ml−1 of carbenicillin and 30 μg ml−1 chloramphenicol until OD600 was approximately 0.7. IPTG was added to a final concentration of 1 mM and cells were induced for 3 h at 30 °C. Cells were pelleted by centrifugation and lysed by sonication. The clarified lysate was purified on Ni-NTA agarose and fractions that were >95% TEV S219V were consolidated and dialyzed against TBS. Enzyme concentrations were calculated from A280 measurements using the reported extinction coefficient of 32,290 M−1 cm−1 (ref. 55).
Yeast library preparation and FACS
Induced cells were pelleted and resuspended in 10 ml of TBS-B (100 mM of Tris pH 7.5, 500 mM of NaCl, 1% BSA). Then, 50 μl of 1 M of MgCl2, 10 μl of 200 mM of H2NGGGK(CoA) and 50 μl of 100 μM of Sfp (10 mM of Tris pH 7.5, 1 mM of EDTA, 10% glycerol) was added to this cell suspension. The Sfp ligation reaction was incubated for 45 min at room temperature. Cells were then pelleted at 2,400g for 10 min and the supernatant was removed. The desired sortase reaction buffer (TBS-BC; 100 mM of Tris pH 7.5, 500 mM of NaCl, 1% BSA, 5 mM of CaCl2 or phosphatidylcholine; human plasma (catalog no.GTX73265; GeneTex) was centrifuged at 21000g for 10 min and passed through a 0.4-μm filter; 5 mM of CaCl2 was then added and the cell pellet resuspended.
Separately, 100× target substrate and negative selection substrates (custom syntheses from GenScript, with the exception of biotinylated LC-Aβ40 and biotinylated LC-Aβ42 obtained from Eri Amyloid Laboratory) were added to Eppendorf tubes. Typically, this involved 3–4 aliquots of varying substrate concentrations such that a range of selection stringencies is represented across the aliquots. Cell suspension was added to the substrates, inverted to mix and incubated for 15–60 min at room temperature. Cells were pelleted and treated with 1 ml of TEV solution (100 μg ml−1 in PBS, 0.5% BSA, 2 mM of EDTA) for 30 min on ice. Cells were pelleted and labeled with antibodies (1:200 streptavidin-phycoerythrin (PE) and 1:250 anti-HA Alexa Fluor488, both from Invitrogen, in PBS, 0.5% BSA and 2 mM of EDTA) for at least 30 min on ice. Cells were pelleted and washed once with 1 ml of PBS, 0.5% BSA and 2 mM of EDTA, then suspended in the same buffer before analysis and sorting on a BD FACS Aria Cell Sorter.
A negative control lacking any biotinylated target substrate was used to draw gates for sortase activity:expression level (PE:fluorescein isothiocyanate (FITC)) (Supplementary Fig. 2). Aliquots that contained target substrate were then analyzed and the number of events in the PE:FITC gate was compared to the negative control. Aliquots that showed a greater than tenfold increase in gated events versus the negative control were considered suitable for sorting. The top 0.5–1.0% of cells were collected from a total number of events at least tenfold greater than the estimated library size.
Cells sorted in active gate were collected in 2 ml SCD-tryptophan-uracil dropout medium + 100 U ml−1 of penicillin, 100 μg ml−1 of streptomycin, 100 μg ml−1 of kanamycin in a 15-ml conical. Collected cells were then divided into 2–4 10 ml SCD-tryptophan-uracil cultures and grown at 30 °C for 2 d before they were induced again for a subsequent sort under more stringent conditions. Increased stringency was most commonly achieved by decreasing target substrate concentration but occasionally by increasing off-target concentration or decreasing reaction times. Cycles of growth, induction and enrichment were iterated until active variants could no longer be isolated using more stringent conditions than those used in the previous cycle, generally about 4–6 times. At this point, the surviving pool was extracted and rediversified to create a library for the next round.
Yeast library collection
After the final FACS screen of a round, yeast were grown to saturation (OD approximately 1.5) in SCD-tryptophan-uracil dropout medium + 100 U ml−1 of penicillin, 100 μg ml−1 of streptomycin and 100 μg ml−1 of kanamycin at 30 °C, then lysed using a Zymo Research Zymoprep II kit according to the manufacturer’s instructions
Isolation of single clones
A portion of the collected plasmid was transformed directly into One Shot Mach1 T1 Phage-Resistant Chemically Competent E. coli cells according to the manufacturer’s instructions (Thermo Fisher Scientific). Then, 36–48 colonies, each bearing a single library member, were picked for rolling circle amplification and subject to Sanger sequencing with the following primers: CA205: 5′-AGGCAATGCAAGGAGTTTTTG-3′; CA232: 5′-CAGTGGGAACAAAGTCGATTTTGTTACATCTAC-3′. Clones of interest were then subcloned into pET29 expression vectors.
Alternatively, at the end of the last sort of a given round, the BD FACS Aria Cell Sorter was switched to plate mode, gates were adjusted to only collect the top 0.1–0.3% of cells and single cells were collected in each well of a 96-well plate. After growing to saturation, these clones were subjected to flow cytometry assays. Top performers were sequenced and then subcloned into pET29 expression vectors.
Reversion mutants
SrtAβ was subcloned into pET29 and used as the PCR template for reactions with the primers in Supplementary Table 3. After USER assembly or KLD ligation, products were transformed into One Shot Mach1 T1 Phage-Resistant Chemically Competent E. coli cells. After sequence verification, the reversion mutants were amplified out of the pET29 backbone with HR primers and transformed into ICY200 with NheI/BamHI-digested pCTCon2CTev vectors in a 5:1 insert:backbone mass ratio to yield yeast bearing single reversion mutants for flow cytometry analysis.
