
Abstract
Severe respiratory infections can result in acute respiratory distress syndrome (ARDS)1. There are no effective pharmacological therapies that have been shown to improve outcomes for patients with ARDS. Although the host inflammatory response limits spread of and eventually clears the pathogen, immunopathology is a major contributor to tissue damage and ARDS1,2. Here we demonstrate that respiratory viral infection induces distinct fibroblast activation states, which we term extracellular matrix (ECM)-synthesizing, damage-responsive and interferon-responsive states. We provide evidence that excess activity of damage-responsive lung fibroblasts drives lethal immunopathology during severe influenza virus infection. By producing ECM-remodelling enzymes—in particular the ECM protease ADAMTS4—and inflammatory cytokines, damage-responsive fibroblasts modify the lung microenvironment to promote robust immune cell infiltration at the expense of lung function. In three cohorts of human participants, the levels of ADAMTS4 in the lower respiratory tract were associated with the severity of infection with seasonal or avian influenza virus. A therapeutic agent that targets the ECM protease activity of damage-responsive lung fibroblasts could provide a promising approach to preserving lung function and improving clinical outcomes following severe respiratory infections.
Similar content being viewed by others


Main
Respiratory infections are a leading cause of morbidity and mortality3. These infections can result in ARDS with pulmonary oedema and hypoxia, causing mild to severe respiratory failure1. Much of the lung damage induced by viral infection is a result of infiltrating immune cells, which kill infected and bystander cells4. Defining the mechanisms that alter the balance between pathogen clearance and immunopathology may enable identification of strategies to improve outcomes following ARDS2. Therapeutic targets include components of the lung ECM, which provides structural support that is critical for lung function and tissue-specific signals to coordinate immune responses5.
The ECM consists of structural proteins as well as proteases and glycosidases that degrade or modify the ECM6. Upon lung injury, ECM proteases are upregulated and remodel the ECM, facilitating the migration of immune cells to sites of inflammation5,7. Non-immune lung cells, including epithelial cells, endothelial cells and fibroblasts, coordinate immune responses and directly mediate lung function8. Through their remodelling activity, specific cell populations can influence the outcome of infection and long-term sequelae, as has been described for myofibroblasts and lung fibrosis9. The role of lung stromal cell populations in coordinating host responses to active respiratory infections has received less attention than their role in late-stage repair following pathogen clearance. While there is extensive literature on how immune cells regulate the host response to respiratory viral infection8, there is less information on the heterogeneity and role of non-haematopoietic cells.
Stromal responses to influenza infection
We sorted live, CD45− lung cells from mice 0, 1, 3 and 6 days after infection with influenza A virus and performed single-cell gene-expression profiling (scGEX) identifying three main populations: fibroblasts, epithelial cells and endothelial cells (Fig. 1a, b, Extended Data Fig. 1a, b). High levels of viral mRNA, indicative of productive infection, were detected primarily in type I pneumocytes, ciliated epithelial cells and type II pneumocytes (Extended Data Fig. 1c). Fibroblasts were particularly dynamic, with multiple transcriptional states emerging following infection (Fig. 1c, d, Extended Data Fig. 1d). To assess the pathways active in each cluster, we performed gene-set enrichment analysis (GSEA), comparing each cluster to a putative baseline cluster (cluster 5) that was present before infection with few differentially expressed genes (Fig. 1c, Extended Data Fig. 2a). We identified three primary functional groups on the basis of gene expression: resting, ECM-synthesizing and inflammatory (Fig. 1c, Extended Data Fig. 2a). ECM-synthesizing fibroblasts (ESFibs) were enriched for genes encoding ECM structural proteins, but lacked inflammatory signatures (Fig. 1d, Extended Data Fig. 2a). Inflammatory fibroblasts exhibited high expression of genes involved in type I interferon, interleukin-6 (IL-6) or NF-κB signalling, whereas resting fibroblasts lacked strong enrichment for pathways involved in either inflammatory responses or synthesis of ECM proteins. (Fig. 1e, Extended Data Fig. 2a).

a, Schematic of sample collection for scGEX. SSC, side scatter. b, t-distributed stochastic neighbour embedding (t-SNE) projection of mouse lung cells based on scGEX. Data from all time points and mice were aggregated (n = 5 (0, 1 and 3 dpi), and n = 4 (6 dpi)). Approximately 40,800 individual cells are represented in the figure. c, t-SNE projection of mouse mesenchymal cells expressing Col1a2. d, t-SNE projection of mouse mesenchymal cells at different time points after infection. e, Summary of GSEA comparing all cells in each cluster (cluster 1, n = 1,188; cluster 2, n = 1,123; cluster 4, n = 892; cluster 6, n = 749; cluster 8, n = 681). βcat, β-catenin; resp., response; ROS, reactive oxygen species; sig., signalling; SMCs, smooth muscle cells. f, Expression of key genes in mouse lung mesenchymal cells. g, Kinetics of fibroblast activation states. Representative flow cytometry for ITGA5, CD9 and BST2 staining. h, Frequency of DRFib and IRFib activation states during infection (0 dpi, n = 5; 3 dpi, n = 4; 6 dpi, n = 3; 9 dpi, n = 4; 12 dpi, n = 4; 21 dpi, n = 5). Data are mean ± s.e.m.
Among inflammatory fibroblasts, we identified two distinct activation states, damage-responsive fibroblasts (DRFibs) and interferon-responsive fibroblasts (IRFibs) (Fig. 1d). DRFibs were enriched for pathways involved in tissue-damage responses, including NF-κB signalling and hypoxia, whereas IRFibs were enriched for type-I-interferon-responsive pathways (Fig. 1e, f, Extended Data Fig. 2b). Analysis of five human lung biopsies identified fibroblast activation states analogous to those defined in mouse lungs (Extended Data Fig. 2c) with clusters enriched for ECM synthesis, NF-κB signalling and type-I interferon signalling (Extended Data Fig. 2c, d).
To validate these fibroblast activation states, we identified genes encoding surface-protein markers that were enriched in DRFibs and IRFibs in mouse and human lungs (Fig. 1f, Extended Data Figs. 2d, 3a, b). Upregulation of Itga5 and downregulation of Cd9 defined the DRFib transcriptional program, whereas upregulation of Bst2 and expression of Cd9 defined IRFibs (Fig. 1g, Extended Data Fig. 3a). In mice, DRFibs increased in frequency and number over the course of infection, reaching a peak around 12 days (Fig. 1g, h, Extended Data Fig. 3c, d). IRFibs emerged rapidly, by day 3, and gradually decreased in frequency (Fig. 1h). ITGA5hiCD9lo fibroblasts expressed high levels of DRFib signature genes (Lox and Adamts4), whereas ITGA5loCD9hi fibroblasts exhibited a resting phenotype, with low expression of infection-induced genes (Extended Data Fig. 3e, f). Primary lung fibroblasts stimulated with IL-1 and TNF, cytokines known to drive NF-κB signalling, acquired a surface phenotype with higher ITGA5 expression, indicative of a damage-responsive activation state (Extended Data Fig. 3g). Analysis of human lung biopsies identified fibroblasts that exhibited surface phenotypes similar to those in mouse lungs. Activated fibroblasts with a damage-responsive profile (ITGA5hiCD9lo) were present in donors who died of respiratory failure, but were noticeably absent from healthy donors (Extended Data Fig. 4a, b).
Regulation of fibroblast activation
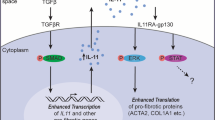
We next sought to identify the upstream stimuli that regulate fibroblast activation states. We tested whether in vitro infection of normal human bronchial epithelial cells (NHBEs) with human seasonal (H3N2) or avian viruses (H5N6 or H7N9) was sufficient to drive inflammatory transcriptional programs in human lung fibroblasts in co-culture (Fig. 2a). Infection of NHBEs induced expression in fibroblasts of genes enriched in inflammatory states, including genes encoding cytokines (IL6) and several matrix proteases (ADAMTS4, MMP3 and MMP13). We also tested the ability of individual cytokines and influenza viruses to stimulate these transcriptional programs in respiratory cells cultured on their own (Fig. 2a, monoculture), comparing them with nasal wash cells from patients infected with influenza virus, which comprise primarily immune cells10. These data demonstrated cell-type-specific expression of ECM-related genes (Extended Data Fig. 5a). In contrast to NHBEs, fibroblasts were highly responsive to stimulation with IL-1 cytokines and TNF, and ECM-related genes were broadly upregulated (Fig. 2a, Extended Data Fig. 5b, c). Nasal wash cells exhibited low levels of expression of ECM-related genes encoding both structural proteins and matrix proteases. However, nasal wash and epithelial cells also exhibited high levels of IL1A, IL1B and TNF expression, indicating that they probably provide immune signals to fibroblasts to regulate ECM-related gene expression (Extended Data Fig. 5a).

a, Average log2-transformed fold change in gene expression relative to mock-infected or mock-treated controls. For co-culture experiments, each column represents a biological replicate. For monoculture experiments, each column represents the average of two independent experiments performed with duplicate stimulations. GMCSF is also known as CSF2. CA09, influenza A/California/04/2009 H1N1; PR8, influenza A/Puerto Rico/8/34 H1N1; Perth, influenza A/Perth/16/2009 H3N2. b, Average log2-transformed fold change in gene expression relative to mock-infected mice (n = 3–5 mice per time point) in mouse lung homogenates. c, Adamts4 expression by cell type from scGEX data. d, Fold change in Adamts4 gene expression relative to bulk CD45+ cells in cells sorted before infection and at 3 dpi. e, Spatial transcriptomics of mouse lung tissue collected 10 days after infection. Scale bar, 1 mm. H&E, haematoxylin and eosin. DRFib signature (Adamts4–Itga5–Lox) represents the combined relative gene expression of Adamts4, Itga5, and Lox.
