
Abstract
Growing evidence suggests that many diseases of aging, including diseases associated with robust changes and adipose deports, may be caused by resident adult stem cell exhaustion due to the process called cellular senescence. Understanding how microRNA pathways can regulate cellular senescence is crucial for the development of novel diagnostic and therapeutic strategies to combat these pathologies. Herein, using integrated transcriptomic and semi-quantitative proteomic analysis, we provide a system level view of the regulation of human adipose-derived stem cell senescence by a subset of mature microRNAs (termed senescence-associated-microRNAs) produced by biogenesis of oncogenic MIR17HG and tumor-suppressive MIR100HG clusters. We demonstrate functional significance of these mature senescence-associated-microRNAs in the process of replicative senescence of human adipose-derived stem cells ex-vivo and define a set of senescence-associated-microRNA gene targets that are able to elicit, modulate and, most importantly, balance intimate connections between oncogenic and senescent events.
Similar content being viewed by others


Introduction
Adipose tissue is not only a storage depot for triglycerides but also a critical functional organ regulating energy homeostasis. It is well known that abnormal fat accumulation and function is associated with adverse health outcomes, including obesity, type II diabetes, cardiovascular and cerebrovascular diseases and ultimately, aging. During adult adipose tissue homeostasis and turnover (tissue maintenance), adipocytes are derived from adipose tissue stem cells (ADSCs), whose origin has been traced to mural cells (also termed pericytes) residing in the perivascular niche via a specialized cell lineage differentiation process.1,2,3 ADSCs are a type of adult stem cell of mesenchymal origin that possess many of the traits common to bone marrow-derived mesenchymal stem cells (BMMSCs). New adipocyte formation is critical for adult homeostatic balance, and adipose tissue maintenance often requires a steady replenishment of cells from stem or progenitor sources.4, 5 However, throughout life it appears that changes in the quantity and quality of ADSCs due to external stimuli, specialized stem cell microenvironment, and/or intrinsic stem cell aging processes, can influence adipose tissue metabolism, turnover rate and regeneration and, surprisingly, also impose restrictions on ADSC immunomodulation properties invoked in settings such as tissue injury, transplantation and autoimmunity.1, 2, 5,6,7,8
Robust aging-related changes in tissue maintenance are thought to be caused by resident adult stem cell exhaustion due to the process called cellular senescence (SEN).9,10,11,12,13 SEN involves signaling, metabolic and cytoskeletal changes resulting in the diminished ability of cells to cope with DNA damage and to maintain the structure and function of chromatin.2, 14 Despite the effort to uncover crosstalk between cellular signaling pathways controlling SEN,15, 16 the full set of regulators involved in its establishment and maintenance are not well defined, and their complex interactions are still poorly understood.
Over the past decade, microRNAs (miRNAs) have emerged as a new dimension of sophisticated genomic regulations in a variety of physiological processes. Once the messenger RNA is targeted by miRNAs, the RNA-induced silencing complex is thought to inhibit protein production either through blocking translation or by reducing messenger RNA stability.17,18,19,20 A given miRNA can target a multitude of different mRNAs, and a given gene target might similarly be targeted by multiple miRNAs. For this reason, miRNAs frequently represent the central nodes of several regulatory networks and may act as rheostats to provide stability and/or fine-tuning to gene expression cascades.21, 22 Different miRNA expression profiles were reported for various cell types undergoing replicative SEN, such as arterial and umbilical vein-derived endothelial cells, replicating CD8(+) T cells, renal proximal tubular epithelial cells and skin fibroblasts,23, 24 indicating that miRNAs might play a major role in orchestrating replicative SEN.
Some miRNAs have been reported to accelerate or inhibit the process of adipocyte differentiation during adipogenesis, thereby influencing the process of cellular SEN as well as impacting the aging process in general.25,26,27,28,29 However, whether the altered miRNA profile is a consequence of SEN or whether it triggers replicative SEN is still a matter of debate. Although many methods have been proposed for miRNA target identification,30, 31 little is known about a specialized cohort of miRNA gene targets that can trigger and/or mediate senescent phenotypes and how downregulation in its expression is linked to restriction of proliferation capacity, diminished DNA damage repair and severe abnormality in the chromatin assembly generally observed upon SEN. Therefore, a better understanding of how miRNA pathways can regulate human adipose-derived stem cell (hADSC) SEN through their gene targets is crucial for the development of novel therapeutic strategies to combat the many diseases of aging, including diseases associated with robust changes and adipose depots.
In this manuscript, we investigate the critical role of two micro-RNA clusters, oncogenic MIR17HG and tumor-suppressive MIR100HG, in the process of replicative SEN of human adipose-derived stem cells (hADSCs) by using an integrated approach that combines RNA sequencing analysis (RNA-seq) and semi-quantitative proteomic analysis. Here, we uncover the complex interactions among several cellular processes biologically relevant for the state of cellular SEN. We provide functional evidence demonstrating that the senescent state of hADSCs is achieved by the combined action of SEN–associated miRNAs (SA-miRNAs), and we identify a set of novel gene targets that are susceptible to these miRNAs. Our data suggests a functional significance of these miRNAs in the complex SEN–associated changes within human adult adipose-derived stem cells.
Results
Replicative SEN of human adult ADSCs is associated with upregulation of a subset of non-coding RNA (ncRNAs)
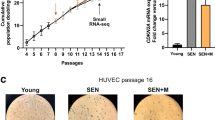
Isolated hADSCs share many characteristics with BMMSCs, their counterparts in bone marrow.32,33,34 Similar to other types of MSCs, hADSCs are not considered to be immortal either in-vivo or ex-vivo. We and others have previously reported that isolated and ex-vivo cultured hADSCs exhibit consistent self-renewing (SR) and, upon approaching replicative SEN (Fig. 1 and Supplementary Figure S1A), cultures accumulate giant non-dividing cells, as determined by incorporation bromodeoxyuridine (BrdU) into DNA (Supplementary Figure S1B), expressing the enzyme lysosomal pH6 SEN-associated β-galactosidase (SA-β-Gal) (Fig. 1a). SEN hADSCs manifest a loss of control for chromatin organization, activate a persistent DNA damage response (DDR) (Supplementary Figure S1B and C) and manifest robust changes in transcriptional activity.13, 35, 36 As hADSCs approached SEN, both mediators of DDR, phosphorylated form of histone variant H2AX (γH2AX),37 and p53 binding protein-1,38 form characteristic persistent DNA damage foci (Supplementary Figure S1B and C). The presence of these foci drastically increased from very rare in SR ADSCs, to almost 90% in hADSCs approaching SEN (hADSCs) triggered by activation of the p53/P21WAF1/Cip1 pathway (Supplementary Figure S1B).13 In corroboration with previous reports, cluster of differentiation (CD) antigen marker characterization revealed that SEN hADSCs robustly express stromal markers CD29, CD44, CD73, CD90, CD105 while staying negative for hematopoietic lineage markers CD31, CD34 and CD4532 (Fig. 1b, Supplementary Figure S2A and S2B), suggesting phenotypical stability of SEN hADSCs.