Minimal mutant
SrtA 4S.6 was subcloned into pET29 and used as the PCR template for two reactions, one with the primers 5′-ATCGTCCGAAC/ideoxyU/ATCAGTTTACCAACCTGCGCGCGGCGAAA AAAGGCAGC-3′ and 5′-AGGGTCAGC/ideoxyU/GTCTATCTTTGCCTTTCTGTTCATCCAGC ACTTCC-3′, the other with the primers 5′-AGCTGACCC/ideoxyU/GGCGACCTGCGATGATTAT AACGTGGAAACCG-3′ and 5′-AGTTCGGACGA/ideoxyU/CAATCGCGGTATGGCCGATAA TGCTAATGTTCTGATCATCCAGGC-3′. USER assembly of these two fragments yielded 4S.6 with S118I, G134R, K177R and V182A mutations. This mutant version of 4S.6 was used as template for two further PCRs, one with the primers 5′-ACCAGCATTTGTAA CG/ideoxyU/GAAACCGACCGCGGTGG-3′ and 5′-AAAAATTTTACTGGTT/ideoxyU/CCC ACACGCCGGTTTCCAC-3′, the other with the primers 5′-AAACCAGTAAAATTTT/ideoxyU/G TGGCGACCGAAGTGAAAGGATCC-3′ and 5′-ACGTTACAAATGCTGG/ideoxyU/CATTTTA TATTTACGGGTTTCGTTGC-3′. USER assembly of these two fragments yielded the minimal mutant 4S.6 with S118I, G134R, R159C, K177R, V182A and R197S mutations. This mutant was then amplified out of the pET29 backbone with HR primers and transformed into ICY200 and ligated into NheI/BamHI-digested pCTCon2CTev by homologous recombination.
Yeast transformation with LiAc/ss carrier DNA/PEG
A 10-ml ICY200 starter culture in YPD (100 U ml−1 of penicillin, 100 μg ml−1 of streptomycin and 50 μg ml−1 of kanamycin) was grown overnight at 30 °C. Cells were centrifuged at 2,500g for 10 min before removal of the supernatant and two washes with 25 ml of water. Cells were resuspended in 1 ml of water and transferred to a 1.5-ml Eppendorf tube. Cells were pelleted and washed once more before being resuspended in 1 ml of water and split into 100-μl aliquots. Aliquots were pelleted and the supernatant was removed. Then, 240 μl of PEG 3550 (50% w/v), 36 μl of LiOAc (1.0 M), 50 μl of single-stranded carrier DNA (2.0 mg ml−1), 34 μl of plasmid DNA or fragments (500–1,000 ng) plus sterile water were added to each cell pellet. Cells were then heat-shocked at 42 °C for 40 min. After heat shock, cells were spun at 2,500g for 10 min, supernatant was removed and the pellet was resuspended in 1 ml of water. Then, 10–100 μl of cell suspension was plated on SCD-tryptophan-uracil dropout plates and grown at 30 °C for 2–3 d (ref. 56).
Flow cytometry assays
Single clones were assayed by flow cytometry in a process similar to a library being prepared for sorting. Once a single clone was obtained via single-cell sorting or lithium acetate transformation, it was grown to saturation in SCD-tryptophan-uracil dropout medium and then induced overnight in SGR. Triglycine was conjugated to the cell surface by Sfp as with a library, with the volume scaled down depending on culture size. Reactions of surface-displayed sortases, TEV cleavage and labeling were carried out as with a library preparation before analysis on a Bio-Rad ZE5 Cell Analyzer57.
Sortase expression and purification
E. coli BL21(DE3) transformed with pET29 sortase expression plasmids was cultured at 37 °C in LB with 50 μg ml−1 of kanamycin until OD600 = 0.5–0.8. IPTG was added to a final concentration of 1 mM and protein expression was induced overnight at 16 °C. Cells were collected by centrifugation and resuspended in lysis buffer (50 mM of Tris pH 8.0, 300 mM of NaCl supplemented with 1 mM of MgCl2, 2 U ml−1 of DNase I (New England Biolabs), 260 nM of aprotinin, 1.2 μM of leupeptin and 1 mM of phenylmethylsulfonyl fluoride). Cells were lysed by sonication and the clarified supernatant was purified on Ni-NTA agarose according to the manufacturer’s instructions. Fractions with >95% purity, as judged by SDS–PAGE, were consolidated and buffer exchanged into 25 mM of Tris pH 7.5, 150 mM of NaCl, 10% glycerol and 1 mM of tris(2-carboxyethyl)phosphine by size-exclusion chromatography in this buffer on a Superdex 200 Increase 10/300 GL column (GE Healthcare Life Sciences). Enzyme concentrations were calculated by reducing agent-compatible bicinchoninic acid protein assay kit (Pierce).
Aβ37-GGGRR cloning
The expression plasmid Aβ42/pET3 was amplified with the primers GGRR-Fwd and GGRR-Rev. The PCR product was ligated with the KLD enzyme mixture (New England Biolabs) and transformed into One Shot Mach1 T1 Phage-Resistant Chemically Competent E. coli cells, from which Aβ37-GGGRR/pET3 was sequence verified and isolated: GGRR-Fwd: 5′-CGCCGTTAATAGGAGCTCGATCCGG-3′; GGRR-Rev: 5′-CCCACCGCCACCAACCATCA-3′.
Aβ expression and purification
E. coli BL21(DE3) transformed with pET3 Aβ expression plasmids (AβM1–40, AβM1–42 or AβM1–37GGGRR) were cultured at 37 °C in LB-carbenicillin until OD600 = 0.5–0.6. IPTG was added to a final concentration of 1 mM (AβM1–40 and AβM1–42) or 0.1 mM (AβM1–37GGGRR) and protein expression was induced for 4 h at 37 °C. For AβM1–40 and AβM1–42, cells were pelleted and lysed by resuspension in 10 mM of Tris-HCl pH 8.0, 1 mM of EDTA and sonication. After lysis, the lysate was centrifuged for 10 min at 18,000g. The supernatant was discarded and pellet was resuspended in 10 mM of Tris-HCl pH 8.0 and 1 mM of EDTA. Sonication, centrifugation and removal of supernatant were repeated to yield an insoluble pellet. For AβM1–37GGGRR, cells were pelleted and lysed using B-PER bacterial protein extraction reagent supplemented with DNase I and lysozyme and then centrifuged for 10 min at 18,000g, with the insoluble pellet retained.
Insoluble pellets were resuspended in 8 M of urea, 10 mM of Tris/HCl pH 8.0 and 1 mM of EDTA and then sonicated. Solubilized inclusion bodies were diluted with 10 mM of Tris-HCl pH 8.0, 1 mM of EDTA and added to pre-equilibrated diethylethanolamine-sepharose. After a 20–30-min incubation, resin was batch-filtered, washed for 5 min with 50 mM of Tris pH 8.5 and then washed again for 5 min with 50 mM of Tris pH 8.5 and 25 mM of NaCl. After washing, recombinant peptides were eluted from resin with 50 mM of Tris pH 8.5 and 125 mM of NaCl and lyophilized44.