ECM proteases in activated fibroblasts
We sought to determine whether any individual protease has a fundamental effector role underlying productive and nonproductive lung repair. We assessed the regulation of ECM-related genes in mouse lung homogenates collected at different times during influenza virus infection (Fig. 2b). Detection of ECM proteases and tissue inhibitors of metalloproteinases (TIMPs) by enzyme-linked immunosorbent assay (ELISA) in the bronchoalveolar lavage (BAL) fluid corresponded with the kinetics of gene expression in lung homogenates (Extended Data Fig. 5d). Among the tested ECM proteases, the expression of ADAMTS4—one of the enzymes that degrade the proteoglycan versican11—was the earliest to be induced, and its expression continued throughout the repair stage (Fig. 2b). On the basis of scGEX analyses, expression of Adamts4 was restricted to non-immune, stromal cells (Fig. 2c). We next sorted immune and non-immune populations before and after infection. Before infection, Adamts4 gene expression was restricted to fibroblasts and endothelial cells. After infection, a significant upregulation of Adamts4 expression was observed only in fibroblasts, which we conclude account for the bulk of Adamts4 expression in infected lungs (Fig. 2d).
To determine the specificity of ADAMTS4 gene expression in human lung disease, we performed a meta-analysis of publicly available scGEX datasets from seven studies of pulmonary fibrosis12,13,14, interstitial lung disease15, allergy and asthma16,17, and viral infection (with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2))18 (Extended Data Fig. 6 a). For reference, we included a study of rheumatoid arthritis, a disease driven by inflammatory fibroblasts19. ADAMTS4-positive cells included mesenchymal cells and endothelial cells, with a smaller number of epithelial and immune cells (Extended Data Fig. 6b, c). Fibroblasts with the highest levels of ADAMTS4 expression exhibited an activated phenotype with enriched expression of genes associated with DRFibs (Extended Data Fig. 6d). Analysis of a subset of data from a study by Habermann et al.12 indicated that expression of ADAMTS4 was markedly increased in inflammatory lung disease compared with healthy control tissue (Extended Data Fig. 6e). Mesenchymal cells expressed the highest levels of ADAMTS4 on a per-cell basis—at least an order of magnitude higher than other cell types, including endothelial cells (Extended Data Fig. 6f). Per-cell levels of ADAMTS4 were low in epithelial and immune cells and did not increase in diseased lungs compared to controls.
To determine how fibroblast responses were organized anatomically, we performed spatial transcriptomics on mouse lung sections collected at the peak of respiratory distress (10 days post-infection (dpi)). Unsupervised clustering of transcriptional profiles identified areas of the lung (red, 0) that overlapped with regions of interstitial inflammation (Fig. 2e, Extended Data Fig. 7a). Genes associated with DRFibs, including Itga5, Lox and Adamts4, were enriched in these regions (Fig. 2e, Extended Data Fig. 7b–d). Adamts4 expression was restricted to areas of interstitial inflammation near the alveoli. Expression of DRFib-associated genes was reduced or absent from bronchial regions (green, 2), areas of dense lymphoid infiltration (teal, 3), and from areas of normal alveolar parenchyma (yellow, 1). Together, these data place DRFibs expressing Adamts4 in the distal airways at a critical inflection point when hosts either begin to recover or their condition worsens.
ADAMTS4 promotes lethal immunopathology
To determine the contribution of ADAMTS4 to infection outcome, we challenged Adamts4−/− and Adamts4+/+ mice with a lethal dose of influenza A virus. Adamts4−/− mice exhibited improved survival compared with Adamts4+/+ controls, independently of the viral burden in the lungs (Fig. 3a, b, Extended Data Fig. 8a, b). Neither the frequency nor total number of infected cells differed substantially between Adamts4−/− and Adamts4+/+ mice, although there were modest cell-type-specific differences (Extended Data Fig. 8c). In contrast to wild-type mice, Adamts4−/− mice were able to maintain lung function, as measured by arterial oxygen saturation, at 6 dpi (Fig. 3c). At 9 dpi, Adamts4−/− mice exhibited a significant decrease in immune cell infiltration and alveolar inflammation, with less extensive lung tissue damage (Fig. 3d). Consistent with reduced damage, Adamts4−/− mice had less total protein in BAL fluid compared with Adamts4+/+ controls and lower levels of key inflammatory mediators (TNF and MCP-1) in lung homogenates (Extended Data Fig. 8d, e). These differences in tissue damage translated to improved lung mechanics in Adamts4−/− mice, as measured by airway resistance and dynamic lung compliance (Fig. 3e, Extended Data Fig. 8f).

a, Survival curves following challenge with influenza A virus (Adamts4+/+ (WT), n = 33; Adamts4−/− (KO), n = 23; log-rank test, two-sided; data pooled from five independent experiments). b, Representative images of lung from infected mice and quantification of viral spread. Areas of active infection are outlined in red (3 dpi: WT, n = 6; KO, n = 5, and 6 dpi: WT, n = 5; KO, n = 6). c, Arterial oxygen saturation (SpO2) at 3 and 6 dpi (WT, n = 5; KO, n = 5). d, Representative images and histological scores at 9 dpi with sublethal challenge (WT, n = 9; KO, n = 9). NP, viral nucleoprotein immunohistochemistry. e, Airway resistance with sublethal challenge (uninfected: WT, n = 8; KO, n = 3, and infected: WT n = 8; KO, n = 9). RI, resistance. f, Quantification of IFNγ+CD8+ T cells from lungs collected at 9 dpi (WT, n = 10; KO, n = 9). g, Quantification of major histocompatibility complex (MHC) tetramer+CD8+ T cells in the lung at 9 dpi (WT, n = 10; KO, n = 10) containing peptides from polymerase basic protein 1 (PB1), polymerase acid protein (PA) and nucleoprotein. h, Representative images of CD3 (green), intact versican (red) and DAPI (blue) staining in lung sections and quantification of CD3+ cells. Three to four fields of view imaged from each individual mouse (uninfected, n = 3; WT, n = 5; KO, n = 4). Scale bar, 50 μm. i, CD8+ T cell migration in the presence of Adamts4 wild-type and knockout fibroblasts. The number of migrated cells is normalized to uncoated control for each genotype (n = 6 biologically independent samples per group). j, CD8+ T cell migration with different fibroblast activation states (n = 4 biologically independent samples per group). PBS, vehicle control (uncoated membrane). Unless otherwise indicated, data are pooled from two independent experiments. Data are mean ± s.e.m. Groups were compared using a two-sided Mann–Whitney U test.
We examined the consequences of ADAMTS4 deficiency on the levels of versican, a major substrate of ADAMTS4. During inflammation, versican is an important component of the provisional matrix, a loose extracellular network that directs cell migration, proliferation and differentiation20; its function depends on its degradation state and interaction with other ECM proteins. Compared with wild-type mice, Adamts4−/− mice exhibited higher levels of intact versican in areas of lung remodelling after infection (Extended Data Fig. 9a, b). The levels of other known versicanases were not changed after infection in wild-type or Adamts4−/− mice (Extended Data Fig. 9c). As CD8+ T cells are one of the main drivers of immunopathology during infection with influenza A virus4,21, we assessed CD8+ T cell responses in the lungs at 9 dpi, the time point with peak T cell responses and lung damage. Adamts4−/− mice had a significantly lower percentage of IFNγ-producing CD8+ T cells (Fig. 3f, Extended Data Fig. 9d, e). The percentage of influenza A virus-specific CD8+ T cells did not differ between wild-type and Adamts4−/− mice, indicating that Adamts4−/− mice were able to mount influenza A virus-specific T cell responses similar to those of wild-type mice (Fig. 3g, Extended Data Fig. 9f). We observed significantly fewer T cells in lung sections of Adamts4−/− mice compared with Adamts4+/+ mice, with T cells being largely absent from areas with high intact versican (Fig. 3h). We next tested whether fibroblast-derived ADAMTS4 could promote T cell migration across a versican barrier (Fig. 3i). Activated CD8+ T cells migrated across uncoated and versican-coated membranes with nearly identical efficiency in the presence of ADAMTS4-sufficient fibroblasts. By contrast, migration across a versican-coated membrane was reduced by more than 40% in the presence of ADAMTS4-deficient fibroblasts. Consistent with DRFibs being the main producers of ADAMTS4, DRFibs promoted migration across a versican barrier, whereas migration was inhibited in the presence of resting fibroblasts and IRFibs (Fig. 3j), highlighting the distinct functions of the different fibroblast activation states.
ADAMTS4 and severe influenza in humans
We next investigated the contribution of ADAMTS4 in the human respiratory tract to severe influenza virus infection in the Paediatric Intensive Care Influenza (PICFLU) network. We analysed samples of endotracheal aspirate (ETA) collected within 72 h of admission to intensive care from a multicentre cohort of 84 paediatric patients with influenza virus infection (Extended Data Fig. 10a). Levels of ECM proteases and TIMPs were analysed in the context of inflammatory mediators22. Controlling for age, ADAMTS4 was positively correlated with levels of IL-1B and TNF, which we identified as upstream regulators of ADAMTS4 expression, and with IFNγ, which was expressed at a higher frequency in CD8+ T cells in ADAMTS4 sufficient mice during influenza A virus infection (Fig. 4a). We next tested whether levels of ADAMTS4 in ETA were associated with the severity of influenza-related disease using three indicators: prolonged multiple organ dysfunction syndrome (PrMODS) or death, prolonged acute hypoxic respiratory failure (PrAHRF) or death, and fewer than ten ventilator-free days (VFDs) (Extended Data Fig. 10b). In each case, the log of the ADAMTS4 concentration was significantly associated with disease severity outcome after controlling for age, gender and baseline health status (PrMODS/death: odds ratio = 3.14, P = 0.003; PrAHRF/death: odds ratio = 2.27, P = 0.018; VFD < 10: odds ratio = 2.95, P = 0.027; Fig. 4b, Extended Data Fig. 10c).

a, Correlation matrix of analytes for the PICFLU cohort. For each pairwise comparison, colour indicates the Spearman correlation coefficient and the size of the box indicates significance level. Analytes are arranged by hierarchical clustering. b, Odds ratios for VFD < 10 with gender, previously healthy, age and steroid treatment as covariates. Box plots of log10(1 + [ADAMTS4]) values for each severity indicator (n = 84 samples from individual patients). Centre line indicates median, box edges indicate first and third quartiles, and whiskers extend to 1.5 times the interquartile range. c, Correlation matrix of analytes from the first available time point for each patient in the Taiwan and Guangzhou cohorts. d, Odds ratio for VFD < 10 with gender and age as potential covariates. Odds ratios in b, d determined using logistic regression with false-discovery rate adjustments for multiple testing; positive (red) or negative (blue) association.
