
Abstract
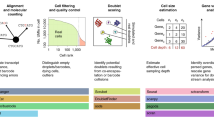
Breakthroughs in the development of high-throughput technologies for profiling transcriptomes at the single-cell level have helped biologists to understand the heterogeneity of cell populations, disease states and developmental lineages. However, these single-cell RNA sequencing (scRNA-seq) technologies generate an extraordinary amount of data, which creates analysis and interpretation challenges. Additionally, scRNA-seq datasets often contain technical sources of noise owing to incomplete RNA capture, PCR amplification biases and/or batch effects specific to the patient or sample. If not addressed, this technical noise can bias the analysis and interpretation of the data. In response to these challenges, a suite of computational tools has been developed to process, analyse and visualize scRNA-seq datasets. Although the specific steps of any given scRNA-seq analysis might differ depending on the biological questions being asked, a core workflow is used in most analyses. Typically, raw sequencing reads are processed into a gene expression matrix that is then normalized and scaled to remove technical noise. Next, cells are grouped according to similarities in their patterns of gene expression, which can be summarized in two or three dimensions for visualization on a scatterplot. These data can then be further analysed to provide an in-depth view of the cell types or developmental trajectories in the sample of interest.
Key points
-
As single-cell RNA sequencing datasets increase in scale and complexity, faster and more efficient computational tools for processing and analysis are required.
-
New computational tools that correct technical and batch effects can unlock additional heterogeneity and enable higher-resolution clustering and trajectory inference.
-
Graph-based methods for clustering and trajectory inference allow for the scalable analysis of large single-cell RNA sequencing datasets.
-
Visualization methods can distort the structure of the data and batch correction methods can reduce cell-type resolution; both methods should therefore be used with care and might require specific parameter tuning for each dataset.
-
High-level biological interpretation, such as cell-type annotation, remains challenging and time-consuming — new automated methods, alongside the creation of single-cell reference atlases, promise to address these issues.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


References
Ramsköld, D., Wang, E. T., Burge, C. B. & Sandberg, R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput. Biol. 5, e1000598 (2009).
Potter, S. S. Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol. 14, 479–492 (2018).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Zheng, G. X. Y. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Rosenberg, A. B. et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 12, eaam8999 (2018).
Cao, J. et al. Comprehensive single cell transcriptional profiling of a multicellular organism by combinatorial indexing. Science 357, 661–667 (2017).
Lake, B. B. et al. A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat. Commun. 10, 2832 (2019).
Combes, A. N., Zappia, L., Er, P. X., Oshlack, A. & Little, M. H. Single-cell analysis reveals congruence between kidney organoids and human fetal kidney. Genome Med. 11, 3 (2019).
Tanay, A. & Regev, A. Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338 (2017).
Chen, C. et al. Single-cell whole-genome analyses by linear amplification via transposon insertion (LIANTI). Science 356, 189–194 (2017).
Smallwood, S. A. et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 11, 817–820 (2014).
Cusanovich, D. A. et al. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015).
Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015).
Chen, S., Lake, B. B. & Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 37, 1452–1457 (2019).
Linker, S. M. et al. Combined single-cell profiling of expression and DNA methylation reveals splicing regulation and heterogeneity. Genome Biol. 20, 30 (2019).
Gu, C., Liu, S., Wu, Q., Zhang, L. & Guo, F. Integrative single-cell analysis of transcriptome, DNA methylome and chromatin accessibility in mouse oocytes. Cell Res. 29, 110–123 (2019).
Amezquita, R. A. et al. Orchestrating single-cell analysis with Bioconductor. Nat. Methods 17, 137–145 (2020). A useful stepwise practical tutorial on how to perform scRNA-seq analysis in the R programming language using the Bioconductor suite of tools.
Lun, A. T. L., Mccarthy, D. J. & Marioni, J. C. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with bioconductor. F1000Res. 5, 2122 (2016).
Luecken, M. D. & Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019). This tutorial discusses scRNA-seq analysis steps using the latest methods developed for each step.
Petukhov, V. et al. Accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 19, 78 (2018).
Melsted, P. et al. Modular and efficient pre-processing of single-cell RNA-seq. Preprint at https://doi.org/10.1101/673285 (2019).
Islam, S. et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 11, 163–166 (2014).