Chemically synthesized Aβ
Aβ1–40 and Aβ1–42 peptides (including biotinylated LC-Aβ40 and biotinylated LC-Aβ42) were synthesized and purified using reverse-phase HPLC by J. I. Elliott at the Eri Amyloid Laboratory. Peptide mass and purity (>99%) were confirmed by reverse-phase HPLC and electrospray ion trap MS.
Isolation of Aβ monomers
Lyophilized Aβ peptides, whether synthetic in origin (Eri Amyloid Laboratory) or produced by recombinant technology, were dissolved in 7 M of guanidium chloride, 50 mM of Tris pH 7.5, 2 mM of EDTA at a concentration of 1 mg ml−1 and incubated overnight. Denatured Aβ was then purified by size-exclusion chromatography using a Superdex 75 300/10 column (GE Healthcare) at a flow rate of 0.5 mL min−1 in alkaline buffer (50 mM of Tris-HCl pH 8.5) to minimize peptide aggregation. Peptide concentration was measured by A275 (ε = 1361 M−1cm−1). Peptide was either used immediately after purification or diluted to 20 μM, aliquoted and frozen at −80 °C for later use28.
Western blot analysis for fetuin-A
Samples of sortase reactions with fetuin-A were added to 4× NuPAGE lithium dodecyl sulfate (LDS) buffer (catalog no. NP0007; Invitrogen), heat-denatured and loaded onto a 4–12% Bis-Tris gel and ran at 160 V for 30 min in MES running buffer. Samples from reactions in plasma were diluted as follows: 20 μl of sample diluted + 30 μl of TBS + 20 μl of LDS buffer, for a total dilution of 3.5×. Gels were transferred to polyvinylidene fluoride membrane via iBlot and the membrane was blocked with SuperBlock Blocking Buffer (catalog no. 37515; Thermo Fisher Scientific) for 1 h at room temperature. The membrane was then incubated with mouse anti-fetuin-A antibody (1:500 dilution in SuperBlock TBS + 0.1% Tween 20; catalog no. ab89227; Abcam) overnight at 4 °C followed by washing in PBS and Tween 20 (PBS + 0.1% Tween 20) 3 times for 5 min each. The secondary antibodies streptavidin-IRDye 800CW (catalog no. 926-32230; LI-COR) and goat anti-mouse-IRDye 680LT (catalog no. 926-68020; LI-COR) (both 1:10,000 dilution in Odyssey Blocking Buffer in PBS (catalog no. 927-40000; LI-COR), 0.1% Tween 20, 0.01% SDS) were applied for 30 min at room temperature in the dark. The membrane was washed with PBS Tween 20 three times for 5 min each, followed by 1 wash with Milli-Q water and imaged on an Odyssey Imager.
Western blot analysis for Aβ
Samples of sortase reactions with Aβ were added to 4× LDS buffer and, without heat denaturation, loaded onto a 4–12% Bis-Tris gel and ran at 160 V for 30 min in MES running buffer. Gels were transferred to a polyvinylidene fluoride membrane via iBlot and the membrane was blocked with SuperBlock Blocking Buffer for 1 h at room temperature. The membrane was then incubated with mouse anti-Aβ 4G8 antibody (1:1,000 dilution in SuperBlock TBS + 0.1% Tween 20; catalog no. 800702; BioLegend) overnight at 4 °C followed by washing in PBS and Tween 20 (PBS + 0.1% Tween 20) 3 times for 5 min each. The secondary antibodies streptavidin-IRDye 800CW and goat anti-mouse-IRDye 680LT (both 1:10,000 dilution in Odyssey Blocking Buffer in PBS, 0.1% Tween 20, 0.01% SDS) were applied for 30 min at room temperature in the dark. The membrane was washed with PBS and Tween 20 three times for 5 min each, followed by one wash with Milli-Q water and imaged on an Odyssey Imager.
Streptavidin pulldown of sortase-labeled plasma proteins
One milliliter of normal human plasma was combined with 10 μl of 1 M of CaCl2, 10 μl of 0.1 M of GGGK(biotin) and 10 μl of 100 μM of sortase 4S.6 or SrtAβ, then incubated at room temperature for 2 h. Then, 100 μl of pre-equilibrated Ni-NTA resin slurry was added to the mixture and incubated at room temperature with shaking for 15 min before being filtered through a 0.2-μm spin filter before dilution to a 10-ml final volume in PBS with EDTA (PBS + 1 mM of EDTA). The solution was concentrated using a 3-kDa molecular weight cutoff spin concentrator for 30 min at 3,500g and a final volume of <1 ml. This sample was diluted with PBS with EDTA to a 10-ml final volume, reconcentrated and rediluted in a total of 6 wash cycles to give an expected small molecule biotin concentration of <1 nM. The concentrated mixture was then incubated with 200 μl of pre-equilibrated MyOne Streptavidin C1 Dynabeads (Invitrogen) with shaking for 30 min before magnetic separation and washing 3 times with PBS + 0.1% Tween 20. Beads were then resuspended in 100 μl of SDS–PAGE loading buffer with 100 μM of free biotin and incubated at 95 °C for 15 min. A 15-μl aliquot was then run on a 4–12% Bis-Tris PAGE gel and visualized by staining with Coomassie Brilliant Blue.
HPLC assay of sortases on LMVGG
Reactions were performed with the Abz-LMVGGK(dinitrophenyl)-CONH2 peptide (custom synthesis from GenScript) fixed at 10 μM. Reaction conditions were 300 mM of Tris pH 7.5, 150 mM of NaCl, 100 mM of H2N-GGG-COOH, 5 mM of CaCl2 and 5% v/v DMSO. Then, 5 μl of 10 μM sortase stock was added to the 45-μl reaction buffer, yielding a final enzyme concentration of 1 μM. Reactions were incubated for 120 min at 22.5 °C. Reactions were quenched with 10 μl of 1 N of HCl. The total volume of each reaction was transferred to HPLC sample vials and ran on analytical reverse-phase Zorax SB-C18 (Agilent Technologies) (2.1 × 150 mm, 5 μm) and chromatographed using a linear gradient 10–56.5% acetonitrile with 0.1% TFA in 0.1% aqueous TFA over 13 min. To calculate the percentage conversion, the ratio of the integrated areas of the GK(Dnp)-CONH2 (room temperature for 6.7 min) and Abz-LMVGG(dinitrophenyl)-CONH2 (room temperature for 11.6 min) A355 peaks were compared directly.