To determine whether these findings translated to adults, we analysed samples from two distinct, primarily adult cohorts: one from Taipei, representing moderate-to-severe cases of seasonal influenza (37 individuals) and one from Guangzhou, representing severe cases of H7N9 avian influenza (16 individuals) and seasonal H1N1 influenza (14 individuals) (Extended Data Fig. 10d). We tested samples from multiple respiratory compartments (ETA, sputum and BAL fluid). Individuals from the Taiwan cohort were sampled at a single time point after symptom onset, whereas most of those from the Guangzhou cohort were sampled at multiple time points.
A correlation matrix of the same analytes indicated similar patterns of ECM protease, TIMP and cytokine regulation in the cohorts of primarily adult patients with influenza (Fig. 4c). We tested whether ADAMTS4 was associated with a particular respiratory compartment, including 131 nasal wash samples from the Taiwan cohort. ADAMTS4 was detectable at very low levels in 5% (6 out of 131) of nasal wash samples tested, but at higher frequencies and concentrations in each of the lower respiratory tract (LRT) compartments, suggesting that the source of ADAMTS4 is localized to the LRT during infection (Extended Data Fig. 10e).
Finally, we tested whether ADAMTS4 is a predictor of disease severity during adult infection with influenza virus. To make the data comparable with the PICFLU cohort, we focused on the earliest available ETA sample, including only the first BAL fluid or sputum sample (in that order) when an ETA sample was not available. The log of the ADAMTS4 concentration was significantly associated with severity of disease, as determined by fewer than ten VFDs (odds ratio = 1.95, P = 0.020) (Fig. 4d). By combining samples from all three cohorts, the log of the ADAMTS4 concentration was associated with VFD values of less than ten, including age and gender as potential covariates (odds ratio = 2.22, P = 3.1 × 10−5) (Extended Data Fig. 10f). On the basis of these results, we propose that the level of ADAMTS4 protein in the LRT is a strong predictor of hypoxic respiratory failure and mortality following severe influenza virus infection across a wide range of ages.
Discussion
Surviving a severe respiratory infection is dependent on a delicate balance between mounting a sufficient immune response to clear the infection and maintaining lung function despite immunity-induced tissue damage. Fibroblasts are located at the interface of the epithelial site of infection and the endothelial entry point of immune cells, well placed to integrate inflammatory signals and coordinate immune responses through modification of the local tissue environment23. In this study we provide evidence that lung fibroblasts are critical for coordinating immune responses at the site of viral infection in the tissue, starting soon after infection and continuing throughout the repair process. During infection, lung fibroblasts respond to damaged epithelial cells, transmit inflammatory signals and modify the ECM to generate a tissue environment that promotes robust immune responses to infection. Recent studies have described both protective24 and pathogenic25,26,27 roles for ECM proteases in the host response to influenza virus. Whereas these studies identified immune cells as the primary sources of ECM proteases, the data presented here indicate that inflammatory fibroblasts integrate danger signals from epithelial cells and resident immune cells to produce diverse inflammatory cytokines, ECM components and degradative enzymes that modify the local tissue environment. In mice, deficiency of ADAMTS4 activity in the lung preserves intact ECM-localized versican and modulates immune cell migration into areas of infection. Previous studies have indicated that intact versican is able to suppress cytotoxic T cell responses and inhibit migration24,28,29,30. Our data from studies with human participants suggest that ADAMTS4 is an important determinant of influenza disease severity across a wide age range. High ADAMTS4 concentrations in endotracheal samples from the paediatric cohort—collected soon after admission to intensive care—were strong predictors of PrAHRF and PrMODS, and in all cohorts, high ADAMTS4 concentrations in LRT samples were associated with fewer ventilator-free days. The role of ADAMTS4 in promoting excessive immunopathology also warrants further investigation in the context of other severe respiratory infections, including infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Understanding the basis for activation phenotypes in fibroblasts will be a critical future direction for these studies. Determining whether these are true differentiation lineages or if the cells can revert back to a resting state will require the development of precise lineage-tracing models. Currently, there are no effective therapeutic agents to treat severe complications of respiratory viral infections such as ARDS, and treatment is supportive1. We propose that targeting the ECM protease activity of damage-responsive fibroblasts could provide clinical benefits, by both limiting excessive inflammation and maintaining the integrity of lung tissue, and that ADAMTS4 activity may be a suitable target for therapeutic intervention.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments. The investigators were blinded to outcome assessment in some experiments as indicated below.
Mice
B6.129P2-Adamts4tm1Dgen/J (Adamts4−/−) mice were obtained from Jackson Laboratories. These mice were backcrossed with C57BL/6J mice for at least 10 generations to generate congenic Adamts4−/− and Adamts4+/+ mice. Mice were housed at 68 °F, 12 h light/dark cycle and approximately 45% humidity.
Studies involving mice were approved by the St Jude Children’s Research Hospital Committee on Care and Use of Animals. All mice that were tested in this study were bred and maintained at the St Jude Children’s Research Hospital under specific-pathogen free conditions.
Single-cell RNA sequencing
Whole lungs were collected from pathogen-free C57BL/6 mice (Taconic Biosciences) at 0, 1, 3, 6 and 21 days after initial infection. Four or five mice for each time point were infected with 2500 EID50 of mouse-adapted influenza A/Puerto Rico/8/1934 (PR8) virus intranasally in 30 μl of 1× PBS31. Lungs were perfused with 10 ml cold 1× PBS injected into the right ventricle of the heart and then minced and digested in lung digestion buffer containing 400 U ml−1 collagenase I (Worthington Biochemical Corporation) and 50 U ml−1 of DNaseI (ThermoFisher) in Hank’s balanced salt solution (HBSS) for 75 min at 37 °C. Digested lungs were filtered through a 100-μm cell strainer and washed with HBSS. Red blood cell were lysed and resuspended at 1 × 106 cells per ml. Cells were incubated with Ghost Dye Violet 510 (Tonbo) (1:100 dilution) and APC anti-mouse CD45.2 (clone 104, Biolegend) (1:100 dilution) for 30 min at room temperature. Live, CD45− cells were sorted into 1.7-ml microfuge tubes and then centrifuged at 400g for 5 min. Cells were then incubated with PE-conjugated anti-mouse H-2 antibody (1:100, clone M1/42, Biolegend) for 30 min on ice.
Human lung biopsy samples from de-identified, deceased patients were obtained through the National Disease Research Interchange (NDRI). Cause of death of these patients was unrelated to respiratory tract infection or lung damage, and they did not have a history of tobacco use or lung cancer. Fresh 5 cm × 5 cm × 5 cm lung biopsy samples were minced and digested in 400 U ml−1 of collagenase I (Worthington Biochemical Corporation) and 50 U ml−1 of DNaseI (ThermoFisher) in HBSS for 90 min at 37 °C. Lung samples were first filtered through sterile gauze and then through a 100-μm cell strainer and washed with HBSS, then red blood cell lysis was performed. Single-cell suspensions of total lung cells were frozen in freeze medium (90% FBS, 10% DMSO) at a concentration of 5 × 106 cells per ml. For single-cell RNA sequencing using 10X Genomics, vials of cells were thawed and incubated with Ghost Dye Violet 510 (Tonbo) (1:100 dilution) and APC anti-human CD45 (1:200) for 30 min at room temperature. Live, CD45− cells and live, CD45+ cells were sorted into 1.7-ml microfuge tubes and then centrifuged at 400g for 5 min. Following sorting, cells were incubated with PE anti-human β2 microglobulin (1:100) and PE anti-human CD45 (1:100) antibodies for 30 min on ice.
For both mouse and human lung samples, cells were washed three times and then incubated with custom-designed hashtag oligos (HTO) conjugated to anti-PE antibodies (Thunder-Link PLUS Oligo Conjugation System, Expedeon), unique for each individual mouse. After incubation, cells were washed three times and then pooled within each time point. Approximately 25,000 cells per sample were loaded onto the Chromium controller (10X Genomics) to partition single cells into gel beads. Single-cell transcriptomic libraries were generated using the 5′ Gene Expression Kit (V2, 10X Genomics) according to the manufacturer’s instructions with the addition of primers to amplify HTOs during cDNA amplification. Sequencing was performed on the Illumina NovaSeq to generate approximately 500 million reads per sample.
Single-cell gene-expression analyses
The 10X gene-expression data were first processed using CellRanger (v.3.0.2, 10X Genomics). Human data were processed using version 3.0.0 of the GRCh38 10X reference, whereas mouse data were processed using the mm10 reference altered to include the PR8 influenza genome. CellRanger was then used to aggregate species-specific samples, normalizing by the number of mapped reads per identified cell. Normalized feature-barcode matrices including both gene expression and HTO counts were then imported into Seurat (v.3.0.0.900) for downstream analysis32.
Data were first filtered by excluding any gene that was not present in at least 0.1% of total called cells and then by excluding cells that exhibited extremes in the species-specific distributions of: the number of genes expressed (mouse: <100 or >5,550; human: <100 or >4,500), the number of mRNA molecules (mouse: >30,000; human: >20,000), or the percentage of expression owed to mitochondrial genes (mouse and human: >7.5%). Gene-expression counts were log-normalized using a scaling factor of 1 × 104, variable features targeted for downstream analysis were identified using the ‘vst’ method with default parameters, and cell-cycle scores were generated for each cell using markers identified elsewhere33. Gene expression was then scaled to regress out the effects of total transcript expression, percent of mitochondrial expression, and cell cycle scores. For first-pass analyses of entire datasets, we selected principal components (PCs) visually using an elbow plot of the PC standard deviations (mouse: 25; human: 19). These PCs were then used for t-SNE dimensionality reduction and cell clustering with Seurat’s shared-nearest-neighbour modularity algorithm to broadly characterize the cell types in the entirety of each sample. Individual cell subsets were annotated using known markers from the literature.