Schematic representation of sample collection for analysis and immunological stability of hADSCs upon replicative SEN. a hADSCs were isolated from healthy donor subcutaneous tissue and passaged ex-vivo as described in materials and methods. Colorimetric detection of SA-β-Gal (10x) in SR and SEN hADSCs is shown. Samples were collected and processed for transcriptomic and proteomic studies as described in material and methods. b The table summarizes immunostability of hADSCs in SR and senescent states, which were assessed by expression of MSC-positive and MSC-negative CD markers, details shown in Supplementary Table S3
We further assessed the changes associated with replicative SEN via transcriptome analysis (RNA-seq) and matching high-resolution LC-MS/MS differential abundance proteome analysis, a technique that can measure global changes in relative protein abundance (Fig. 1a) as described in materials and methods.39, 40
Comparative transcriptomic analysis (RNA-seq) between SR and SEN hADSCs revealed a number of ncRNAs that are upregulated in SEN compared to SR hADSCs (Fig. 2a and b, Supplementary Figure S3A, S3B and materials and methods). Differentially expressed ncRNAs are identified as those that have high levels of fold change (log2 SEN/SR) and significant differences in the normalized number of reads (dRPKM SEN-SR) as shown in Supplementary Figure S3B. The 216 ncRNAs upregulated upon SEN have been identified (shown in red in the upper right quadrant of Fig. 2a). Three out of four upregulated ncRNA loci (Fig. 2b) encode polycistronic transcripts that could be processed to yield multiple miRNAs (Fig. 2b): chr11:MIR100HG (encoding mir-125b1, mir-let7a-2, mir-100), chr13: MIR17HG (encoding mir-17, mir-18a, mir-19a, mir-20a, mir-19b-1, mir-92a-1) and chr22: MIRLET7BHG (encoding mir-3619, mir-let7a-3, mir-4763, mir-let-7b).

MiRNA clusters and SEN-associated miRNA (SA-miRNAs) discovered via RNA-seq analysis and experimentally validated by qPCR. a Differential expression of non-coding RNA genes in SR versus SEN hADSCs revealed by RNA-seq analysis. Fold-change values (log2 SEN/SR) are shown on the x-axis and RPKM differences (log2 SEN-SR) are shown on the y-axis. SEN upregulated non-coding RNA genes are shown in red (upper right quadrant). b Genomic locations and locus names for SEN upregulated miRNA gene clusters revealed by RNA-seq analysis. c Graphical representation of oncogenic MIR17HG locus and qPCR analysis of mature mirRNA expression in SR (blue bar) and senescent (SEN, red bar) states of hADSCs. Relative expression of either passenger strand mature miRNAs (depicted in the graphs as -3p) or guide strand mature miRNAs (depicted in the graphs as -5p) to U6 small RNA was measured. Data are shown as fold change (ΔΔCτ) The mean ± SD from three independent experiments is shown. The statistical difference was evaluated by Student’s t-test and P-value (p) related to experimental measurements and are listed under the graphs, where ***p < 0.001, **p < 0.01. d Graphical representation of tumor-suppressive MIR100HG locus and qPCR analysis of mature miRNA expression in SR (blue bar) and senescent (SEN, red bar) states of hADSCs. Relative expression of either passenger strand mature miRNAs (depicted in the graphs as -3p) or guide strand mature miRNAs (depicted in the graphs as -5p) to U6 small RNA was measured. Data are shown as fold change (ΔΔCτ) The mean ± SD from three independent experiments is shown. The statistical difference was evaluated by Student’s t-test and P-values (p) related to experimental measurements are listed under the graphs, where ***p < 0.001.
Numerous studies have been done on these ncRNA loci, reporting the potent phenotypes induced by their genetic perturbations and by overexpression/deletion of these loci-associated miRNAs, thus placing these genetic regions at the center of numerous cellular and developmental pathways.41,42,43 MIR17HG and MIR100HG clusters and their paralogs have been placed at the nexus of opposing activities during malignancy development: oncogenic41, 44 and tumor-suppressive,42, 45, 46 respectively. Similarly, the MIRLET7BGH cluster has been identified as a potent tumor suppressor and a regulator of cellular SEN, functionally linked to neural stem cell age-related decline through the regulation of p16(Ink4a).43, 47,48,49
Dynamic and differential expression of mature miRNAs from MIR17HG and MIR100HG clusters upon replicative SEN of hADSCs
In the context of this study, we further focused on upregulation of functionally antagonistic MIR17HG and MIR100HG miRNA-bearing loci upon senescence. The human chromosome 13 MIR17HG cluster (800bp) encodes six tightly grouped miRNAs with four distinct “seed” sequences31, 50: mir-17, mir-18a, mir-19a, mir-20a, mir-19b1, and mir-92a1 (schematically shown in Fig. 2c). The miRNAs from this locus have been designated as onco-miRNAs because of their importance in cell transformation and tumorigenesis.41, 46 The chromosome 11 MIR100HG cluster homes three miRNAs (mir-125b1, mir-let7a-2, mir-100) situated within a comparable genomic distance (Fig. 2d).
The miRNAs are frequently transcribed together as polycistronic primary transcripts that are processed into multiple individual mature miRNAs.51 To identify specific miRNA production from these clusters in SEN hADSCs, we examined the abundance of mature miRNAs originating from both guide strand (mir-5p) and passenger strand (mir-3p/ mir*) by the MystiCq miRNA qPCR assay system as described in materials and methods.
Analysis of the MIR17HG cluster has revealed that only mature guide strand miRNAs: mir-17-5p, mir-18a-5p, mir-20a-5p, mir-19b1-5p and mir-92a1-5p, are detected in both SR and SEN hADSCs (Fig. 2c). No mature passenger strands: mir-17-3p, miR-18a-3p, miR-20a-3p, mir-19b1-3p and mir-92a1-3p, have been observed in the tested samples. Contrary to that, only mature passenger strand miRNA for mir-19a-3p is robustly recorded by real-time PCR (Fig. 2c). We have observed a statistically significant SEN-related increase in production of mature miRNAs in accordance with their corresponding primary non-coding transcripts MIR17HG: miR-17-5p (p < 0.001), miR-18a-5p (p < 0.01), miR-20a-5p (p < 0.01), mir-92a1-5p (p < 0.001) and mir-19a-3p (p < 0.01) (Fig. 2c and Supplementary Figure S4A). No significant change in the mature mir-19b1-5p has been detected upon replicative SEN of hADSCs (Fig. 2c).