HPLC assay of sortases on Aβ40
Reactions were performed with 20 µM of Aβ40 and 1 mM of GGGK(dinitrophenyl) in 50 mM of Tris pH 8.5, 150 mM of NaCl and 5 mM of CaCl2. Then, 5 µM of SrtAβ was added to this mixture and incubated at room temperature overnight. Reactions were quenched with 10 μl of 1 N of HCl. The total volume of each reaction was transferred to HPLC sample vials and ran on analytical reverse-phase Zorax SB-C18 (2.1 × 150 mm, 5 μm) and chromatographed using a linear gradient 10–56.5% acetonitrile with 0.1% TFA in 0.1% aqueous TFA over 13 min. To calculate the percentage conversion, the ratio of the integrated areas of the GGGK(dinitrophenyl) (room temperature for 8.2 min) and Aβ37-GGGK(dinitrophenyl) (room temperature for 12.6 min) A355 peaks were compared directly.
Kinetic assay of sortases on LPESG
Assays to determine Kcat and Km LPESG were performed in 300 mM of Tris pH 7.5, 150 mM of NaCl, 5 mM of CaCl2, 5% v/v DMSO and 10 mM of glycine-glycine-glycine-COOH (GGG). The concentration of the LPESG peptide substrate ranged from 62.5 μM to 4 mM and enzyme concentrations ranged from 100 nM to 1000 nM. Reactions were initiated by adding enzyme and incubated at 22.5 °C for 7 min (sortase 4S.6) or 2 h (SrtAβ) before quenching with 0.2 volumes of 5 M of HCl. Then, 5–10 nmol of peptide from the quenched reactions were injected onto an analytical reverse-phase Eclipse XDB-C18 HPLC column (4.6 × 150 mm, 5 μm; Agilent Technologies) and chromatographed using a linear gradient of 10–65% acetonitrile with 0.1% TFA in 0.1% aqueous TFA over 13 min. Retention times under these conditions for the Abz-LPESGK(dinitrophenyl)-CONH2 substrate and the released GK(dinitrophenyl) peptide were 12.8 and 10.4 min, respectively. To calculate the percentage conversion, the ratio of the integrated areas of the GK(dinitrophenyl)-CONH2 and Abz-LPESGK(dinitrophenyl)-CONH2 peptide A355 peaks were compared directly. To determine Kcat and Km LPESG, reaction rates were fitted to the Michaelis–Menten equation in Prism v7 (GraphPad Software).
Kinetic assay of sortases on LMVGG
Assays to determine Kcat and Km LMVGG were performed in 300 mM of Tris pH 7.5, 150 mM of NaCl, 5 mM of CaCl2, 5% v/v DMSO and 10 mM glycine-glycine-glycine-COOH (GGG). The concentration of the Abz-LMVGG(dinitrophenyl)-CONH2 peptide substrate ranged from 10 to 200 μM with an enzyme concentration of 1 μM. Reactions were conducted in 96-well half area black/clear flat bottom plates (Corning) and initiated by adding the enzyme. Plates were incubated at 24 °C and monitored for increases in fluorescence (excitation wavelength = 317 nm, emission wavelength = 420 nm) in a Tecan plate reader for 2 h. Changes in fluorescence were converted to molar velocities using the calibration curves of Abz-LMVGG(dinitrophenyl)-CONH2 and a 1:1 mixture of free aminobenzoic acid and dinitrophenyl. Inner filter quenching effects were corrected using Fcorr = Fobs × antilog[(Aex + Aem)/2], where Fcorr is the corrected fluorescence value, Fobs is the observed fluorescence value, Aex is the absorbance at 317 nm and Aabs is the absorbance at 420 nm. To determine Kcat and Km LMVGG, initial velocities were fitted to the Michaelis–Menten equation in Prism v7.
Semisynthesis of AβM1–37-GGGK(biotin)
Freshly purified AβM1–40 monomers (120 μM in 50 mM of Tris pH 8.5) were supplemented with 150 mM of NaCl, 5 mM of CaCl2 and 1 mM of tris(2-carboxyethyl)phosphine and reacted overnight at room temperature with 50 μM of SrtAβ and 1 mM of GGGK(biotin). After desalting in a 3-kDa molecular weight cutoff spin filter, the reaction mixture was lyophilized and then dissolved in 7 M of guanidium chloride, 50 mM of Tris pH 7.5 and 2 mM of EDTA and ran on a Kinetex C18 100 Å (150 × 30 mm, 5 μm; Phenomenex) column. The acetonitrile concentration was increased from 10 to 35% over the first 5 min, 35–38% over the next 6 min and then from 38 to 90% over the next 5 min. The major peak eluted at 12.8 min. This was confirmed to be AβM1–37-GGGK(biotin) by LC–MS (m/z = 4,731.98 observed, 4,730.27 expected) and the product was lyophilized and stored at −20 °C for later use.
Streptavidin capture ELISA for detection of biotinylated Aβ
AβM1–37-GGGK(biotin) standards were prepared in diluent (TBS + 0.1% Tween 20 + 1% BSA) in a range of concentrations from 20 nM to 312 pM. Samples were diluted as necessary in this same diluent. Preblocked Streptavidin Coated High Capacity plates (clear, 96-well; Pierce) were washed twice with TBS and Tween 20 (TBS + 0.1% Tween 20) before the addition of standards and any samples. Biotinylated material was captured by streptavidin at room temperature for 2 h. Plates were washed three times with TBS and Tween 20. The, 100 μl of mouse anti-Aβ clone 4G8 (1:2,000 in diluent) was added to each well and incubated at room temperature for 1 h. After 3 TBS and Tween 20 washes, each well was treated with 100 μl of goat anti-mouse IgG HRP conjugate (1:4,000 in diluent; catalog no. A-10668; Thermo Fisher Scientific) for 30 min at room temperature. Plates were washed 4 times with TBS and Tween 20 before adding 50 μl of 3,3′,5,5′-tetramethylbenzidine (TMB) (catalog no. 34028; Thermo Fisher Scientific). Wells were allowed to develop until saturation and then quenched with 50 μl of 2 M of H2SO4. The absorbance of each well at 450 nm was then measured using a Tecan Plate Reader. The standard curve was fitted to a four-parameter logistics curve by Solver in Excel (Microsoft Office 365) and used to calculate the concentration of biotinylated Aβ in the present samples.