Smith, T. & Sudbery, I. UMI-tools: modelling sequencing errors in unique molecular identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
van den Brink, S. et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936 (2017).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e4 (2019).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
McCarthy, D. J., Campbell, K. R., Lun, A. T. L. & Wills, Q. F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186 (2017).
Wagner, A., Regev, A. & Yosef, N. Uncovering the vectors of cellular states with single cell genomics. Nat. Publ. Gr. 34, 1–53 (2016).
Vallejos, C. A., Risso, D., Scialdone, A., Dudoit, S. & Marioni, J. C. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nat. Methods 14, 565–571 (2017).
L. Lun, A. T., Bach, K. & Marioni, J. C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol. 17, 75 (2016).
Bacher, R. et al. SCnorm: robust normalization of single-cell RNA-seq data. Nat. Methods 14, 584–586 (2017).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Fan, J. et al. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat. Methods 13, 241–244 (2016).
Brennecke, P. et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 10, 1093–1095 (2013).
Barkas, N. et al. pagoda2: a package for analyzing and interactively exploring large single-cell RNA-seq datasets. GitHub https://github.com/hms-dbmi/pagoda2 (2018).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019).
Risso, D., Perraudeau, F., Gribkova, S., Dudoit, S. & Vert, J. P. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat. Commun. 9, 284 (2018).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Eraslan, G., Simon, L. M., Mircea, M., Mueller, N. S. & Theis, F. J. DCA: single cell RNA-seq denoising using a deep count autoencoder. Nat. Commun. 10, 390 (2019).
Yip, S. H., Sham, P. C. & Wang, J. Evaluation of tools for highly variable gene discovery from single-cell RNA-seq data. Brief. Bioinform. 20, 1583–1589 (2018). A benchmark analysis of methods available for selecting over-dispersed genes.
Leek, J. T. et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 11, 733–739 (2010).
Haghverdi, L., Lun, A. T. L., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
Barkas, N. et al. Joint analysis of heterogeneous single-cell RNA-seq dataset collections. Nat. Methods 16, 695–698 (2019).
Tran, H. T. N. et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12 (2020). A benchmark study of methods available for batch correction during analysis of scRNA-seq data.
Leek, J. T. Svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 42, e161 (2014).
Stuart, T. & Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 20, 257–272 (2019).
Hie, B., Bryson, B. & Berger, B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 37, 685–691 (2019).
Kiselev, V. Y., Andrews, T. S. & Hemberg, M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 20, 273–282 (2019).
Saelens, W., Cannoodt, R., Todorov HelenaSaeys, Y., Todorov, H. & Saeys, Y. A comparison of single-cell trajectory inference methods: towards more accurate and robust tools. Nat. Biotechnol. 37, 547–554 (2019). A benchmark analysis of methods for single-cell trajectory inference.
Bellman, R. On the theory of dynamic programming. Proc. Natl Acad. Sci. USA 38, 716–719 (1952).
Sun, S., Zhu, J., Ma, Y. & Zhou, X. Accuracy, robustness and scalability of dimensionality reduction methods for single-cell RNA-seq analysis. Genome Biol. 20, 269 (2019). A benchmark study of methods used for dimensionality reduction of scRNA-seq data.
Abdi, H. & Williams, L. J. Principal component analysis. Chemom. Intell. Lab. Syst. 2, 433–459 (2010).
Pierson, E. & Yau, C. ZIFA: dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol. 16, 241 (2015).
Buettner, F., Pratanwanich, N., McCarthy, D. J., Marioni, J. C. & Stegle, O. f-scLVM: scalable and versatile factor analysis for single-cell RNA-seq. Genome Biol. 18, 212 (2017).
Lee, D. D. & Seung, H. S. Learning the parts of objects by non-negative matrix factorization. Nature 401, 788–791 (1999).
Lin, X. & Boutros, P. C. Optimization and expansion of non-negative matrix factorization. BMC Bioinformatics 21, 7 (2020).
Roweis, S. T. & Saul, L. K. Nonlinear dimensionality reduction by locally linear embedding. Science 290, 2323–2326 (2000).
Angerer, P. et al. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243 (2015).
Welch, J. D., Hartemink, A. J. & Prins, J. F. SLICER: inferring branched, nonlinear cellular trajectories from single cell RNA-seq data. Genome Biol. 17, 106 (2016).
Eraslan, G., Avsec, Ž., Gagneur, J. & Theis, F. J. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 20, 389–403 (2019).