The MIR100HG cluster has given rise to two guide strand mature miRNAs: mir-125b1-5p and mir-100-5p in SEN hADSCs. No mature passenger strands: mir-125b1-3p and mir-100-3p have been detected in our samples. Contrary to this notion, both guide mir-let7a-2-5p and passenger mir-let7a-2-3p have been detected in both SR and SEN conditions, where the balance in the stability/maturation preference of guide mir-let7a-2-5p is shifted upon SEN, favoring the production of passenger strand mir-let7a-2-3p (greater than 25-fold upregulation shown in Fig. 2c and Supplementary Figure S3A). Interestingly, this switch in the mature strand selection for mir-let7a-2 in SEN hADSCs is not due to an increase in AGO3 protein expression as was reported previously.52 The level of endogenous AGO3 protein does not seem to change significantly with replicative SEN (Supplementary Figure S4B).
Together, these data provide evidence that SEN of hADSCs correlates with a dramatic upregulation of the subset of mature miRNAs from the MIR100HG and MIR17HG clusters, and for some of them, such as mir-let7a-2, a notable shift in the maturation equilibrium between guide and passenger strands of miRNA has been observed. These abundantly upregulated mature miRNAs are called herein: SEN-associated micro RNAs (SA-miRNAs).
Identification of the gene targets of SA-miRNAs by combined transcriptome and proteome analysis
Despite a plethora of available miRNA target prediction algorithms, it remains a challenge to predict the potential target genes of a given miRNA. A number of these prediction algorithms use sequence, contextual, structural and/or evolutionary constraints and rely on subsequent validation of the targets by assessment of mRNA expression level at the large scale.53 However, transcriptional analysis of miRNA target genes does not fully reveal the extent to which miRNAs can exert control on protein expression levels, which have a tendency to change more dramatically than mRNA levels.31, 54, 55
Next, we have undertaken an integrated approach (illustrated in Fig. 1, Supplementary Figure S3C, S5 and described in materials and methods) to simultaneously explore two mechanisms by which SA-miRNAs might exert their functional effects: (1) mRNA degradation (Fig. 3a-left side of the cartoon), and (2) inhibition of protein translation without triggering mRNA decline (translational repression; Fig. 3a-right side of the cartoon).

Differential expression of mRNA and protein targets of SA-miRNAs. a Schematic showing the mRNA degradation versus translational repression modes of miRNA regulation. b Differential expression of protein-coding mRNAs in SR versus SEN hADSCs. Fold-change values (log2 SEN/SR) are shown on the x-axis, and RPKM differences (log2 SEN-SR) are shown on the y-axis. SEN downregulated protein-coding mRNAs are shown in green (lower left quadrant). c Heatmap depicting differential expression of proteins in SR versus SEN hADSCs. Normalized protein expression levels are shown for three replicate samples each of SR versus SEN hADSCs (see z-score color scale). SEN downregulated proteins are shown in the lower right quadrant. d A flowchart illustrating the approach to identifying downregulated SA-miRNA targets is shown along with a Venn-diagram indicating the numbers of genes or proteins identified via each method and the numbers identified by multiple methods
Since we wished to relate SA-miRNAs to the downregulation of their target genes at the level of mRNA and/or protein expression, we have focused our analysis on SEN-downregulated mRNAs and proteins. SEN-downregulated mRNAs are characterized as those that have low levels of fold change (log2 SEN/SR) and the smallest values for the difference in the normalized number of reads (dRPKM SEN-SR). There are a total of 937 SEN-downregulated mRNAs that have been identified in this way (shown in green in the lower left quadrant of Fig. 3b). SEN-downregulated proteins have been identified by comparing protein expression levels across SR versus SEN replicate samples. There are 986 proteins that have shown significantly lower levels of expression among SEN replicates compared to SR replicates (shown in blue in the lower right quadrant of Fig. 3c).
Having identified SEN downregulated mRNAs and proteins in this way, we then applied the mirSVR prediction algorithm (Supplementary Figure S6) to search for potential target genes of SA-miRNAs (Fig. 2c and d) among these mRNAs and proteins (materials and methods). The mirSVR algorithm has been chosen because it combines multiple sources of information for miRNA target prediction, including gene expression data from miRNA transfection experiments, thereby allowing for a lower rate of false positive predictions (Supplementary Methods). We have undertaken a conservative approach to miRNA prediction by only selecting targets with a score <−0.2. Among 8367 targets predicted by mirSVR, 389 mRNAs, and 418 proteins have been downregulated upon SEN of hADSCs (shown as Venn diagram in Fig. 3d).
Collectively, the SA-miRNA target genes captured by this approach may represent numerous biological pathways relevant to the establishment and/or maintenance of the SEN phenotype pathways in hADSCs.
Validity and sensitivity of the integrated transcriptome and proteome approach for the identification of SA-miRNA target genes
To verify the validity and sensitivity of our integrated approach for the identification of SA-miRNA target genes, we evaluated gene expression of two previously established targets of mature mir-100 from the MIR100HG locus, the HOXA1 and SMARCA5 genes.42, 56 Mir-100 directly targets these genes in mammary epithelial cells, imposing epithelial-to-mesenchymal transition (EMT) through downregulation of their expression (Supplementary Figure S7A). Consistent with published findings, our LC-MS/MS proteomic data demonstrates that the protein expression levels of both SMARCA5 and HOXA1 are significantly reduced upon SEN of hADSCs (Supplementary Figure S7B) in accordance with endogenous upregulation of mir-100-5p (Fig. 2d). Notably, both SMARCA5 and HOXA1 mRNA levels in SEN cells do not show significant downregulation when compared to SR cells, thus suggesting that mir-100-5p operates via the translational repression pathway illustrated in Fig. 3a (right panel). These findings provide a proof-of-principle that our approach is valid and reliable for deciphering targets of SA-miRNA action.
Destabilization of mRNA and translational repression through SA-miRNAs upon SEN of hADSCs
Our transcriptome analysis has revealed 389 downregulated mRNA representing SEN-associated degradation targets of SA-miRNAs in hADSCs (Fig. 3d). Representative heatmaps of transcriptional changes of SA-miRNA mRNA targets for individual SA-miRNA are shown in Fig. 4a and Supplementary Figure S8A, and their expression levels are shown in Supplementary Table S1.