For more detailed analyses of fibroblast differentiation states, we generated a subset of the identified fibroblast clusters, identified the top variable features within this subset, and re-scaled those data as described above. For the mouse fibroblast data, we also regressed out the effects of days post infection to more precisely characterize the variance among fibroblast clusters and distinguish cellular responses to experimental infection. After repeating principal component analysis, principal components were scored for significance (false-discovery rate (FDR)-adjusted P < 0.05) using random permutations as implemented in Seurat. Differential gene expression was assessed among clusters for all genes expressed in at least 1% of cells within a cluster using a generalized linear hurdle model that incorporates both expression frequency and abundance34. For pairwise cluster comparisons of interest, genes were then ranked as a function of the product of their average log fold change, the absolute value of the difference in percent expression, and the inverse of the scaled, FDR-adjusted P value. These gene-rank lists were then analysed using preranked GSEA35.
Meta-analysis of publicly available human single-cell gene-expression datasets
Human single-cell gene-expression data from published reports (Extended Data Fig. 6a) were obtained from online data repositories. From ref. 17 we were only able to obtain data from four healthy controls, as other data, including the data from patients with asthma, were not publicly available. Each dataset was filtered to exclude cells with more than 20% mitochondrial content, fewer than 200 genes, or fewer than 200 RNA molecules before being normalized, scaled, and assessed for variable features using the SCTransform wrapper in Seurat32. Focal analyses of specific datasets were conducted on these resulting data objects. We also compiled the datasets for meta-analysis using reference-based integration with 3,000 integration features, with data from ref. 12 as the reference, again using the SCTransform normalization method. Downstream analyses were focused on the subset of cells that contained at least one ADAMTS4 transcript. Dimensionality reduction and clustering were conducted as described previously, but in this case using the integration assay. Differential gene-expression analyses were conducted on the RNA assay of the integrated object using default parameters in Seurat.
Flow cytometry for lung stromal cells and FACS
At the indicated time point after infection, lungs were perfused with 10 ml cold 1× PBS injecting into the right ventricle of the heart and then minced and digested in lung digestion buffer containing 400 U ml−1 of collagenase I (Worthington Biochemical Corporation) and 50 U ml−1 of DNaseI (ThermoFisher) in HBSS for 30 min at 37 °C. Digested lungs were filtered through a 100-μm cell strainer and washed with HBSS. Red blood cell lysis was performed, and cells were washed and resuspended in fluorescence-activated cell sorting (FACS) buffer (1% FBS and 2 mM EDTA in 1× PBS). CD45+ cells were depleted using mouse CD45 Microbeads (Miltenyi) according to the manufacturer’s instructions. In brief, cells were incubated with microbeads for 15 min on ice, washed with FACS buffer, and resuspended in 500 μl of buffer. CD45+ cells were then depleted using a Miltenyi LS column, and the flow through (CD45− cells) were collected. For antibody staining, 1 × 106 cells were plated and then incubated with Fc block for 10 min at room temperature. Cells were washed one time with FACS buffer and then resuspended in 100 μl of an antibody cocktail containing the following: Ghost Dye Violet 510 (1:100), BV605 anti-mouse CD45 (1:200), BV421 anti-mouse EpCAM (1:200), PerCP/Cy5.5 anti-mouse CD31 (1:200), FITC anti-mouse BST2 (1:100), PE anti-mouse CD9 (1:100) and APC anti-mouse CD49e (1:100). Cells were incubated with the antibody cocktail for 30 min at room temperature and then washed twice with FACS buffer before resuspending in a final volume of 150 μl. For single-cell suspensions from human lung biopsy samples, cells were washed one time with FACS buffer and then resuspended in 100 μl of an antibody cocktail containing the following: Ghost Dye Violet 510 (1:100), BV605 anti-human CD45 (1:100), BV785 anti-human EpCAM (1:100), APC Fire 750 anti-human CD31 (1:100), PE/Cy7 anti-human CD140a (1:100), APC anti-human CD49e (1:100) and PE anti-human CD9 (1:100). Cells were incubated with the antibody cocktail for 30 min at room temperature and then washed twice with FAC buffer before resuspending in a final volume of 150 μl. Cells were analysed using a BD LSRII Fortessa and BD FACSDIVA software (Becton Dickinson). Data were analysed using FlowJo software (FlowJo).
High-throughput quantitative PCR
For in vitro stimulation of primary mouse lung fibroblasts, fibroblasts were isolated from C57BL6/J. Lungs were collected, minced, and then digested in lung digestion buffer containing 0.1% collagenase I (Worthington Biochemical Corporation) and 2.4 U ml−1 dispase (Thermo Fisher) for 90 min at 37 °C. Digested lungs were filtered through a 100-μm cell strainer and then washed with 15 ml of 0.05 M EDTA in 1× PBS. Cells were centrifuged at 400g for 10 min and then resuspended in complete Dulbecco’s modified Eagle medium (cDMEM) and plated in a T75 tissue-culture flask (Corning). For cytokine and virus stimulations, 2 × 105 cells were plated in each well of a 24-well plate 24 h before stimulation. Fibroblasts were stimulated with the following mouse cytokines: IL-1B (10 ng ml−1), IL-1A (10 ng ml−1), IL-33 (10 ng ml−1), IL-18 (10 ng ml−1), TNF (200 ng ml−1), IFNα (1,000 U ml−1), IL-6 (40 ng ml−1), TGFβ (2 ng ml−1), IL-17A (50 ng ml−1), IL-27 (50 ng ml−1), and GM-CSF (40 ng ml−1). For the virus stimulations, cells were infected at a multiplicity of infection (MOI) of 2. Virus inoculum was incubated with the cells for 1 h at 37 °C. The following influenza viruses were used for the stimulations: A/Puerto Rico/8/1934 (PR8), A/California/04/2009 (CA09) and A/Perth/16/09 (Perth). The supernatant was removed, and cells were washed three times with cDMEM. The wells were replenished with 1 ml of cDMEM, and the cells were incubated at 37 °C for 24 h. To collect BAL fluid, a catheter was inserted into the trachea, and 3 ml of 1× PBS was used to wash the lungs. BAL fluid was centrifuged at 400g for 5 min to pellet cells, and then the cells were resuspended in 350 μl Trizol reagent.
For co-culture experiments, normal human lung fibroblasts (Lonza) were plated at a density of 2,500 cells per cm2 in the basal chamber of a 24-well plate and cultured in fibroblast growth medium (Lonza) overnight at 37 °C and 5% CO2. NHBEs were seeded at a density of 10,000 cells per cm2 in 12-mm Transwell inserts (Corning) coated with human placental collagen and then added to plates containing fibroblasts. Co-cultures were cultured in bronchial epithelial growth medium (BEGM) (Lonza) at 37 °C and 5% CO2 for 7–10 days. Co-cultures were then taken to the air–liquid interface and basolateral medium was exchanged for medium consisting of 50% DMEM and 50% BEGM supplemented with the BEGM BulletKit (Lonza). Medium was changed every 2–3 days until the cells were fully differentiated (2–3 weeks). For infections, NHBEs were inoculated at a MOI of 2 using the following influenza A virus strains: H3N2 A/Perth/16/09, H5N6 A/Oriental Magpie Robin/Hong Kong/6154/2015, and H7N9 A/Hong Kong/125/2017. Infections with avian strains were performed in an ABSL3+ facility.
For monoculture experiments, NHBEs from a single donor were plated in 24-well Transwell plates (Corning) and cultured in bronchial epithelial basal medium (BEBM) (Lonza) supplemented with bovine pituitary extract, insulin, hydrocortisone, gentamicin-amphotericin, transferrin, triiodothyronine, epinephrine, epidermal growth factor and retinoic acid until wells were confluent and cells formed tight junctions. Supernatant was then removed from the apical side of the Transwell and cells were taken into the air–liquid interface for 2–3 weeks. NHBEs were then stimulated with the following human cytokines adding each factor to medium in the basal chamber: IL-1B (10 ng ml−1), IL-1A (10 ng ml−1), IL-33 (10 ng ml−1), IL-18 (10 ng ml−1), TNF (200 ng ml−1), IFNα (1,000 U ml−1), IL-6 (40 ng ml−1), TGF-β (2 ng ml−1), IL-17A (50 ng ml−1), IL-27 (50 ng ml−1), and GM-CSF (40 ng ml−1). For virus stimulations, cells were infected at an MOI of 2. Virus inoculum was incubated with the cells in the apical chamber for 1 h at 37 °C. The supernatant was removed, and cells were washed three times with BEBM, and incubated at 37 °C for 24 h. Normal human bronchial fibroblasts (NHBFs) (Matek) were plated in 24-well plates at a density of 2 × 105 cells per well and cultured in cDMEM. NHBFs were stimulated with the cytokines and viruses listed above. Following incubation, the supernatant was removed and cells were collected in 350 μl Trizol Reagent (Thermo Fisher). RNeasy spin columns (Qiagen) were then used to isolate RNA from the samples according to the manufacturer’s instructions. Following RNA isolation, the amount of RNA input was normalized (200 ng), and cDNA was generated using the iScript cDNA synthesis kit (Biorad). cDNA was then prepared for quantitative PCR (qPCR) using the Fluidigm Biomark (Fluidigm). Data were collected using the Fluidigm Biomark data collection software (Fluidigm). Exon-spanning qPCR primers were designed to target a panel of 96 ECM-related genes for humans (Supplementary Table 1) and mice (Supplementary Table 2).
For whole lung homogenates from mice, lungs were collected at 0, 1, 3, 6, 9, 12, 15, 30 and 40 days after infection. C57BL6/J mice were infected with 2500 EID50 PR8. Whole lungs were dissected and homogenized in 750 μl Trizol reagent using the TissueLyser II (Qiagen). RNA was isolated according to the manufacturer’s protocol.