Deng, Y., Bao, F., Dai, Q., Wu, L. F. & Altschuler, S. J. Scalable analysis of cell-type composition from single-cell transcriptomics using deep recurrent learning. Nat. Methods 16, 311–314 (2019).
Svensson, V. Droplet scRNA-seq is not zero-inflated. Nat Biotechnol. 38, 147–150 (2020).
Wagner, F., Yan, Y. & Yanai, I. K-nearest neighbor smoothing for single-cell RNA-seq data. Preprint at https://doi.org/10.1101/217737 (2017).
van Dijk, D. et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729.e27 (2018).
Huang, M. et al. SAVER: gene expression recovery for single-cell RNA sequencing. Nat. Methods 15, 539–542 (2018).
Lin, P., Troup, M. & Ho, J. W. K. CIDR: ultrafast and accurate clustering through imputation for single cell RNA-seq data. Genome Biol. 18, 59 (2017).
Li, W. V. & Li, J. J. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat. Commun. 9, 997 (2018).
Andrews, T. S. & Hemberg, M. False signals induced by single-cell imputation. F1000Res. 7, 1740 (2019).
Lloyd, S. P. Least squares quantization in PCM. IEEE Trans. Inf. Theory 28, 129–137 (1982).
Žurauskiene, J. & Yau, C. pcaReduce: hierarchical clustering of single cell transcriptional profiles. BMC Bioinformatics 17, 140 (2016).
Zeisel, A. et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015).
Zeisel, A. et al. Molecular architecture of the mouse nervous system. Cell 174, 999–1014.e22 (2018).
Duò, A., Robinson, M. D. & Soneson, C. A systematic performance evaluation of clustering methods for single-cell RNA-seq data. F1000Res. 7, 1141 (2018). A benchmark analysis of methods available for clustering in scRNA-seq data analysis.
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, P10008 (2008).
Traag, V. A., Waltman, L. & van Eck, N. J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019).
Kiselev, V. Y. et al. SC3: consensus clustering of single-cell RNA-seq data. Nat. Methods 14, 483–486 (2017).
Li, H. et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 49, 708–718 (2017).
Combes, A. N. et al. Single cell analysis of the developing mouse kidney provides deeper insight into marker gene expression and ligand-receptor crosstalk. Development 146, dev178673 (2019).
Qiu, X. et al. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 14, 309–315 (2017).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Wolf, F. A. et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59 (2019).
La Manno, G. et al. RNA velocity of single cells. Nature 560, 494–498 (2018).
Wu, Y., Tamayo, P. & Zhang, K. Visualizing and interpreting single-cell gene expression datasets with similarity weighted nonnegative embedding. Cell Syst. 7, 656–666.e4 (2018).
van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
McInnes, L. & Healy, J. UMAP: uniform manifold approximation and projection for dimension reduction. Preprint at https://arxiv.org/abs/1802.03426 (2018).
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 37, 38–44 (2018).
Wattenberg, M., Viegas, F. & Johnson, I. How to use t-SNE effectively. Distill https://doi.org/10.23915/distill.00002 (2016).
van der Maaten, L. Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res. 15, 3221–3245 (2014).
Kobak, D. & Linderman, G. C. UMAP does not preserve global structure any better than t-SNE when using the same initialization. Preprint at https://doi.org/10.1101/2019.12.19.877522 (2019).
Kobak, D. & Berens, P. The art of using t-SNE for single-cell transcriptomics. Nat. Commun. 10, 5416 (2019).
Moon, K. R. et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 37, 1482–1492 (2019).
Ding, J., Condon, A. & Shah, S. P. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat. Commun. 9, 2002 (2018).
Abdelaal, T. et al. A comparison of automatic cell identification methods for single-cell RNA-sequencing data. Genome Biol. 20, 194 (2019). A benchmark study of methods available for automated cell-type classification in scRNA-seq data.
Soneson, C. & Robinson, M. D. Bias, robustness and scalability in single-cell differential expression analysis. Nat. Methods 15, 255–261 (2018).
Lun, A. T. L. & Marioni, J. C. Overcoming confounding plate effects in differential expression analyses of single-cell RNA-seq data. Biostatistics 18, 451–464 (2017).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Suykens, J. A. K. & Vandewalle, J. Indefinite kernels in least squares support vector machines and principal component analysis. Neural Process. Lett. 43, 162–172 (2017).