Downregulation of SA-miRNAs target genes via mRNA degradation in SEN. a SEN downregulated protein coding mRNAs targeted by SA-associated miRNAs. Differential expression levels are quantified by the Euclidean distance as described in the electronic supplementary materials and Supplementary Figure S2. b Coordinated regulation of SEN downregulated protein-coding mRNAs by multiple SA-miRNAs. Individual miRNAs are shown and color-coded according to their miRNA gene cluster. c, d Coordinated regulation of NAP1L1 UTRs by SA-miRNAs. Schematic diagrams of predicted target sites of SA-miRNAs in the two distal portions of NAP1L1 UTRs: A portion from 2713 to 3062 relative to the transcriptional start site TSS (c) and a portion from 3362 to 5037 relative to the TSS (d). Repression of luciferase reporters bearing the UTRs (pmirGLO-NAP1L1) and corresponding control luciferase vector pmirGLO by mimic SA-miRNAs (n = 3, mean = +/−SD, two-tailed, type 2, Student t-test, compared to the control vector pmirGLO). P-value (p) related to experimental measurements are listed over the graphs, where ***p < 0.001, **p < 0.01
Similarly, 418 out of 8367 targets predicted by mirSVR targets have shown evidence of downregulation at the level of protein expression based on the results of the LC-MS/MS proteomic analysis (Figs. 3d and 5). The heatmaps of the representative targets of the individual SA-miRNAs are shown in Fig. 5a, Supplementary S9A and detailed in Supplementary Table S2.

Downregulation of proteins via SA-miRNA-based translational repression in SEN. a SEN downregulated proteins targeted by SEN upregulated miRNAs. Differential protein expression is quantified by the Students’ t-test (-log10 P-values shown) as described in the electronic supplementary materials. b Coordinated regulation of SEN downregulated proteins by multiple SA-associated miRNAs. Individual miRNAs are shown and color-coded according to their miRNA gene cluster. c Coordinated regulation of SMARCD2 UTR by SA-miRNAs. Schematic diagrams of the predicted target sites of SA-miRNAs in the (1913-2438) portion of SMARCD2 UTR. d, e Coordinated regulation of USP6 UTRs by SA-miRNAs. Schematic diagrams of the predicted target sites of SA-miRNAs in the two distal portions of USP6 UTRs: a portion from 6220 to 6895 relatively to the transcriptional start site TSS (d) and a portion from 7420 to 7945 relatively to the TSS (e). Repression of luciferase reporters bearing the UTRs (pmirGLO-SMARCD2 and pmirGLO-USP6,) and corresponding control luciferase vector pmirGLO by mimic SA-miRNAs (n = 3, mean = +/−SD, two-tailed, type 2, Student t-test, compared to the control vector pmirGLO). P-value (p) related to experimental measurements are listed over the graphs, where ***p < 0.001, **p < 0.01, *p < 0.05.
Interestingly, both downregulated mRNA (Fig. 4a) and numerous SA-miRNA targets associated with translation repression have shown a trend towards a potential co-regulation by two or more of the SA-mRNAs (Figs. 4b and 5b, Supplementary S8B and S9B). These results, in accordance with previously reported data, suggest that under physiological conditions many of the SA-miRNA target genes might be subjected to concurrent regulation by multiple co-expressing miRNAs from clusters with opposing biological roles: oncogenic versus tumor-suppressive.31, 57 This observation raises the following question: what function does the targeting of mRNA by multiple miRNAs from the same cluster or from clusters with opposing biological activity serve?
Combinatorial regulation of SA-miRNA gene targets NAP1L1, SMARCD2 and USP6 by the miRNA from oncogenic MIR17HG and tumor-suppressive MIR100HG clusters
To answer this question, we tested the hypothesis that co-expression of multiple miRNAs induces stronger downregulation of their common targets. We have focused on three SA-miRNA target genes that exemplify the following co-targeting arrangements: (1) a critical chromatin chaperone, NAP1L158 targeted by multiple miRNAs from antagonistic MIG17HG and MIR100HG clusters (Fig. 4b), (2) a component of the chromatin remodeling complex, barrier to autointegration factor complex, SMARCD2/BAF60B59, 60 targeted by a single miRNA from antagonistic clusters (Fig. 5b), (3) a potent oncogene, USP6/TRE1761, 62 targeted by multiple miRNAs with two distinct “seed” sequences from the same cluster (Fig. 5b).
To demonstrate the regulatory effects of mir-let-7a-5p, 18a-5p and mir-19a-3p, mir-19a-5p on NAP1L1, SMARCD2 and USP6 expression we performed in vitro luciferase assays (materials and methods). For this purpose, corresponding 3′UTRs of: (1) NAP1L1 (3′UTR 2713-3062 and a portion of 3′UTR 3362-5037) shown in Fig. 4c and d, (2) USP6 (portions of 3′UTR 6220-6895 and 3′UTR 7420-7945) shown in Fig. 5d and e, and (3) 3′UTR 1913-2438 of SMARCD2 (Fig. 5c) genes were cloned into the pGL3-promoter vector, immediately downstream of the luciferase gene. These reporter constructs were transfected into 293T cells lacking endogenous expression of mature mir-let-7p-2-3p, mir-18a-5p, mir-19a-3p or mir-125b-5p miRNAs, either alone or in combination with synthetic small, double-stranded RNA molecules designed to mimic endogenous mature miRNA molecules, mimic miRNA (Sigma, St. Louis, MO), as previously described.63, 64 To validate our hypothesis the mimic miRNAs were transfected either alone or in combinations as described in materials and methods and shown in Figs. 4c, d and 5c, d, e.
We observed that luciferase activity in cells transfected with pGL3-NAP1L1-3′UTR was significantly reduced as compared with cells transfected with the control pGL3 vector only by mir-let-7a-5p (47%) and mir-19a-3p (81%) as shown in Fig. 4c and d, respectively. No significant downregulation of luciferase activeity of pGL3-NAP1L1-3′UTR was observed when the mir-18a-5p mimic was used (Fig. 4c); although, the mir-18a-5p mimic can efficiently downregulate luciferase activity of pGL3-USP6 -3′UTR (65%) in similar experiments shown in Fig. 5e. This suggests that NAP1L1 is efficiently targeted by mir-let-7a-3p and mir-19a-3p, but not mir-18a-5p, which originates from the same cluster, MIR17HG. Analysis of pGL3-SMARCD2-3′UTR revealed a similar trend and confirmed that SMARCD2 is a target of two SA-miRNAs: mir-19a-3p and mir-125b-5p (Fig. 5c).
In a similar experiment, we tested two composite 3′UTR parts of the USP6 gene (Fig. 5d and e). The 3′UTR of the USP6 gene responded with statistical significance to only one miRNA from the MIR17HG cluster. Transient transfection of the mimic of mir-18a-5p resulted in a 65% downregulation of luciferase activity (Fig. 5e), while transfection of the mimic of mir-19a-3p showed no significant change (Fig. 5d). These findings argue for the preferential use of SA-miRNAs originating from the same cluster for the concurrent regulation of the same genes.