Spatial transcriptomics
C57BL/6J (Jackson Laboratories) were infected intranasally with 2,500 50% egg infective dose (EID50) of influenza A virus PR8 in 30 μl of 1× PBS. Lungs were dissected from the mice 10 days after infection. Sections obtained from four distinct mice were imaged and processed for spatially resolved gene expression using the Visium Spatial Transcriptomics kit (10x Genomics). Lungs were inflated with 300 μl of 50% OCT and 50% PBS v/v and immediately snap-frozen in OCT using isopentane that was cooled in a liquid nitrogen bath. For each lung, the same left lobe was collected and stored at −80 °C until cryosectioning. For cryosectioning, samples were equilibrated to −22 °C. Exploratory sections were stained with a fluorescent lectin and DAPI to determine areas of immune infiltration and remodelling, and then blocks were trimmed to 5 × 5 mm to encompass normal and remodelled areas before sectioning directly onto the spatial transcriptomics kit slide. Tissue permeabilization was optimized at 12 min on independent lung sections. Libraries were sequenced on the Illumina NovaSeq platform at 28 × 120 base pairs, and resulting data were processed using SpaceRanger (v.1.0.0, 10X Genomics) with manual alignment of fiducial markers, manual tissue identification, and r2-length trimmed to 91 base pairs. Downstream analyses were conducted with Seurat (v.3.1.4.9901)32.
Adamts4 −/− and Adamts4 +/+ survival experiments
Mice were anaesthetized with 2,2,2-tribromoethanol by intraperitoneal injection and infected intranasally with 6,000 EID50 of PR8 in 30 μl 1× PBS. Following infection, mice were monitored twice daily and their weight was recorded. Mice were euthanized if they become severely moribund based on body index score and substantial weight loss31. Righting reflex of severely moribund mice was assessed to determine whether immediate euthanasia was necessary. Mice that lost more than 20% of their body weight were placed on their backs, and if unable to right themselves within 10 s, were euthanized. Mice were identified by ear tag numbers, and investigators assessing morbidity were blinded to the genotype.
Plaque titres
BAL fluid was used to obtain viral titres inoculating Madin–Darby Canine Kidney (MDCK) cells. MDCKs were obtained from and authenticated by the American Type Culture Collection (ATCC). The cells were not tested for mycoplasma contamination. MDCKs were plated in 6-well tissue culture plates at a density of 4 × 105 cells per well. Twenty-four hours after plating, cells were washed three times with 1× PBS and then incubated with 1 ml of BAL fluid, after six tenfold dilutions in serum-free MEM. Cells were incubated with inoculum for 10 min at 4 °C and then for 50 min at 37 °C. Inoculum was removed and 3 ml of MEM containing 0.9% agarose and of trypsin containing 1 mg ml−1 TPCK was overlaid onto the cell monolayer. Plates were then incubated at 37 °C for 72 h, and then plaques were counted after staining the cells with crystal violet.
Pulse oximetry
Mice were inoculated intranasally with 6,000 EID50 of influenza A virus (IAV) PR8 and arterial oxygen saturation was measured using the MouseOx system (STARR Life Sciences). Following infection, fur was removed around the neck of the mouse, and arterial oxygen saturation (SpO2) was measured using a throat collar sensor. After acclimatization, oxygen saturation measurements were recorded for 3 min, and measurements were averaged over the measurement period.
Histology and morphometry viral-spread analysis
Lungs were dissected at 3 and 6 days after infection with 2,500 EID50 of PR8. Lungs were inflated with 1 ml of 10% neutral-buffered formalin, and then placed in a 15-ml conical tube containing a further 3 ml of formalin. Lungs were incubated in formalin at room temperature for at least 72 h to ensure complete fixation of the tissue. Lungs were embedded in paraffin blocks and then sectioned onto glass slides. Serial tissue sections were stained with haematoxylin and eosin for histology. Immunohistochemical labelling of viral antigen was completed by using a primary goat polyclonal antibody (US Biological) against influenza A, USSR (H1N1) at 1:1,000 and a secondary biotinylated donkey anti-goat antibody (sc-2042; Santa Cruz Biotechnology) at 1:200 on tissue sections subjected to antigen retrieval for 30 min at 98 °C. The extent of virus spread was quantified by first capturing digital images of whole-lung sections stained for viral antigen using an Aperio ScanScope XT Slide Scanner (Aperio Technologies) and then manually outlining fields with the alveolar areas containing virus antigen-positive pneumocytes. The percentage of each lung field with infection/lesions was calculated using the Aperio ImageScope software. For histologic grading of lesions, a pathologist blinded to treatment group identity evaluated pulmonary lesions in haematoxylin and eosin-stained histologic sections and assigned scores on the basis of their severity and extent as follows: 0, no lesions; 1, minimal, focal to multifocal, barely detectable; 15, mild, multifocal, small but conspicuous; 40, moderate, multifocal, prominent; 80, marked, multifocal coalescing, lobar; 100, severe, diffuse, with extensive disruption of normal architecture and function.
Assessment of IAV-infected cells by flow cytometry
Lungs were perfused with 10 ml cold 1× PBS injected into the right ventricle of the heart and then minced and digested in lung digestion buffer containing 400 U ml−1 collagenase I (Worthington Biochemical Corporation) and 50 U/ml of DNaseI (ThermoFisher) in HBSS for 30 min at 37 °C. Digested lungs were filtered through a 100-μm cell strainer and washed with HBSS. Red blood cell lysis was performed, and cells were washed and resuspended in FACS buffer. Cells were washed and then incubated with Trustain FcX anti-mouse CD16/CD32 (Fc Block) at a dilution of 1:100 for 10 min at room temperature. For surface staining, cells were then washed and incubated in 100 μl of FACS buffer (1% FBS, 1 mM EDTA in 1× PBS) containing Ghost Dye Violet 510 (1:100), PE/Cy7 anti-mouse CD45.2 (1:200), PacBlue anti-mouse CD11c (1:100), APC/Cy7 anti-mouse CD11b (1:100), BV650 anti-mouse Gr-1 (1:200), APC anti-mouse CD326 (EpCAM) (1:100), PE anti-mouse CD90.2 (1:100), PerCP/Cy5.5 anti-mouse CD31 (1:100) for 30 min at room temperature. For intracellular staining, cells were fixed and membranes permeabilized by incubating in 100 μl fixation–permeabilization solution (BD Biosciences) for 20 min on ice. Cells were washed with permeabilization buffer and then incubated with FITC anti-influenza A (1:100).
Assessment of CD8+ T cell responses
In order to assess immune cell populations by flow cytometry, whole lungs were dissected, minced, and incubated with spleen dissociation medium (STEMCELL) for 30 min at 37 °C. Red blood cell lysis was performed, and cells were washed with HBSS and counted using a Vi-CELL XR cell counter (Beckman Coulter). For ex vivo stimulation and intracellular cytokine staining, 1 × 106 total lung cells were plated in a 96-well plate for each condition. Cells were stimulated with Cell Stimulation Cocktail (Biolegend), containing PMA, ionomycin and brefeldin A, at a dilution of 1:500 or with influenza peptides at a concentration of 1 μM for 4 h. Following stimulation, cells were washed and then incubated with Trustain FcX anti-mouse CD16/CD32 (Fc Block) at a dilution of 1:100 for 10 min at room temperature. For surface staining, cells were then washed and incubated in 100 μl FACS buffer (1% FBS, 1 mM EDTA in 1× PBS) containing Live/Dead Aqua (1:100), APC anti-mouse CD45.2 (1:100), FITC anti-mouse CD3 (1:100), BV650 anti-mouse CD8 (1:200) and APC/Cy7 anti-mouse CD4 (1:200) for 30 min at room temperature. For intracellular staining, cells were fixed and membranes permeabilized by incubating in 100 μl fixation–permeabilization solution (BD Biosciences) for 20 min on ice. Cells were washed with permeabilization buffer and then incubated with PE/Cy7 anti-mouse IFNγ (1:100) for 30 min on ice.
For IAV peptide:MHC tetramer staining, 1 × 106 total lung cells were plated in a 96-well plate for each sample. Cells were incubated with APC PB1 (residues 703–711) tetramer (1:750), PE PA (residues 224–233) tetramer (1:750), and BV785 NP (residues 366–374) tetramer (1:250) for 1 h on ice. Then, cells were surface stained with FITC anti-mouse CD3 (1:100), BV650 anti-mouse CD8 (1:200), APC/Cy7 anti-mouse CD4 (1:200) for 30 min on ice.
Lung tissue immunofluorescence
Lungs were dissected 9 days after infection with 2,500 EID50 of PR8. Lungs were inflated with 1 ml fixative containing 2% paraformaldehyde, 0.1% Triton X-100 and 1% DMSO and then transferred to a 15 ml conical tube containing 3 ml of fixative for 24 h before cryoprotection with 30% sucrose in PBS for an additional 24 h. Tissues were cryosectioned at 10 μm thickness and blocked in buffer comprised of PBS containing 2% bovine serum albumin and 5% donkey serum. Tissues were stained overnight in blocking buffer containing 4 μg ml−1 rat anti-CD3 (Biolegend, clone 17A2) and 4 μg ml−1 rabbit anti-mouse versican GAG beta domain (Millipore Sigma, AB1033). Sections were washed in PBS before incubation with CF488-conjugated donkey anti-rat IgG (Biotium, 20027, lot 16C0301, 2 μg ml−1), CF568-conjugated donkey anti-rabbit IgG (Biotium, 20098, lot 19C0110, 2 μg ml−1) and 10 μM DAPI for 1 h at room temperature. Slides were washed with PBS and mounted with Prolong Diamond Hardset Mounting Medium (ThermoFisher). High-resolution images were acquired using a Marianis spinning disk confocal microscope (Intelligent Imaging Innovations) equipped with a 40× 1.3NA Plan-Neofluar objective, 405-nm, 488-nm and 561-nm laser lines and Prime 95B CMOS camera (Photometrics), and analysed using Slidebook software (Intelligent Imaging Innovations). For quantification of CD3+ cells, 3–4 fields of view in the areas of tissue remodelling were collected for each lung.
Lung function test
For lung function studies, mice were infected with 2,500 EID50 of PR8. At 10 dpi, mice were anaesthetized with 2,2,2-tribromoethanol by intraperitoneal injection and a tracheal tube was inserted. Airway resistance and dynamic lung compliance were measured using the Buxco Finepointe Resistance and Compliance system (Data Sciences International). Data were collected using Buxco Finepointe software (Data Sciences International). Mice were mechanically ventilated at a rate of 140 breaths per minute. Five methacholine challenges were performed at doses of 1.5625, 3.125, 6.25, 12.5 and 25 mg ml−1, administering the methacholine in 0.010 ml PBS. Following each methacholine challenge, resistance and compliance were measured for 3 min allowing the mice to recover for 1 min after each measurement. Measurements for resistance and compliance were averaged over the 3 min measurement period for each methacholine challenge and baseline measurements.