m266 antibody capture ELISA for detection of biotinylated Aβ
Nunc MaxiSorp plates (96-well, clear; Thermo Fisher Scientific) were incubated overnight with 100 μl of 3 μg ml−1 of anti-Aβ antibody m266. The next day, plates were washed three times with TBS and Tween 20 and blocked for 2 h with 5% MSD Blocker A (Meso Scale Diagnostics) in TBS and Tween 20. AβM1–37-GGGK(biotin) standards were prepared in diluent (1% MSD Blocker A in TBS and Tween 20) in a range of concentrations from 2.5 ng ml−1 to 39 pg ml−1. Samples were diluted as necessary in this diluent. Plates were washed three times with TBS and Tween 20 before the addition of standards, samples and blanks in triplicate. After 2 h of capture, plates were washed three times with TBS and Tween 20. Then, 100 μl of HRP-conjugated streptavidin (1:100 in diluent; catalog no. 890803; R&D Systems) was added to each well for 30 min. After 4 TBS and Tween 20 washes, wells were developed with 50 μl of TMB substrate solution (N301; Thermo Fisher Scientific) and quenched with 2 M of H2SO4. The absorbance at 450 nm of each well was then measured with a Molecular Devices plate reader. The standard curve was fitted to a four-parameter logistics curve by Solver in Excel (Microsoft Office 365) and used to calculate the concentration of biotinylated Aβ present in samples.
ELISA assays for Aβ40 and Aβ42
Nunc MaxiSorp plates (96-well, clear) were incubated overnight with 100 μl of 3 μg ml−1 of anti-Aβ antibody m266. The next day, plates were washed three times with TBS and Tween 20 and blocked for 2 h with 5% MSD Blocker A in TBS and Tween 20. Aβ40 and Aβ42 standards were prepared in diluent (1% MSD Blocker A in TBS and Tween 20) in a range of concentrations from 2.5 ng ml−1 to 39 pg ml−1. Samples were diluted as necessary in the same diluent. Plates were washed three times with TBS and Tween 20 before adding standards, samples and blanks in triplicate. After a 2-h capture, plates were washed and secondary antibodies (1:2,500 biotinylated 21F12 for Aβ42, 1:4,000 biotinylated 2G3 for Aβ40) were added for 2 h. After another set of washes, HRP-conjugated streptavidin (1:100) was added for 30 min. Plates were then washed, developed with TMB and quenched with H2SO4. The absorbance at 450 nm of each well was measured with a Molecular Devices plate reader. The standard curves were fitted to four-parameter logistics curve by Solver in Excel (Microsoft Office 365) and used to calculate the concentrations of Aβ40 and Aβ42 present in the samples40.
CSF and plasma samples
Human CSF specimens were obtained in accordance with local clinical regulations approved by the Partners institutional review board (Walsh, no. BWH2017P0000259). All samples were from the Biobank at Partners HealthCare in Boston. Donors had no history of diseases of the central nervous system. Donor demographic information can be found in Supplementary Table 4. Pooled human plasma was purchased from GeneTex (catalog no. GTX73265).
CSF labeling with GGG using SrtAβ
Aliquots of CSF collected from 10 different patients were supplemented with 5 mM of CaCl2 and treated for 1 h with 5 μM of SrtAβ and 500 μM of GGG. Reactions were quenched by adding 5 mM of EDTA and diluted twofold with 1% MSD Blocker A in TBS and Tween 20. Part of the sample was set aside for Aβ42 measurement, while the rest was diluted fivefold (total dilution = tenfold) for Aβ40 measurement. Untreated aliquots from the same patients were diluted similarly. Aβ40 and Aβ42 were captured by the anti-Aβ antibody m266 and detected with C-terminal-specific antibodies as described above.
CSF labeling with GGGK(biotin) using SrtAβ
Aliquots of CSF collected from 10 different patients were supplemented with 5 mM of CaCl2 and treated for 2 h with 5 μM of SrtAβ and 500 μM of GGGK(biotin). Reactions were quenched by adding 5 mM of EDTA and all samples (full reactions, no SrtAβ control, no GGGK(biotin) control and untreated) were diluted tenfold with 1% MSD Blocker A in TBS and Tween 20. Biotinylated Aβ was captured by the anti-Aβ antibody m266 and detected without secondary antibody using HRP-conjugated streptavidin as described above.
Semisynthesis of AβM1–37-GGGRR
Immediately after elution from diethylethanolamine resin in 50 mM of Tris pH 8.5 + 125 mM of NaCl, recombinant Aβ42 (20 ml of an estimated concentration of 40 μM = 3–4 mg) was supplemented with 5 mM of CaCl2 and 5 mM of DTT and treated overnight at room temperature with 20 μM of SrtAβ and 200 μM of GGGRR. The reaction mixture was concentrated to 1 ml in a 3-kDa molecular weight cutoff spin concentrator, diluted to 20 ml with Milli-Q water to reduce the salt concentration and then concentrated back to 1 ml and lyophilized. The lyophilized reaction mixture was then denatured overnight in 7 M of guanidium chloride, 50 mM of Tris pH 7.5 and 2 mM of EDTA and ran on a Zorbax 300SB-C18 (9.4 × 250 mm, 5 μm; Agilent Technologies) column. After 5 min at 10% acetonitrile with 0.1% TFA in 0.1% aqueous TFA, the acetonitrile concentration was increased to 30% over 5 min and then to 50% over 20 min. AβM1–37-GGGRR was eluted at 17.5 min. AβM1–37-GGGRR identity was confirmed by LC–MS (m/z = 4,689.85 observed, 4,688.30 expected) and the fraction containing it was lyophilized and stored at −20 °C for later use.
ThT assay
Aβ peptides were denatured and size-exclusion chromatography-isolated in 20 mM of sodium phosphate pH 8.0. Concentrations were determined by A275 and stock solutions of 20.2 μM of peptide in elution buffer were prepared. Then, 10 μl of ThT (2 mM in water) was added to 990 μl of each stock solution, yielding 1 ml of 20 μM of peptide and 20 μM of ThT. Then, 20 μM of ThT in elution buffer was used as diluent to make 10 μM of peptide samples. Samples were aliquoted 120 μl per well to a sterile Nunc F96 MicroWell Black and White Polystyrene Plate (catalog no. 237105; Thermo Fisher Scientific). A Molecular Devices plate reader was used to follow change in fluorescence (435 excitation/480 emission) over 48–60 h45.