We also tested the ability of pairs of miRNAs to synergistically regulate mutual targets in order to facilitate more efficient target repression, a phenomenon known as cooperating miRNAs.31 Although each single SA-miRNA efficiently downregulated the NAP1L1, SMARCD2 and USP6 UTRs in transient transfection experiments (Figs. 4c and d, 5c and d), data has shown that simultaneous transfection of multiple micro-RNA mimics targeting the same UTR does not increase the efficiency of target downregulation in all of the tested reporter assay combinations. This argues against the implicit assumption that a stronger downregulation of common gene targets could be achieved by multiple simultaneously co-expressing miRNAs, thus leading to a larger response of the target to miRNA perturbation.
These data suggest that the concept of miRNA cooperativity might imply a much more sophisticated mechanism of regulation of miRNA targets than was initially anticipated. For example, selective, physiologically-relevant expression of cooperating miRNAs could be adopted by cells to facilitate distinctive and fine-tuned gene expression patterns to meet the requirements of different biological scenarios and this phenomenon is unlikely to be appropriately tested in transient transfection experiments.
Network-based functional enrichment analysis of SA-miRNA targets
Since clustered SA-miRNAs are co-expressed at different levels upon SEN in hADSCs (Fig. 2c and d), one might expect that they jointly regulate distinct molecular pathways not only by co-targeting individual genes but also by targeting differential components of the same pathways as previously shown for Drosophila melanogaster.44, 65 With this in mind, we developed a network-based functional enrichment analysis method to visually elucidate the potential roles of, and interactions among, integrated molecular networks of functionally related gene targets of SA-miRNAs in hADSCs (see materials and methods for details on the network analysis).
SA-miRNA targets that were found to be downregulated at the mRNA (Fig. 4 and Table S1) or protein level (Fig. 5 and Supplementary Table S2), were interrogated based on their functional annotations and used to seed the network analysis.66 Four categories of particular interest have been identified as relevant to the establishment and maintenance of the senescent phenotype: cell cycle, chromatin, transcription/translation and histone methyltransferases. To identify functional interactions among the corresponding SA-miRNA gene targets from these four categories, we then linked these genes in a network by edges that represent known relationships between the genes based on a variety of functional interactions, such as physical protein-protein interactions, gene co-expression and text mining co-relationships. Genes that do not have any direct known relationships of this kind are transitively linked via the minimum number of possible intermediate gene nodes, some of which are not targets of SA-miRNAs but have been downregulated in SEN hADSCs (Fig. 6 open circles). The intermediate nodes, which were not initially identified as miRNA targets or downregulated upon SEN, are the so-called Steiner nodes shown in gray in Fig. 6 and described in materials and methods.

Functional relationships and enrichment of SA-miRNAs targets genes. Four functional categories of genes were evaluated for their relationships and functional enrichment using a network-based approach as described in the experimental procedures. The network nodes represent genes and are color-coded based on their functional category. The color key shows function. Gene nodes are labeled in regards to miRNA seed presence (see node label key in the Figure). Edges represent annotated protein relationships from the STRING database. Black solid edges represent connections of the sub-network minimal spanning trees (i.e. Steiner trees), dark gray dashed edges show additional sub-network connections, and light gray dashed edges represent connections between function-specific sub-networks. P-values indicate the extent to which each function-specific sub-network is enriched for genes from that particular functional category. Steiner nodes are shown in gray. Downregulated genes, which are not targeted by SA-miRNAs are shown with colored rim based on their functional category.
The network functional enrichment analysis resulted in the elucidation of four clearly defined function-specific sub-networks, each of which corresponds to a distinct functional category, along with the inter-relationships between these functional groups (Fig. 6). The coalescence of genes with the same function into discrete sub-networks supports their close functional relationships and tight interactions, and the statistical significance of the functional enrichment within these groups is represented by P-values (Fig. 6) determined via simulation of random Steiner networks with the same number of genes from that particular functional category as described in experimental procedures. The P-values represent the probability of reconstructing sub-networks of the observed sizes, or smaller, by chance; in other words, they provide significance levels for the observed functional coherence of the sub-network.
Considering that we started with less than 1% of the total human genes, the large network of functionally related clustered genes identified by this method is surprising but immediately apparent. These data indicate that SA-miRNAs jointly regulate molecular pathways not only by co-targeting individual genes but also by targeting different components of the pathways that interconnect and could be relevant to SEN of hADSCs.
Functional significance of SA-miRNAs from the MIR17HG and MIR100HG clusters in establishment of the senescent phenotype in hADSCs
To support our hypothesis that SA-miRNAs originating from MIR17HG and MIR100HG clusters may coordinately regulate gene targets that play key functional roles in the transition of hADSCs from self-renewal to replicative SEN, we next investigated whether transient delivery of SA-miRNAs to SR ADSCs is sufficient to cause senescent phenotypes. Our data indicates that delivery of the miRNA mimics of SA-miRNAs from either MIR100HG or MIR17HG clusters separately does not result in a senescent phenotype as detected by SA-β-Gal (Fig. 7a). And are negative for additional SEN-associated markers such as immunostaining with anti-P21WAF1/Cip1 antibodies (Supplementary Figure S7F and G) and persistent SEN-associated DNA damage foci (γ−H2AX staining) (Supplementary Figure S7F and G). FITC-labeled random RNAs have been used as a control for the transfection efficiency in all of these experiments. The transfection efficiency ranges from 50 to 60% (Fig. 7d). When SA-miRNAs from both clusters were transfected into SR hADSCs, about 40% of the cells became marked by expression of SA-β-Gal (Fig. 7a), about 98% of FITC positive (FITC+) cells contained senescent-associated γ−H2AX (Fig. 7b and S1B and C), marked by proliferation arrest as detected P21WAF1/Cip1 staining and quantified by BrdU incorporation in FITC + cells (Fig. 7b and Supplementary Figure S1B). The senescent phenotype can be achieved by a range of concentrations (5pM or 10pM combined SA-miRNA mimics) widely used by others in these types of experiments (Supplementary Figure S7C).