Pliner, H. A., Shendure, J. & Trapnell, C. Supervised classification enables rapid annotation of cell atlases. Nat. Methods 16, 983–986 (2019).
Kiselev, V. Y., Yiu, A. & Hemberg, M. scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods 15, 359–362 (2017).
Alquicira-Hernandez, J., Sathe, A., Ji, H. P., Nguyen, Q. & Powell, J. E. ScPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 20, 264 (2019).
Regev, A. et al. The human cell atlas. eLife 6, e27041 (2017).
Stoeckius, M. et al. Cell hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224 (2018).
Denisenko, E. et al. Systematic bias assessment in solid tissue 10x scRNA-seq workflows. Preprint at https://doi.org/10.1101/832444 (2019).
Lake, B. et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590 (2016).
Krishnaswami, S. R. et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat. Protoc. 11, 499–524 (2016).
Acknowledgements
The authors were supported by NIH grants U01MH098977, R01HL123755, U54HL145608, UH3DK114933 and R01HG009285.
Author information
Authors and Affiliations
Contributions
All authors researched data for the article, wrote the manuscript, made substantial contributions to discussions of the content and reviewed or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
Y.W. declares no competing interests. K.Z. is a co-founder, equity holder, scientific advisory board member and paid consultant of Singlera Genomics, which has no commercial interests related to this article.
Additional information
Peer review information
Nature Reviews Nephrology thanks B. J. Aronow and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
Broad Institute online single-cell data browser: https://portals.broadinstitute.org/single_cell
EMBL-EBI online single-cell data browser: https://www.ebi.ac.uk/gxa/sc/home
UCSC online single-cell data browser: https://cells.ucsc.edu/
Glossary
- FASTQ file
-
A text file that stores DNA sequences and their associated quality metrics and metadata; a single sequence in a FASTQ file is called a ‘read’.
- Counts matrix
-
An integer matrix (that is, numerical data arranged in a set of columns and rows) in which the columns typically correspond to cells, whereas the rows correspond to genes; each entry represents the number of molecules of that gene expressed in that cell.
- Total counts
-
The total number of reads or UMIs in a given cell.
- Size factor
-
An estimate of how much variation in sequencing depth or RNA capture efficiency affects the overall quantification of gene expression in a cell.
- Over-dispersed genes
-
Genes that show a greater than expected variance between cells given their average expression, which suggests that they are expressed in a cell-type-specific manner.
- Regression model
-
A model that compares the relationship between two variables. In the context of single-cell RNA sequencing, regression can assess relationships between observed gene expression, and technical and/or biological factors.
- Mutual nearest neighbours
-
(MNNs). Cells from different batches that belong to each other’s set of k-nearest neighbours (that is, cells with the most similar gene expression patterns).
- Dimensionality reduction
-
Summarizing a large set of variables with a smaller set of variables, while retaining as much information as possible.
- Embedding
-
The set of variables that remains after running some form of dimensional reduction.
- Dropout
-
The absence of a detectable gene or transcript in a cell.
- Classification
-
A machine learning task in which an algorithm learns the relevant features that distinguish the different classes of a training dataset to predict the classes of an unknown test dataset.
- Cell hashing
-
A technique that attaches unique molecular barcodes to multiple batches of samples for pooling and processing in one batch, which not only improves the experimental throughput but also reduces technical batch differences.
Rights and permissions
About this article

Cite this article
Wu, Y., Zhang, K. Tools for the analysis of high-dimensional single-cell RNA sequencing data. Nat Rev Nephrol 16, 408–421 (2020). https://doi.org/10.1038/s41581-020-0262-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41581-020-0262-0
This article is cited by
-
Overcoming barriers to single-cell RNA sequencing adoption in low- and middle-income countries
European Journal of Human Genetics (2024)
-
Advancements in Single-Cell RNA Sequencing Research for Neurological Diseases
Molecular Neurobiology (2024)
-
Single-Cell RNA Sequencing: Technological Progress and Biomedical Application in Cancer Research
Molecular Biotechnology (2023)
-
Recent developments in application of single-cell RNA sequencing in the tumour immune microenvironment and cancer therapy
Military Medical Research (2022)
-
Chord: an ensemble machine learning algorithm to identify doublets in single-cell RNA sequencing data
Communications Biology (2022)