T cell migration assays
For migration assays, CD8+ T cells were isolated from spleens of C57BL/6J mice. To enrich for CD8+ T cells, samples were magnetically depleted using a cocktail of the following biotinylated antibodies: anti-mouse B220 (1:50), anti-mouse MHC class II (1:50), anti-mouse CD11b (1:100), anti-mouse CD49b (1:200), and anti-mouse CD4 (1:100). Up to 109 total spleen cells were incubated with the antibody cocktail for 15 min on ice. Cells were washed with FACS buffer and then incubated with Miltenyi Streptavidin microbeads (1:25) for 15 min on ice. The cells were centrifuged at 500g for 5 min and then resuspended in 1 ml FACS buffer. The sample was added to a pre-equilibrated Miltenyi LS column and the flow-through enriched by CD8+ T cells was collected. CD8+ T cells were then activated with anti-mouse CD3e (1 μg ml−1) and anti-mouse CD28 (3 μg ml−1) antibodies in complete RPMI at 37 °C for 72 h. Forty-eight hours before the migration assay, primary mouse lung fibroblasts, which were plated at a concentration of 105 cells per well in a 24-well plate the previous night, were stimulated with IL-1B (10 ng ml−1) and TNF (200 ng ml−1). For activation states (resting, DRFib, IRFib), sorted fibroblasts were plated at a density of 104 cells per well 48 h before the migration assay. 6.5 mm polycarbonate Transwell inserts with 8.0-μm pore size (Corning) were coated with 10 μg ml−1 versican purified from bovine aorta29 for 2 h at 37 °C. Versican was aspirated and Transwell inserts were transferred to the 24-well plate with stimulated fibroblasts. To test migration, 105 activated CD8+ T cells were added to the apical chamber of the Transwell in a volume of 100 μl complete DMEM. Cells were allowed to migrate to the basal chamber for 4 h at 37 °C. After 4 h, supernatants from the basal chamber were collected and the number of lymphocytes were manually counted.
Quantification of proteins in mouse lung homogenates and bronchoalveolar lavage fluid
Cytokine levels in lung tissue homogenates were measured using LEGENDplex cytokine bead arrays (Biolegend). Lungs were dissected from PR8-infected mice at 6 dpi and homogenized in 500 μl of PBS. Homogenized lung samples were centrifuged at 4,000 rpm for 10 min, and 25 μl of undiluted supernatant was tested in the assay. The assay was performed according to the manufacturer’s instructions.
BAL washes were performed using 500 μl of PBS. Matrix protease levels in mouse BAL fluid were quantified using ELISA or a multiplexed, Luminex-based fluorescent assay. A custom Luminex kit (R&D Systems) was used to assay the following analytes: MMP-2, MMP-8, MMP-9, MMP-12, TIMP-1 and TIMP-4. Duoset ELISA kits (R&D Systems) were used to assay the following analytes: ADAMTS4, MMP-3, MMP-13, and TIMP-3.
Human cohorts
For the PICFLU cohort, the study was approved by the lead site Boston Children’s Hospital (IRB X08-11-0534) and then at each enrolling site’s IRB (enrolling sites are listed in acknowledgements) and written informed consent was obtained from at least one parent or guardian. Patients were excluded before enrolment if they had an underlying condition predisposing them to severe influenza infection as has been described previously36,37. Additional cohort information is presented in Extended Data Fig. 10a. Endotracheal aspirate samples were obtained using standardized methods with 1.5 ml of sterile normal saline down the endotracheal tube then 3–4 breaths before obtaining the sample with 2 suctioning attempts aiming to collect at least 2 ml of aspirate. The sample was put on ice and transferred immediately to the laboratory for processing. One aliquot was immediately frozen (−80 °C) and the rest was centrifuged. The supernatant was aliquoted and frozen at −80 °C.
For the Guangzhou cohort, the study was approved by the ethics committee of the First Affiliated Hospital of Guangzhou Medical University (no: 2016-78), and informed consent was obtained from all patients or their guardians. Additional cohort information is presented in Extended Data Fig. 10c.
For the Taiwan cohort, the human participants’ protocol was reviewed and approved by the Johns Hopkins School of Medicine and the Chang Gung Memorial Hospital Institutional Review Boards (IRB00091667). Informed consent was obtained from all patients or their guardians. This protocol allowed for the collection of residual BAL and sputum samples that were initially collected for clinical purposes from patients with influenza (Extended Data Fig. 10c).
For lung samples from de-identified, deceased donors, the tissues used were procured by the NDRI with support from NIH grant U42OD11158. All NDRI consent forms and protocols were reviewed and approved by the Institutional Review Board at the University of Pennsylvania. Research involving these tissues was determined to not involve human participants by the Institutional Review Board at St Jude Children’s Research Hospital.
Quantification of matrix protease and cytokine/chemokine protein levels in human samples
Cytokine and chemokine levels were measured using Milliplex Human Cytokine/Chemokine Magnetic Bead Panel—Premixed 41 plex. MMP and TIMP protein levels were quantified using the following Milliplex bead-based multiplexed assay: Human MMP Magnetic Bead Panel 1 (MMP-3, MMP-12, MMP-13), Human MMP Magnetic Bead Panel 2 (MMP-1, MMP-2, MMP-7, MMP-9, MMP-10) and Human TIMP Magnetic Bead Panel 2 (TIMP-1, TIMP-2, TIMP-3, TIMP-4). ADAMTS4 and ADAMTS5 were measured using Duoset ELISA kits (R&D Systems).
Cytokine correlation matrices
Correlation matrices were generated using samples analysed in severity analyses, with log10-transformed cytokine levels. To control for potential effects of age and day of sample collection (days after symptom onset), we computed partial pairwise Spearman correlations and assessed their statistical significance using the R psych package (https://cran.r-project.org/web/packages/psych/index.html). Correlations were visualized using the R corrplot package (https://github.com/cran/corrplot/blob/master/R/corrplot.R), and correlations with P < 0.05 after FDR adjustment were excluded.
Severity analyses
For analysis of association of ADAMTS4 levels with severity of infection in the PICFlu cohort, we used three distinct indicators of severity: PrMODS or death, PrAHRF or death, and VFDs less than 10. In survivors, ‘prolonged’ was defined as being present on or after day 7 of admission to paediatric intensive care or being on extracorporeal membrane oxygenation on or after day 7. PrAHRF was defined as partial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2) ratio less than 200 and mechanical ventilator support, which are criteria for moderate-severe ARDS1. VFDs were defined as days alive and free of invasive or non-invasive mechanical ventilation up to 28 days38. Owing to the bimodal distribution of VFDs, we used a cut-off of 10 VFDs to classify cases as severe (Extended Data Fig. 10b) ADAMTS4 was modelled as a predictor of these categorical outcomes using logistic regressions in R that explicitly included gender, age, baseline health (‘previously healthy’), and steroid administration. FDR adjustments for multiple testing were applied independently to each model. Patients were considered ‘previously healthy’ if they were, upon admission, otherwise healthy, on no prescription medications, without underlying medical conditions, and not dependent on any medical devices before initial admission to the hospital for flu. No violations of the model assumptions were found in any reported results after specifically checking for: (1) the assumption that the relationship between continuous predictor variables and the logit of the outcome is linear, (2) evidence of extreme values in continuous predictors (as indicated by standard residuals with an absolute value over three), and (3) evidence of multicollinearity among variables (as indicated by variance inflation factors (VIFs)). Odds ratios were visualized using the sjPlot package in R (https://strengejacke.github.io/sjPlot/).
Sequence data used for analyses
Hallmark gene sets from the Molecular Signatures Database were used for GSEA (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). The following publicly available datasets were used for meta-analysis: http://shaleklab.com/wp-content/uploads/2018/08/20180822_PolypAll_cleaned_rawdata.txt.zip; http://shaleklab.com/wp-content/uploads/2018/06/20180822_PolypAll_cleaned_metadata.txt; https://browser.immport.org/browser?path=SDY998%2FResultFiles%2FRNA_sequencing_result, GSE145926_RAW.tar (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145926); GSE135893_barcodes.tsv.gz, GSE135893_genes.tsv.gz, and GSE135893_IPF_metadata.csv.gz (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135893); GSE130148_raw_counts.csv.gz and GSE130148_barcodes_cell_types.txt.gz (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130148); GSE122960_RAW.tar (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE122960); GSE128169_RAW.tar (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128169); and GSE132771_RAW.tar (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132771).
Statistical analyses
For experiments involving mice, statistical analyses were performed using Graphpad Prism v.7.0d software. For analysis of human samples, statistical analyses were performed in R statistical software v.3.5.0 and as detailed in the sections above.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Cohort cytokine and ADAMTS4 data are available in Supplementary Tables 3, 4. Single-cell and spatial gene-expression data are available on the NCBI Short Read Archive under BioProjects PRJNA612345 and PRJNA613670. Source data are provided with this paper.
Code availability
All analyses were conducted using publicly available software as detailed in the methods. All code is available upon request.
Change history
18 November 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Matthay, M. A. et al. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 5, 18 (2019).
Soares, M. P., Teixeira, L. & Moita, L. F. Disease tolerance and immunity in host protection against infection. Nat. Rev. Immunol. 17, 83–96 (2017).
Forum of International Respiratory Societies. The Global Impact of Respiratory Disease 2nd edn (European Respiratory Society, 2017).
Duan, S. & Thomas, P. G. Balancing immune protection and immune pathology by CD8+ T-cell responses to influenza infection. Front. Immunol. 7, 25 (2016).
Bonnans, C., Chou, J. & Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 15, 786–801 (2014).
Hynes, R. O. & Naba, A. Overview of the matrisome—an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 4, a004903 (2012).
Sorokin, L. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 10, 712–713 (2010).
Iwasaki, A. & Pillai, P. S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 14, 315–328 (2014).
El Agha, E. et al. Two-way conversion between lipogenic and myogenic fibroblastic phenotypes marks the progression and resolution of lung fibrosis. Cell Stem Cell 20, 261–273 (2017).