SA-miRNAs from oncogenic MIR17HG and tumor-suppressive MIR100HG clusters function to establish senescent phenotype in hADSCs. a Percentage of SA-β-Gal positive cells among the total amount of cells counted after transient transfection of the mimics of the SA-miRNAs from either the MIR17HG (mir-17-5p, mir-18a-5p, mir-19a-3p, mir-20a-5p and mir-92a1-5p) or the MIR100HG (mir-125b1-5p, mir-1let7a-2-3p, mir-100-5p) clusters separately or after simultaneous transfection by a full set of the SA-miRNA mimics from both clusters in SR hADSCs. SA-miRNA mimics were transfected with FITC-labeled control to account for the transfection efficiency as described in material and methods and electronic supplementary materials. The transfection efficiency for each combination is shown in (Supplementary Figure S8D) and is expressed as a percentage of green cells among the total DAPI-positive cells (n) counted under the fluorescent microscope. b Quantitation of the BrdU incorporation in SA-miRNA mimics-transfected SR hADSCs (FITC+) 72 hrs post-transfection presented as FITC+ BrdU+ (red bar). Quantification of cells positive for SEN-associated markers P21WAF1/Cip1 and γH2AX persistent DDR focal staining is performed under the same conditions in FITC+ (red bars) and FITC− cells. Results were plotted on the graphs as the averages of three independent experiments (biological replicates n = 3) with the standard deviation of data. Cell counted in each experiments: BrdU staining n 1 = 440, n 2 = 279, n 3 = 334; γH2AX staining n 1 = 117, n 2 = 109, n 3 = 133; P21WAF1/Cip1 staining n 1 = 118, n 2 = 135, n 3 = 90. P-values (p) were calculated as Students’ two-tailed test: BrdU labeling experiment ***p < 0.001; γH2AX staining experiment ***p < 0.001; P21WAF1/Cip1 staining **p < 0.01. c The panels demonstrate direct influences of SA-miRNAs on gene transcription. Expression of the direct SA-miRNA target genes was measured by qPCR analysis in SR hADSCs (SR, blue bars) and SR cells transiently transfected with a full set of the SA-miRNA mimics (SR + miRNA, red bars). d The panel demonstrates indirect influences of the SA-miRNAs on gene transcription. Expression of genes previously shown to be downregulated in replicative senescent hADSCs but not identified as SA-miRNA targets was measured by qPCR analysis in SR hADSC cells (SR, blue bars) and in SR cells transiently transfected with the full set of SA-miRNA mimics (SR + miRNA, red bars). RNA was isolated from the cells 48hrs post-transfection. Samples were normalized against β-actin. Mean expression levels ± SEM (n = 3) are shown as fold change (ΔΔCτ). e Normalized mean protein expression levels ± SEM (n = 3) in SR (blue bars) and SEN (red bars) hADSCs are shown for SA-miRNA direct and indirect target genes. Statistical differences for the qPCR mRNA (c) and (d) and protein (e) expression comparisons were evaluated by Student’s t-test, where ***p < 0.0001, **p < 0.01, *p < 0.05.
The senescent phenotype under these conditions is similar to replicative SEN as demonstrated by downregulation of a handful of randomly selected genes (Fig. 7c, d and e). In SR hADSCs transfected with a full set of SA-miRNA mimics, we observed downregulation of endogenous mRNA from the enriched functional network that represents SA-miRNA target genes such as SUZ12, NAP1L1, SMARCD2, SAP18, IGF2BP3, CHD2 and CHD4 (Figs. 6 and 7c), as well as a number of the genes not targeted by SA-miRNAs but, nevertheless, shown to be downregulated upon replicative SEN, such as SMARCA1, CHD8, HDAC3, HDAC5 and HDAC9 (Fig. 7d and Supplementary Tables S3 and S4). Our functional test suggests that even transient delivery of a full set of mature SA-miRNA mimics in SR hADSCs is necessary and sufficient to trigger a cascade of primary (through downregulation of SA-miRNA targets) and secondary (downregulation of downstream genes) events resulting in the senescent phenotype of hADSCs.
These results further support the observation that SA-miRNAs from the MIR17HG and MIR100HG clusters with reported opposing biological activities (Fig. 2c and d) are functionally linked to the process of establishment of the cellular SEN phenotype in hADSCs and identify the direct targets through which these SA-miRNAs could exert their regulatory roles.
Discussion
Cellular senescence involves a coordination of an extremely broad range of processes from telomere homeostasis, genome instability and DNA damage, to drastic changes in chromatin organization, inflammatory signaling and metabolic, cytoskeletal and paracrine changes.67 Due to the profound secretory phenotype of senescent cells, their impact on organs and tissues is far from neutral. The role of senescence in the aging of adult stem cells is tightly linked to tissue maintenance and homeostasis and often viewed as an irreversible barrier to immortalization and tumorigenesis under the assumption that senescence evolved to suppress tumorigenesis.68, 69 This view has been intensely debated in recent years.70, 71 Contrary to the hypothesis that senescence and tumorigenicity are always permanently connected and mutually exclusive, recent data monitoring p16INK4a in mice indicate that the activation of this hallmark of senescence is, in fact, a characteristic of all emerging cancers,72 thus suggesting that cellular senescence might be a quasi-stable and/or plastic cellular state prone to cancerogenesis rather than a cancer preventive mechanism.
It has been proposed that in addition to protein-coding oncogenes and tumor suppressor genes, one has to take into account miRNAs and their regulatory networks in order to understand the complex processes underlying malignant transformation.73,74,75 Concurrently, there is a considerable interest in the development of senescence-targeted cancer and metabolic therapeutics. This goal suggests the need for a system-level view of the regulation of senescence in order to identify not only reliable markers of senescence that will translate into human settings, but also to understand the intimate connections between oncogenic events and senescence better. Such approach will allow for defining a set of genetic targets that can elicit, modulate and, most importantly, balance mechanisms involved in tumor initiation, growth and progression. Our study contributes exactly to such initiative and provides measurable and reliable markers (composing SA-miRNAs and their target genes) that can be further deployed to identify mechanistic underpinnings of oncogenic and tumor-suppressive balance. Such resource has not been previously available and technically challenging to obtain.