Negative contrast transmission electron microscopy of Aβ fibrils
Samples of AβM1–37-GGGRR (n = 6) and AβM1–42 (n = 2) lacking ThT were included alongside ThT-containing samples in the assay described above. Following aggregation, these samples were applied to carbon-coated Formvar grids, left for 1 min, fixed with glutaraldehyde, washed with Milli-Q water and wicked dry with filter paper. Then, 2% uranyl acetate was added and incubated for 2 min. The grid was wicked dry and allowed to air-dry for 10 min. Grids were stored in a sealed container and viewed under a Tecnai G2 BIOTWIN electron transmission microscope operated at 80 kV. All reagents were supplied by Electron Microscopy Sciences47.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Boutureira, O. & Bernardes, G. J. L. Advances in chemical protein modification. Chem. Rev. 115, 2174–2195 (2015).
Chin, J. W. Expanding and reprogramming the genetic code of cells and animals. Annu. Rev. Biochem. 83, 379–408 (2014).
Noren, C. J., Anthony-Cahill, S. J., Griffith, M. C. & Schultz, P. G. A general method for site-specific incorporation of unnatural amino acids into proteins. Science 244, 182–188 (1989).
Shah, N. H. & Muir, T. W. Inteins: nature’s gift to protein chemists. Chem. Sci. 5, 446–461 (2014).
Zhang, C. et al. π-Clamp-mediated cysteine conjugation. Nat. Chem. 8, 120–128 (2016).
Zhang, Y., Park, K-Y., Suazo, K. F. & Distefano, M. D. Recent progress in enzymatic labelling techniques and their applications. Chem. Soc. Rev. 47, 9106–9136 (2018).
Paterson, G. K. & Mitchell, T. J. The biology of Gram-positive sortase enzymes. Trends Microbiol. 12, 89–95 (2004).
Mazmanian, S. K., Liu, G., Ton-That, H. & Schneewind, O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 285, 760–763 (1999).
Ton-That, H., Liu, G., Mazmanian, S. K., Faull, K. F. & Schneewind, O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl Acad. Sci. USA 96, 12424–12429 (1999).
Kruger, R. G. et al. Analysis of the substrate specificity of the Staphylococcus aureus sortase transpeptidase SrtA. Biochemistry 43, 1541–1551 (2004).
Glasgow, J. E., Salit, M. L. & Cochran, J. R. In vivo site-specific protein tagging with diverse amines using an engineered sortase variant. J. Am. Chem. Soc. 138, 7496–7499 (2016).
Popp, M. W., Antos, J. M., Grotenbreg, G. M., Spooner, E. & Ploegh, H. L. Sortagging: a versatile method for protein labeling. Nat. Chem. Biol. 3, 707–708 (2007).
Antos, J. M. et al. A straight path to circular proteins. J. Biol. Chem. 284, 16028–16036 (2009).
Antos, J. M. et al. Site-specific N- and C-terminal labeling of a single polypeptide using sortases of different specificity. J. Am. Chem. Soc. 131, 10800–10801 (2009).
Freiburger, L. et al. Efficient segmental isotope labeling of multi-domain proteins using Sortase A. J. Biomol. NMR 63, 1–8 (2015).
Bartels, L., Ploegh, H. L., Spits, H. & Wagner, K. Preparation of bispecific antibody-protein adducts by site-specific chemo-enzymatic conjugation. Methods 154, 93–101 (2019).
Harmand, T. J. et al. One-pot dual labeling of IgG 1 and preparation of C-to-C fusion proteins through a combination of sortase A and butelase 1. Bioconjug. Chem. 29, 3245–3249 (2018).
Piotukh, K. et al. Directed evolution of sortase A mutants with altered substrate selectivity profiles. J. Am. Chem. Soc. 133, 17536–17539 (2011).
Chen, L. et al. Improved variants of SrtA for site-specific conjugation on antibodies and proteins with high efficiency. Sci. Rep. 6, 31899 (2016).
Chen, I., Dorr, B. M. & Liu, D. R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl Acad. Sci. USA 108, 11399–11404 (2011).
Dorr, B. M., Ham, H. O., An, C., Chaikof, E. L. & Liu, D. R. Reprogramming the specificity of sortase enzymes. Proc. Natl Acad. Sci. USA 111, 13343–13348 (2014).
Selkoe, D. J. & Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608 (2016).
Walsh, D. M., Hartley, D. M. & Selkoe, D. J. The many faces of Aβ: structures and activity. Curr. Med. Chem. Immunol. Endocr. Metab. Agents 3, 277–291 (2003).
Chen, G.-F. et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235 (2017).
Brothers, H. M., Gosztyla, M. L. & Robinson, S. R. The physiological roles of amyloid-β peptide hint at new ways to treat Alzheimer’s disease. Front. Aging Neurosci. 10, 118 (2018).
O’Brien, R. J. & Wong, P. C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204 (2011).
Roche, J., Shen, Y., Lee, J. H., Ying, J. & Bax, A. Monomeric Aβ1–40 and Aβ1–42 peptides in solution adopt very similar Ramachandran map distributions that closely resemble random coil. Biochemistry 55, 762–775 (2016).
O’Malley, T. T. et al. Aβ dimers differ from monomers in structural propensity, aggregation paths and population of synaptotoxic assemblies. Biochem. J. 461, 413–426 (2014).
Boder, E. T. & Wittrup, K. D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 15, 553–557 (1997).
Feldhaus, M. J. et al. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat. Biotechnol. 21, 163–170 (2003).
Angelini, A. et al. in Yeast Surface Display: Methods, Protocols, and Applications (ed. Liu, B.) 3–36 (Humana Press, New York, NY, 2015).
Marraffini, L. A., Ton-That, H., Zong, Y., Narayana, S. V. L. & Schneewind, O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus: a conserved arginine residue is required for efficient catalysis of sortase A. J. Biol. Chem. 279, 37763–37770 (2004).
Frankel, B. A., Tong, Y., Bentley, M. L., Fitzgerald, M. C. & McCafferty, D. G. Mutational analysis of active site residues in the Staphylococcus aureus transpeptidase SrtA. Biochemistry 46, 7269–7278 (2007).
Zong, Y., Bice, T. W., Ton-That, H., Schneewind, O. & Narayana, S. V. L. Crystal structures of Staphylococcus aureus sortase A and its substrate complex. J. Biol. Chem. 279, 31383–31389 (2004).
Suree, N. et al. The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized. J. Biol. Chem. 284, 24465–24477 (2009).
Kruger, R. G., Dostal, P. & McCafferty, D. G. Development of a high-performance liquid chromatography assay and revision of kinetic parameters for the Staphylococcus aureus sortase transpeptidase SrtA. Anal. Biochem. 326, 42–48 (2004).