Oshansky, C. M. et al. Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am. J. Respir. Crit. Care Med. 189, 449–462 (2014).
Kelwick, R., Desanlis, I., Wheeler, G. N. & Edwards, D. R. The ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) family. Genome Biol. 16, 113 (2015).
Habermann, A. C. et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 6, eaba1972 (2020)
Tsukui, T. et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat. Commun. 11, 1920 (2020).
Reyfman, P. A. et al. Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 199, 1517–1536 (2019).
Valenzi, E. et al. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann. Rheum. Dis. 78, 1379–1387 (2019).
Ordovas-Montanes, J. et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature 560, 649–654 (2018).
Vieira Braga, F. A. et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 25, 1153–1163 (2019).
Liao, M. et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26, 842–844 (2020).
Zhang, F. et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 20, 928–942 (2019).
Wight, T. N. Provisional matrix: A role for versican and hyaluronan. Matrix Biol. 60-61, 38–56 (2017).
La Gruta, N. L., Kedzierska, K., Stambas, J. & Doherty, P. C. A question of self-preservation: immunopathology in influenza virus infection. Immunol. Cell Biol. 85, 85–92 (2007).
Fiore-Gartland, A. et al. Cytokine profiles of severe influenza virus-related complications in children. Front. Immunol. 8, 1423 (2017).
Wynn, T. A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 208, 1339–1350 (2011).
McMahon, M. et al. ADAMTS5 is a critical regulator of virus-specific T cell immunity. PLoS Biol. 14, e1002580 (2016).
Bradley, L. M., Douglass, M. F., Chatterjee, D., Akira, S. & Baaten, B. J. G. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog. 8, e1002641 (2012).
Rojas-Quintero, J. et al. Matrix metalloproteinase-9 deficiency protects mice from severe influenza A viral infection. JCI Insight 3, 21 (2018).
Talmi-Frank, D. et al. Extracellular matrix proteolysis by MT1–MMP contributes to influenza-related tissue damage and mortality. Cell Host Microbe 20, 458–470 (2016).
Chang, M. Y. et al. Versican is produced by Trif- and type I interferon-dependent signaling in macrophages and contributes to fine control of innate immunity in lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 313, L1069–L1086 (2017).
Evanko, S. P., Potter-Perigo, S., Bollyky, P. L., Nepom, G. T. & Wight, T. N. Hyaluronan and versican in the control of human T-lymphocyte adhesion and migration. Matrix Biol. 31, 90–100 (2012).
Hope, C. et al. Immunoregulatory roles of versican proteolysis in the myeloma microenvironment. Blood 128, 680–685 (2016).
Sanders, C. J., Johnson, B., Frevert, C. W. & Thomas, P. G. In Mouse Models of Innate Immunity: Methods and Protocols (ed. Allen, I. C.) 177–188 (Humana Press, 2013).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Finak, G. et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 278 (2015).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Hall, M. W. et al. Innate immune function and mortality in critically ill children with influenza: a multicenter study. Crit. Care Med. 41, 224–236 (2013).
Revelle, W. Procedures for Psychological, Psychometric, and Personality Research R package (Comprehensive R Archive Network, 2019).
Schoenfeld, D. A. & Bernard, G. R. Statistical evaluation of ventilator-free days as an efficacy measure in clinical trials of treatments for acute respiratory distress syndrome. Crit. Care Med. 30, 1772–1777 (2002).
Acknowledgements
We thank the participants in each of the cohorts included in this study; H. Zhou, L.-A. Van De Velde, A. Souquette, G. Lennon and D. You for technical assistance in conducting experiments; T. Flerlage for critical reading of the manuscript; and the St Jude Animal Resource Center for the care of the animals used in this study. We acknowledge the use of tissues procured by the NDRI with support from NIH grant U42OD11158. Schematics in figures were made using an academic license of Biorender software. This work was funded by the National Institute of Allergy and Infectious Diseases under HHS contract HHSN27220140006C-OPT18I for the St Jude Center of Excellence for Influenza Research and Surveillance (P.G.T), JHCEIRS contract HHSN272201400007C, the National Institutes of Health R01AI084011 and R21HD095228 (A.G.R.), the National Institutes of Health and the National Natural Science Foundation of China under NIH-NSFC joint project 5R01AI128805-02, the National Natural Science Foundation of China under 81761128014, and ALSAC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Consortia
Contributions
Conceptualization: D.F.B. and P.G.T. Formal analysis: D.F.B., J.C.C. and E.K.A. Investigation: D.F.B., X.J.G., J.C.C., R.B., C.J.S., S.A.C., C.S.G., P.V., N.K.L., Y.W., W.G., T.P.F., N.W., A.G.R., M.M.N., P.M.M. and T.N. Methods development: J.C.C. and C.S.G. Resources: S.S.-C., T.N.W., K.S.-S., A.P., K.-F.C., R.E.R., X.L., Y.L, Z.Y. and N.Z. Data and sample curation: J.C.C., M.M.N., A.G.R. and T.N. Writing, original draft: D.F.B. Writing, review and editing: D.F.B., J.C.C., E.K.A., W.G., T.N.W., K.S.-S., R.E.R., M.M.N., A.G.R. and P.G.T. Visualization: D.F.B., J.C.C., E.K.A. and C.S.G. Supervision: P.G.T. Funding Acquisition: R.J.W., Z.Y., N.Z., A.G.R., P.G.T.
Corresponding author
Ethics declarations
Competing interests
P.G.T. is a consultant for Cytoagents Inc. A patent has been filed on work related to but not directly based on the findings in this manuscript. The authors declare no other competing interests.
Additional information
Peer review information Nature thanks Neil Henderson, Michael Matthay and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Single-cell gene-expression profiling of CD45− lung cells during severe influenza virus infection in mice.
a, Cell hashing of biological replicates for each time point. t-SNE of CD45- cells from mouse lungs at all time points. Cells from individual mice (n = 5 for time points 0, 1, and 3 dpi, and n = 4 for 6 dpi) were labelled with DNA-tagged antibodies (JCChto 10, 11, 12, 13, and 14) and pooled within time points. Pooled cells for each time point were run on separate 10x Genomics gel bead reactions. Cells are colour-coded according to hashtag oligo ID as indicated at the right of the graph. ‘Unknown’ (purple) indicates that an insufficient number of hashtags were sequenced in order to determine the ID. Cells are also symbol-coded according to time point after infection: 0 dpi (circle), 1 dpi (triangle), 3 dpi (square), 6 dpi (cross). b, Feature plots of signature genes used to identify mouse lung cell populations. The following genes were used to identify general populations of cells: immune cells (Ptprc), epithelial cells (Epcam), endothelial cells (Pecam1, Lyve1, Vwf), and mesenchymal cells (Col1a2). The following genes were used to identify specific cell populations within the main types of cells: T cells (Cd3e), B cells (Cd19), myeloid cells (Fcgr1), neutrophils (Mmp9), type I pneumocytes (Akap5), type II pneumocytes (Sftpc), ciliated epithelial cells (Foxj1), club cells (Scgb1a1), lymphatic endothelial cells (Pecam1, Thy1), fibroblasts (Pdgfra), pericytes and smooth muscle cells (Acta2), mesothelial cells (Msln), and platelets (Pf4). c, Violin plot of expression of influenza A PR8 transcripts by scGEX in non-immune lung cells. d, Overlay of days post-infection on t-SNE clustering of mouse lung cells.
Extended Data Fig. 2 Summary of gene-set enrichment analysis of mouse and human fibroblast populations.
a, Relative expression of the indicated gene sets in individual mouse fibroblasts and smooth muscle cells. Gene sets representing ECM structural proteins (EXTRACELLULAR_MATRIX1 and COLLAGEN1) inflammatory cytokine responses (HALLMARK_IL6_STAT3_SIGNALING1, GO_REGULATION_OF_TYPE_I_INTERFERON_MEDIATED_SIGNALING_PATHWAY, HINATA_NFKB_IMMU_INF1), and myogenesis (HALLMARK_TGF_BETA_SIGNALING1, HALLMARK_MYOGENESIS1) are plotted. b, Pairwise comparison of selected hallmark gene sets between mouse inflammatory fibroblast clusters 4 (DRFibs) and 6 (IRFibs). Each bar represents the normalized enrichment score (NES) from GSEA comparing all cells in cluster 4 (n = 892 cells) and cluster 6 (n = 749 cells). c, t-SNE clustering of human COL1A2 expressing mesenchymal cells from five human lung biopsy samples. Relative expression of the indicated gene sets in individual human fibroblasts and smooth muscle cells. Gene sets representing ECM structural proteins (EXTRACELLULAR_MATRIX1 and COLLAGEN1) inflammatory cytokine responses (HALLMARK_IL6_STAT3_SIGNALING1,HALLMARK_INTERFERON_ALPHA_RESPONSE1, HINATA_NFKB_IMMU_INF1), and myogenesis (HALLMARK_TGF_BETA_SIGNALING1, HALLMARK_MYOGENESIS1) are plotted. d, Dot plots of key genes related to cytokine/chemokine induction and ECM-production and modification in human lung mesenchymal cells.
Extended Data Fig. 3 Validation of inflammatory fibroblasts transcriptional states by flow cytometry.
a, b, Feature plots of Itga5/ITGA5 and Cd9/CD9 in mouse and human mesenchymal cells expressing Col1a2/COL1A2 from scGEX data. Legend indicates relative expression within each dataset. c, Gating strategy for identifying inflammatory fibroblast transcriptional states based on ITGA5 (CD49e) and CD9 staining. d, Total cell number for IRFibs (blue) and DRFibs (green) in the lungs during IAV infection. e, Gene expression in sorted ITGA5hiCD9lo, ITGA5lo CD9hi, and bulk CD45- cells as measured by qPCR. Each data point represents an independent biological replicate (CD45-, n = 5; ITGA5hiCD9lo, n = 4, ITGA5lo CD9hi, n = 4). Error bars represent standard error of the mean. f, Principal component analysis of sorted fibroblast populations and bulk CD45- cells from mouse lungs. g, In vitro stimulation of mouse lung fibroblasts with IL-1/TNF. Representative flow plots of ITGA5 staining before and after stimulation. Charts at bottom indicate percentage of ITGA5hi fibroblasts (P = 2 × 10−6)) and median fluorescence intensity (MFI) for ITGA5 (P = 0.002). Each data point represents an independent biological replicate (unstim, n = 3; IL1/TNF, n = 3). For statistical analysis, groups were compared using a two-sided Student’s t-test.