In this study, we not only have identified a set of SA-miRNAs originating from oncogenic MIR17HG and tumor-suppressive MIR100HG clusters as potent controllers of complex and coordinated interactions among several cellular sub-processes associated with cellular senescence. Importantly, we have demonstrated functional significance of these SA-miRNAs in establishing senescent phenotype in adult adipose-derived stem cells (Fig. 7a and b, and Supplementary Figure S7E, F and G). In addition, our study functionally defines a set of gene-targets regulated by these SA-miRNAs. These target genes are linked to interconnected biological networks that control cell fate switches and, most importantly, our data suggest that balancing of these network components might be the cause and consequence of SA-miRNA–target interactions. In our proposed model of the action of SA-miRNAs, it remains unclear if forced overexpression of SA-miRNAs in SR hADSCs has physiologically relevant consequences and fully recapitulates the complexity of the hADSCs senescent phenotype in the long run. However, even in transient transfection experiments, the introduction of a set of mimic miRNAs representing SA-miRNAs produces similar to replicative SEN transcriptional outcomes, such as miRNA-induced decay of the direct SA-miRNA targets (Figs. 6 and 7c, e, and Supplementary Tables S1 and S2) and an adequate downstream response recorded as downregulation of the indirect SA-miRNA targets (Fig. 7d and e and Supplementary Table S3). Therefore, we believe that our approach accurately and comprehensively identifies SA-miRNA targets as well as functionally supports the critical role of SA-miRNAs in establishing early stages of cellular senescence. This regulatory network of miRNAs is probably highly dynamic and sensitive to external signaling. Recently, other methods were developed to capture miRNA-target interactions, however, there is still no consensus as to which approach is the most accurate and appropriate for readout of biological function.31
While our study does not uncover the underlying molecular mechanisms of how SA-miRNA–target interactions balance the network, particularly when miRNA expression upon SEN originates from the clusters with seemingly opposing biological roles in oncogenesis, one can put forth the following hypothesis, that similar to miRNAs and transcriptional factors that often form feed forward loops,31, 76,77,78 the SA-miRNAs in hADSCs have evolved to provide a precise target gene expression among critical SEN-associated sub-processes, such as networks controlling cell cycle, transcription/translation and, ultimately, chromatin assembly.70, 79 In our view, the direct effects of oncogenic SA-miRNAs on target genes might be counter-balanced by the action of tumor-suppressive SA-miRNAs on a set of different targets within the same or an interconnected network, thus buffering stochastic fluctuations73 in order to increase the precision of the expression of the entire regulatory network.
Relevant to this discussion is an example of malignant soft tissue tumors-rare, but aggressive malignancies. Liposarcoma is the most common soft tissue sarcoma in adults characterized by a high rate of local recurrence and high metastatic potential. It has been a subject of intensive debate that the local tumor microenvironment, cell-cell, and cell-stromal interactions are inherent in serving biochemical functions. Malignant cells perpetually stimulate host stromal, vascular and tissue-specific adult stem cells to conduct physiological invasion.73, 74 Senescent cells through their specific transcriptional programs and senescence-associated secretory phenotype might create a permissive “field” for the malignant cell to grow (reviewed in).80 On the other hand, various secreted by the growing tumor signaling molecules, such as cytokines, chemo-tactics and growth factors, also are can modulate the local environment and, in their turn, can feedback on the senescent cells (paracrine loop). Therefore, it is theoretically possible that a shift in the delicate balance between the oncogenic and tumor-suppressive events imposed by SA-miRNAs, as well as transcriptional cascades targeted by these SA-miRNAs could come to play when adipose tissue homeostasis is perturbed in disease. To support such assumption, recent study elegantly demonstrated that, indeed, sarcoma itself, prior to sarcoma invasion, can impose miRNA expression changes in pre-adipocytes and other mesechymal stromal cells through the paracrine effectors.81 It is plausible that similar events could be imposed on senescent ADSCs. Therefore, we speculate that the sequence of oncogenic events that can push senescent cells into a cancerous transformation may depend on the prevalence of autocrine or paracrine signals, which when received by senescent cells can shift the balance in levels of mature miRNAs. Such shift in the balancing act of microRNAs might enable underlying changing in epigenetic regulation, similar to the proposed role of the EMT and its reversal mesenchymal-epithelial transition in controlling or avoiding cancer.70 These autocrine or paracrine signaling upon senescence of hADSCs are subject to further investigations.
We are aware of the argument that miRNA’s impact on individual targets might depend on many dynamic factors, such as cellular localization of miRNAs and their targets, their relative concentrations, and the context-specific effects of other regulators, including transcriptional factors and RNA-binding proteins.82,83,84,85 Despite these arguments, the utility of our study is in the computational and functional identification of comprehensive and interconnected SA-miRNA-target regulatory networks describing the phenotypic manifestation of hADSC- SEN. Although cumulative studies of miRNA expression in different models of cellular senescence suggest that miRNA signatures of senescence are likely to be cell-type and trigger dependent with a common core of effectors, we believe that an adequate signature of senescence for adult adipose-derived stem cells would consist of a number of well-established markers, among which, SA-miRNAs and their direct targets may well be a useful addition for assessment of adipose tissue metabolism and regeneration. The SA-miRNAs identified in this study are particularly promising considering that the endogenous circulating miRNAs in human serum86 and other body fluids, such as cerebrospinal fluid, colostrum, peritoneal fluid, saliva, seminal fluid and urine87 are highly stable. Our study provides a matrix of potentially measurable markers of replicative SEN in hADSCs with a viable hypothesis that the coordinated action of these SA-miRNAs provide for the balance between driving and restraining tumor progression and may be further exploited to define correlations between these senescence-associated miRNAs and other relevant markers of disease progression as well as responses to therapeutic treatments.
Materials and methods
Isolation and culture of hMSCs
Human adipose derived stem cells were isolated from human subcutaneous white adipose tissue collected during liposuction procedures. The lipoaspirate was suspended in Hank’s buffered salt solution (Life Technology), 3.5% Bovine Serum Albumin (BSA, Sigma), 1% Collagenase Type II (Sigma) in 1:3 w/v ratio and shaken at 37 °C for 50 min. The cells were filtered through a 70 μm mesh cell strainer (BD Falcon #352350), treated with red blood cell lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.3), and expanded ex-vivo in DMEM/F12 complete medium (DMEM/F12, 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin; Life technology) in 10% CO2 at 37 °C and passaged at 80% confluency, changing medium every 72–96 h. Cumulative population doublings were calculated by summing the population doublings (PD = log(N/N0) × 3.33, where N0 is the number of cells plated in the flask, and N is the number of cells harvested at this passage) across multiple passages as a function of the number of days it was grown in culture.
Surface marker characterization
Five × 105 cells either SR (PD8) or SEN (PD40) each were incubated for 30 min on ice in the dark with fluorochrome-conjugated antibodies (CD31, CD44, CD45 and CD105; Invitrogen) in PBS with 1% BSA (Sigma), washed and analyzed in a Guava EasyCyte Mini System (Guava Technologies, Millipore). Data analysis was done with FlowJo software (Tree Star, Ashland, OR).
SA-β-Gal staining
A SA-β-Gal activity assay was done according to manufacturer’s instructions (BioVision). The cells were washed with PBS and fixed with fixation solution for 15 min at room temperature. The cells were washed with PBS twice and X-Gal staining solution was added with a staining supplement per well and incubated overnight at 37 °C. The cells were washed twice with PBS, and the images were captured using a microscope (Nikon, TE300, DXM1200 Digital Camera, Japan).