Ton-That, H., Mazmanian, S. K., Faull, K. F. & Schneewind, O. Anchoring of surface proteins to the cell wall of Staphylococcus aureus. Sortase catalyzed in vitro transpeptidation reaction using LPXTG peptide and NH2-Gly3 substrates. J. Biol. Chem. 275, 9876–9881 (2000).
Pawlowski, M., Meuth, S. G. & Duning, T. Cerebrospinal fluid biomarkers in Alzheimer’s disease: from brain starch to bench and bedside. Diagnostics (Basel) 7, 42 (2017).
Baranello, R. J. et al. Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr. Alzheimer Res. 12, 32–46 (2015).
Hong, W. et al. Diffusible, highly bioactive oligomers represent a critical minority of soluble Aβ in Alzheimer’s disease brain. Acta Neuropathol. 136, 19–40 (2018).
Mengel, D. et al. Dynamics of plasma biomarkers in Down syndrome: the relative levels of Aβ42 decrease with age, whereas NT1 tau and NfL increase. Alzheimers Res. Ther. 12, 27 (2020).
Isaacs, A. M., Senn, D. B., Yuan, M., Shine, J. P. & Yankner, B. A. Acceleration of amyloid β-peptide aggregation by physiological concentrations of calcium. J. Biol. Chem. 281, 27916–27923 (2006).
Colvin, M. T. et al. Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J. Am. Chem. Soc. 138, 9663–9674 (2016).
Walsh, D. M. et al. A facile method for expression and purification of the Alzheimer’s disease-associated amyloid β-peptide. FEBS J. 276, 1266–1281 (2009).
O’Malley, T. T., Witbold, W. M. 3rd, Linse, S. & Walsh, D. M. The aggregation paths and products of Aβ42 dimers are distinct from those of the Aβ42 monomer. Biochemistry 55, 6150–6161 (2016).
Bacon, K., Burroughs, M., Blain, A., Menegatti, S. & Rao, B. M. Screening yeast display libraries against magnetized yeast cell targets enables efficient isolation of membrane protein binders. ACS Comb. Sci. 21, 817–832 (2019).
Nguyen, G. K. et al. Butelase 1 is an Asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 10, 732–738 (2014).
Yang, R. et al. Engineering a catalytically efficient recombinant protein ligase. J. Am. Chem. Soc. 139, 5351–5358 (2017).
Nikghalb, K. D. et al. Expanding the scope of sortase‐mediated ligations by using sortase homologues. Chembiochem 19, 185–195 (2018).
Tang, S., Xuan, B., Ye, X., Huang, Z. & Qian, Z. A modular vaccine development platform based on sortase-mediated site-specific tagging of antigens onto virus-like particles. Sci. Rep. 6, 25741 (2016).
Meyer, A. J., Ellefson, J. W. & Ellington, A. D. Library generation by gene shuffling. Curr. Protoc. Mol. Biol. 105, Unit 15.12 (2014).
Chao, G. et al. Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768 (2006).
Killenberg, P. G. & Dukes, D. F. Coenzyme A derivatives of bile acids-chemical synthesis, purification, and utilization in enzymic preparation of taurine conjugates. J. Lipid Res. 17, 451–455 (1976).
Mofid, M. R., Finking, R., Essen, L. O. & Marahiel, M. A. Structure-based mutational analysis of the 4′-phosphopantetheinyl transferases Sfp from Bacillus subtilis: carrier protein recognition and reaction mechanism. Biochemistry 43, 4128–4136 (2004).
Tropea, J. E., Cherry, S. & Waugh, D. S. Expression and purification of soluble His6-tagged TEV protease. Methods Mol. Biol. 498, 297–307 (2009).
Gietz, R. D. & Schiestl, R. H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2, 31–34 (2007).
Walsh, D. M., Lomakin, A., Benedek, G. B., Condron, M. M. & Teplow, D. B. Amyloid β-protein fibrillogenesis: detection of a protofibrillar intermediate. J. Biol. Chem. 272, 22364–22372 (1997).
Acknowledgements
We thank T. O’Malley and G. Newby for helpful comments. We thank A. Vieira for assistance with editing this manuscript. This work was supported by NIH grant nos. R01EB022376, R35GM118062 and R01AG046275, and by the Howard Hughes Medical Institute. D.M.W. is an Alzheimer’s Association Zenith Fellow.
Author information
Authors and Affiliations
Contributions
C.J.P., C.A. and D.R.L. designed the research, performed the experiments and analyzed the data. A.D. helped perform and analyze the CSF ELISA and aggregation experiments. B.M.D. provided the starting sortase library. D.M.W. designed and supervised the CSF ELISA and aggregation experiments. C.J.P. and D.R.L. wrote the manuscript. D.R.L. supervised the research.
Corresponding author
Ethics declarations
Competing interests
C.J.P., C.A., B.M.D. and D.R.L. have filed patent applications on sortase evolution.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Gating strategy for SrtAβ evolution.
Following loading of cell surfaces with triglycine, the library is split into several aliquots that are treated with varying amounts of positive selection substrate. Surface-displayed sortase reactions, TEV removal of sortase, and staining are conducted in parallel on these aliquots. The aliquots are then analyzed before sorting. Populations are gated first on the basis of size (FSC vs. SSC), and then successful induction is confirmed on a FITC histogram. FITC vs PE dot plots are made for each aliquot, and a polygon gate is drawn that includes <0.1% of the negative control (no positive selection substrate) aliquot while roughly matching its slope. This gate is then applied to the other aliquots. The aliquot with the least amount of positive selection substrate that shows >10-fold increase in gated events over the negative control is used for sorting. In the case of this representative data from round 11, that would be the 200 nM Btn-LMVGG aliquot.
Extended Data Fig. 2 Flow cytometry analysis of library activity on LMVGG over the course of evolution.
Stocks of yeast libraries remaining at the end of each round (R) were regrown, induced, and subjected to cell surface sortase reactions. Each library was incubated for 1 hour with 1 μM Btn-LMVGG. All libraries were assayed for LMVGG activity in TBS, while the libraries that survived negative selection against fetuin A and other plasma proteins (R9-16) were also assayed in human plasma. Data shown are the mean ± standard deviation of three replicates. Activity:expression is the transpeptidation activity (PE) to sortase expression (FITC) ratio. Overall, the trend towards improved LMVGG activity per unit expression is strong, indicative of a successful evolution campaign.