Extended Data Fig. 4 Assessment of fibroblast cell surface phenotype in human lung samples.
a, Gating strategy for identifying live, CD45−EpCAM−CD31−PDGFRA+ cells in human lung samples. b, Each flow plot represents cells gated on live, CD45−EpCAM−CD31−PDGFRA+ from an individual sample as indicated in the gating strategy. Resting fibroblasts (ITGA5loCD9hi) and damage-responsive fibroblasts (ITGA5hiCD9lo) are labelled on representative flow plots.
Extended Data Fig. 5 Regulation of ECM-related gene expression in human and mouse respiratory cells following cytokine stimulation.
a, Heat map representing ∆Ct values (normalized to ACTB) in NHBFs and normal human bronchial epithelial cells (NHBEs) stimulated for 24 h with the indicated cytokines or viruses, and nasal wash cells (NWCs). Green shading highlights stimulation with IL-1 cytokines, while blue shading indicates stimulation with indicated viruses. ECM-related genes are grouped by unsupervised, hierarchical clustering. b, Heat map representing log2-transformed fold-change in differentiation normal human bronchial epithelial cells (NHBEs) stimulated with the indicated cytokines/viruses. Values are relative to untreated NHBEs. c, Heat map representing log2-transformed fold-change in gene expression relative to untreated cells as determined by the ∆∆Ct method. Matrix protease genes (y-axis) and treated cell samples (x-axis) were grouped by unsupervised, hierarchical clustering. d, Assessment of ECM protease and TIMP protein levels in bronchoalveolar lavage fluid collected from mice. Heat maps of log2-transformed fold-change values relative to mock-infectedmice for each of the indicated analytes over the course of IAV infection. Protein levels in the BAL (top) are compared to log2-transformed fold-change values relative to mock-infected mice for gene expression as measured by qPCR in whole lung homogenates. The kinetics for individual analytes plotting absolute protein concentrations are presented at the right (0 dpi, n = 5 mice; 3 dpi, n = 5; 6 dpi, n = 5; 9 dpi, n = 5; 12 dpi, n = 4; 21 dpi, n = 5). Error bars represent s.e.m.
Extended Data Fig. 6 Meta-analysis of publicly available human disease single-cell gene-expression datasets.
a, Table of datasets from eight distinct studies (seven from respiratory disease and one from rheumatoid arthritis), which were aggregated for meta-analysis. Cells expressing at least one transcript of ADAMTS4 were identified. b, c, Uniform manifold approximation and projection (UMAP) clustering of ADAMTS4+ cells with overlay of study or of unsupervised clustering by cell type. d, Volcano plot of differentially expressed genes in cluster 14. Differentially expressed genes are based on comparison to all cells in the dataset. Each data point represents a gene and is colour-coded according to whether the gene was above a threshold for adjusted P value (<10−3, blue), log2-transformed fold-change (>|2|, green), adjusted P value and log2-transformed fold-change (red), or not significant (grey). e, Expression level of ADAMTS4 in the pulmonary fibrosis dataset from Habermann et al.12 by disease state. f, Expression level of ADAMTS4 in the Habermann et al.12 dataset by cell type (endothelial, epithelial, immune, mesenchymal) and disease state.
Extended Data Fig. 7 Spatial transcriptomics of lung sections from mice collected 10 days after infection.
a, H&E staining (left) and unsupervised clustering analysis (right) are presented for four lung sections from four individual mice, representing four biologically independent samples. Similar results were obtained across the four independent samples. ‘Ident’ indicates the identity of each lung region determined by unsupervised clustering of transcriptional profiles. b, Relative gene expression of key fibroblast-associated genes (Itga5, Lox, Cd9, Timp3) in a representative lung section. c, Summary of Gene Ontology (GO) analysis of lung regions (clusters) determined by unsupervised clustering. The chart indicates the lung region (‘Cluster’), the GO pathway (‘GO’), fold enrichment of the indicated pathway in the indicated region (‘Fold Enrichment’), and the P value using false discovery rate to assess statistical significance (‘FDR’). d, Average log fold change in expression among all spots pertaining to lung regions (n = 4 independent lung sections from four individual mice). Enrichment determined using two-sided Wilcoxon rank sum test with FDR adjustment. Asterisk (*) indicates that adjusted P < 0.00001 (exact P values in Source Data).
Extended Data Fig. 8 Assessment of IAV infection in Adamts4+/+ and Adamts4−/− mice.
a, Weight loss curves of Adamts4+/+ and KO mice following lethal IAV challenge with 6,000 EID50 PR8. Data points represent mean ± s.d. (WT, n = 27; KO, n = 16). Data are pooled from 5 independent experiments. b, Viral plaque titres in BAL fluid and lung tissue homogenates at 6 dpi. Each data point represents a biologically independent mouse (BAL: WT, n = 12; KO, n = 11; lung tissue: WT, n = 5; KO, n = 5). Groups were compared using a two-sided Mann–Whitney U test. Error bars represent s.e.m. c, Cell-type specific infection based on IAV nucleoprotein (NP) positivity as assessed by flow cytometry and gating strategy for identifying populations for cell-type specific infection. Populations were identified using the following markers: Epithelial (CD45−EpCAM+), Endothelial (CD45−CD31+), Fibroblasts (CD45−EpCAM−CD90+), Monocytes (CD45+CD11c−CD11b+Gr-1lo), Neutrophils (CD45+CD11c−CD11b+Gr-1hi). Each data point represents a biologically independent mouse (uninfected, n = 2; WT, n = 7; KO, n = 7). Groups were compared using a two-sided Mann–Whitney U test. Error bars represent s.e.m. d, Total protein concentration in BAL fluid as measured by BCA assay at 6 dpi. Each data point represents a biologically independent mouse (uninfected, n = 8; WT, n = 7; KO, n = 5). Groups were compared using a two-sided Mann–Whitney U test. Error bars represent s.e.m. e, Cytokine concentrations in BAL fluid at 6 dpi as measured by multiplexed cytokine bead arrays (WT, n = 8; Adamts4−/−, n = 8). f, Measurement of dynamic lung compliance at 10 days after infection following sublethal challenge with IAV (Uninfected: WT, n = 8; KO = 3, Infected: WT n = 8, KO, n = 9). Error bars represent s.e.m. For statistical analysis, groups were compared using a two-sided Mann–Whitney U test.
Extended Data Fig. 9 Versican accumulates in the lung during severe IAV infection in mice and affects localization and responses of T cells.
a, Representative fluorescence microscopy images of VCAN staining at 0, 3, and 6 days after infection from two independent experiments. Scale bar, 200 μm. Quantification of VCAN staining is presented at the right. Area-under-curve was determined from the average of five intensity profiles of fluorescence per image. Each data point represents average of intensity profiles for each individual mouse (0 dpi, n = 6, 3 dpi, n = 8; 6 dpi, n = 6). Error bars represent s.e.m. b, Representative images of intact VCAN staining in lung sections from Adamts4+/+ and Adamts4−/− collect 9 days after infection from two independent experiments. Note that images are also presented in Fig. 3h with CD3 staining. Scale bar, 50 μm. c, Heat map representing log2-transformed fold change values in gene expression for each genotype relative to mock-infected controls. Whole lungs were collected 9 days after infection. d, Gating strategy for identifying CD8+ T cells in lungs of IAV-infected mice. CD8+ T cells were identified as CD45+CD3+CD8+. e, Frequency of IFNγ+ in ADAMTS4 WT and KO mice at 9 and 12 days after infection. Each data point represents a biologically independent animal (9 dpi: WT, n = 5; KO, n = 5; 12 dpi: WT, n = 5; KO, n = 4). f, Total cell number of tetramer+ CD8+ T cells in the lung at 9 days after infection (WT, n = 10; KO, n = 10). Error bars represent s.e.m.
Extended Data Fig. 10 Association of ADAMTS4 with clinical measures from three independent human cohorts.
a, Demographic information for the Paediatric Intensive Care Influenza (PICFLU) cohort. b, Histogram of VFDS for the PICFlu cohort. c, Odds ratios for PRMODS and PRAHRF, as well as gender, previously healthy, age, and steroids as covariates. d, Demographic information for influenza infection cohorts in Taiwan and Guangzhou. e, Compartmentalization of ADAMTS-4 in samples from upper (nasal wash) and lower (sputum, BALF, ETA) respiratory tracts (NW, n = 131; sputum, n = 48; BALF, n = 25; ETA, n = 34). Box plots were generated for samples in which ADAMTS4 protein was detectable. Center line indicates median, bottom and top of boxes indicate first and third quartiles, and whiskers extend to 1.5 times the interquartile range. f, Odds ratios for VFDS <10, including gender and age as covariates, in combined analysis of three human cohorts. Odds ratios (c, f) determined using logistic regression with FDR adjustments for multiple testing.
Supplementary information
Supplementary Table
Supplemental Table 1. List of qPCR primers used to detect human genes.
Supplementary Table
Supplemental Table 2. List of qPCR primers used to detect mouse genes.
Supplementary Table
Supplemental Table 3. Protein data for PICFLU human cohort.
Supplementary Table
Supplemental Table 4. Protein data for Taipei/Guangzhou human cohorts.
Rights and permissions
About this article

Cite this article
Boyd, D.F., Allen, E.K., Randolph, A.G. et al. Exuberant fibroblast activity compromises lung function via ADAMTS4. Nature 587, 466–471 (2020). https://doi.org/10.1038/s41586-020-2877-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2877-5
This article is cited by
-
Acute lung injury: a view from the perspective of necroptosis
Inflammation Research (2024)
-
Single-cell lineage tracing with endogenous markers
Biophysical Reviews (2024)
-
Necroptosis blockade prevents lung injury in severe influenza
Nature (2024)
-
Innate immune responses in pneumonia
Pneumonia (2023)
-
Advances in spatial transcriptomics and related data analysis strategies
Journal of Translational Medicine (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