Extended Data Fig. 3 Flow cytometry analysis of round 8 single clones.
At the end of round 8 sort 5 (8.5), single cells were sorted into a 96-well plate, grown to saturation, and induced. After attachment of GGGK-CoA via Sfp ligation, the clonal populations were given an hour to react with 500 nM Btn-LMVGG or 500 nM Btn-LPVGG. Flow cytometry analysis revealed that clone 8.5-H3 possessed the best combination of high activity on the LMVGG substrate with low activity on the LPVGG substrate. Activity is defined as fold-increase in PE signal over a negative control (0 nM Btn-LMVGG) aliquot of each variant.
Extended Data Fig. 4 HPLC assay of LMVGG activity.
In this assay, sortase-mediated transpeptidation of a chromophore-linked substrate (Abz-LMVGGK-Dnp) liberates free GK-Dnp, the formation of which can be followed by HPLC1. The round 2 consensus sequence (R2, 4 S.6 + R94Y, S118I, A122W, D124G, G134R, and V189F mutations) did not show detectable activity in this assay. The first variants with detectable activity on LMVGG in this assay emerged from round 4; the most active of these variants (R4) displayed modest activity on Abz-LMVGGK-Dnp, with 1 μM of enzyme converting 5.8% of 10 μM substrate to product in two hours. Notably, this variant contained the V182A, T196S, and R197S mutations. By round 8, activity in this assay roughly doubled, with 1 μM of clone 8.5-H3 converting 10.4% of 10 μM substrate to product in two hours.
Extended Data Fig. 5 Evolved sortase labels Aβ alloforms with C-termini that extend to Val40 and beyond.
SrtA 8.5-H3 (10 μM) was incubated with GGGK(Btn) (100 μM) and different alloforms of Aβ (Aβ38, Aβ40, Aβ42, or Aβ43, 10 μM each) for 2 hours in DPBS + 2 mM CaCl2. Labeling was observed for Aβ40, Aβ42, and Aβ43. Aβ38 is not labeled, which is expected given that the final glycine of the LMVGG recognition sequence is not amidated in this alloform. This experiment was repeated three times independently with similar results.
Extended Data Fig. 6 Kinetic analysis of sortase 4S.6 and SrtAβ.
a, Sortase 4S.6 shows kcat = 0.36 s−1 (95% CI= 0.22 to 0.96 s-1) and KM = 610 μM (95% CI = 90 to 5550 μM) on its cognate substrate LPESG in an established HPLC assay for sortase transpeptidation activity. SrtAβ activity was detectable in this assay, but at levels too low to accurately determine its kinetic parameters on LPESG. Attempts to establish kinetic parameters for LMVGG in the HPLC assay were complicated by this substrate’s limited solubility in the reaction buffer. (b) A more sensitive fluorescence method was used to detect smaller amounts of turnover at lower substrate concentrations. Importantly, the solubility of the LMVGG substrate is within a range where inner filter quenching effects can be corrected. Using this method, SrtAβ shows kcat = 0.018 s−1 (95% CI = 0.015 to 0.023 s−1) and KM = 128 μM (95% CI = 87 to 198 μM) on LMVGG. Sortase 4S.6 activity on LMVGG was not detectable in this assay. Points and error bars represent the average of three replicates ± one standard deviation.
Extended Data Fig. 7 Aβ40 labeling in human plasma.
a, Aβ40 (5 μM) added to TBS or plasma was labeled with GGGK(Btn) (50 μM) by SrtAβ (1.5 μM) for two hours. Product was captured on a streptavidin coated plate and detected using the anti-Aβ antibody 4G8. The reaction was 1.6-fold more efficient in TBS than in plasma. b, Aβ40 (5 μM) added to human plasma was labeled in the presence of various concentrations of GGGK(Btn) by an evolved sortase variant from round 14 (5 μM). After 2 hours, biotinylated product was undetectable in the reaction with a 10-fold excess of GGGK(Btn), whereas almost all of the Aβ40 was biotinylated when a 1000-fold excess of GGGK(Btn) was used. c, Aβ40 (5 μM or 5 nM) added to plasma was labeled with GGGK(Btn) (500 μM) by SrtAβ (1 μM) for two hours. No product was detected in the absence of enzyme or when using the starting enzyme 4S.6 in place of SrtAβ (* indicates below limit of detection). Each column represents the average of at least three replicates. Error bars represent one standard deviation.
Extended Data Fig. 8 Aggregation of AβM1-42 compared to AβM1- 37GGGRR.
The aggregation of recombinant Aβ42 and recombinant Aβ37GGGRR monomers was monitored by thioflavin T binding. Curves were fitted to each replicate (n = 3 for both peptides at both concentrations) by the Boltzmann equation. The aggregation of recombinant Aβ37GGGRR monomers is retarded relative to recombinant Aβ42, with an average t1/2 = 24.3 hours at 20 μM and 38.4 hours at 10 μM (compared to 1.25 hours and 1.46 hours for recombinant Aβ42 at 20 μM and 10 μM, respectively). These results are consistent with those observed for semi-synthetic Aβ37GGGRR created by using SrtAβ to label recombinant Aβ42 with GGGRR.
Extended Data Fig. 9 Electron microscopy of amyloid fibrils.
End-point samples from the ThT time-course study of AβM1-42 and AβM1-37GGGRR show marked differences in ultra-structure. Scale bars represent 800 nm. The most apparent difference between the two sets of fibrils is size. 61 of 76 AβM1-37GGGRR structures measured were less than 600 nm long, with an average length of 380 nm. In contrast, the average length of AβM1-42 fibrils was ~7 times larger at 2670 nm (n = 83). Aggregation and electron microscopy of amyloid fibrils was performed twice with similar results.
Supplementary information
Supplementary Information
Supplementary Tables 1–4 and Figs. 1–5.
Source data
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blot.
Source Data Extended Data Fig. 6
Statistical source data
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Rights and permissions
About this article

Cite this article
Podracky, C.J., An, C., DeSousa, A. et al. Laboratory evolution of a sortase enzyme that modifies amyloid-β protein. Nat Chem Biol 17, 317–325 (2021). https://doi.org/10.1038/s41589-020-00706-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41589-020-00706-1
This article is cited by
-
Recent advances in chemical protein synthesis: method developments and biological applications
Science China Chemistry (2024)
-
Nature-inspired protein ligation and its applications
Nature Reviews Chemistry (2023)
-
Desperately seeking sortase
Nature Chemical Biology (2021)