Proteomic analysis and transcriptome analysis with RNA-seq
The details are given in Supplementary Experimental Procedures.
miRNA target identification
The mRNA targets of differentially expressed miRNAs were identified using the program mirSVR. This program was chosen because it combines miRNA-mRNA binding site sequence analysis with several additional sources of contextual information, including gene expression data from miRNA transfection experiments, in order to make target predictions (Supplementary Figure S6). Accordingly, mirSVR has been shown to yield a relatively low rate of false positive predictions for miRNA target identification.88 mirSVR also provides scores to rank the predicted targets, and, for this study, targets with a score <−0.2 were selected for further analysis.
Network-based functional enrichment analysis
The details of network analysis are given in Supplementary Experimental Procedures.
RT-PCR
Total cellular RNA was extracted from cells using the TRIzol reagent™ (Life Technologies) according to manufacturer’s instructions. The miRNA was isolated using a mirPremier miRNA isolation kit (Sigma-Aldrich), RNA and miRNA were quantified with a NanoDrop ND-2000 Spectrophotometer (Thermo Scientific). The cDNA was synthesized by adding the purified RNA and oligo(dT) primers by using Superscript III reverse transcriptase (Life Technologies). Primers were designed by Primer3 software and shown in Supplementary Table S1. For miRNA cDNA synthesis, the Mystic miRNA cDNA synthesis Mix kit (Sigma-Aldrich) was used. All miRNA assay primers were bought from Sigma-Aldrich.
Real-time quantitative PCR
Quantification of mRNA and miRNA expression for candidate genes was performed by real-time quantitative PCR (qRT-PCR) using the LightCycler® 480 Real-Time PCR System (Roche). Total RNA and miRNA was reverse transcribed by using the high capacity superscript III reverse transcriptase (Life Technologies) and the Mystic miRNA cDNA synthesis Mix kit (Sigma-Aldrich), respectively. Primers were designed by primer3 software (Supplementary Table S5). All miRNA assay primers were bought from Sigma-Aldrich. qRT-PCR reactions were performed with the power SYBR® green PCR master mix and the mystic miRNA SYBR green qPCR ReadyMix in a MicroAmp optical 96-well reaction plate. The PCR amplification of total RNA was performed in a LightCycler® 480 Real-Time PCR System (Roche) using the following program: Cycle 1, 95 °C for 10 min. Cycle 2, 40 cycles of 95 °C for 15 sec, 60 °C for 60 sec. Cycle threshold (CT) values were automatically obtained. Relative expression values of RNA were obtained by normalizing CT values of the mRNA genes in comparison with CT values of the endogenous control (beta-actin) using the CT method. The PCR amplification of miRNA was performed in a LightCycler® 480 Real-Time PCR System (Roche) using the following program: Cycle 1, 95 °C for 2 min. Cycle 2, 40 cycles of 95 °C for 5 sec, 60 °C for 30 sec. Relative expression values of miRNA were obtained by normalizing CT values of the miRNA genes in comparison with CT values of the endogenous control (U6) using the CT method.
Luciferase assay
The luciferase reporter constructs were built as previously described.63 NAP1L1-1 (350bp, 2713-3062) and NAP1L1-2 (675bp, 3362-5037) from the 3′ UTR of human NAP1L1 gene, USP6-1 (675bp, 6220-6895) and USP6-2 (527bp, 7420-7945) from the 3′UTR of human USP6 gene and SMARCD2 (525bp, 1913-2438) from the 3′ UTR of human SMARCD2 gene were amplified using the primer sets (shown in Supplementary Experimental Procedures). Purified PCR products were cloned into multiple cloning sites of the pmirGLO dual-luciferase miRNA target expression vector (Promega) downstream of the firefly luciferase gene. The primer sequences were flanked by SacI and SalI sites to generate pmirGLO-NAP1L1-1, pmirGLO-NAP1L1-2, pmirGLO-SMARCD2, pmirGLO-USP6-1 and pmirGLO-USP6-2. Details are given in Supplementary Experimental Procedures.
Mimic miRNA transfection studies
The hADSCs (SR (PD8)) were seeded on 4-well slides at a density of 1 × 104 cells/well 1 day before transfection with 5 and 10 pmol each of different miRNA mimics to SA-miRNA using Fugene 6 (Promega). 48 h after transfection, SA-β-Gal staining was performed according to manufacturer’s instructions (BioVision), RNA extraction and the subsequent real-time qPCR were performed to detect target gene expression.
Statistical analysis
Data points from individual assays represent mean ± SEM. The statistical significance between two conditions was assessed by a two-tailed unpaired t-test. * p < 0.05, ** p < 0.01, *** p < 0.001, and n.s. represents p ≥ 0.05.
Data access
The raw data files are being passed to NCBI’s Sequence Read Archive (SRA).Link from GEO records GSE77284 study at: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE77284. A description of the data formats, software tools to manipulate these data formats, and all codes implementing the statistical models described herein can also be found in the supplementary information on line.
References
Jiang, Y., Berry, D. C., Tang, W. & Graff, J. M. Independent stem cell lineages regulate adipose organogenesis and adipose homeostasis. Cell Rep. 9, 1007–1022 (2014).
Katz, A. J., Tholpady, A., Tholpady, S. S., Shang, H. & Ogle, R. C. Cell surface and transcriptional characterization of human adipose-derived adherent stromal (hADAS) cells. Stem Cells 23, 412–423 (2005).
Gimble, J. & Guilak, F. Adipose-derived adult stem cells: isolation, characterization, and differentiation potential. Cytotherapy 5, 362–369 (2003).
Spalding, K. L. et al. Dynamics of fat cell turnover in humans. Nature 453, 783–787 (2008).
Weissman, I. L. Stem cells: units of development, units of regeneration, and units in evolution. Cell 100, 157–168 (2000).
Niu, P. et al. Transcriptional profiling of interleukin-2-primed human adipose derived mesenchymal stem cells revealed dramatic changes in stem cells response imposed by replicative senescence. Oncotarget 6, 17938–17957 (2015).
Singer, N. G. & Caplan, A. I. Mesenchymal stem cells: mechanisms of inflammation. Annu. Rev. Pathol. 6, 457–478 (2011).
Ben-Ami, E., Berrih-Aknin, S. & Miller, A. Mesenchymal stem cells as an immunomodulatory therapeutic strategy for autoimmune diseases. Autoimmun. Rev. 10, 410–415 (2011).
Signer, R. A. & Morrison, S. J. Mechanisms that regulate stem cell aging and life span. Cell Stem Cell 12, 152–165 (2013).
Rao, M. S. & Mattson, M. P. Stem cells and aging: expanding the possibilities. Mech. Ageing Dev. 122, 713–734 (2001).
Trosko, J. E. Role of diet and nutrition on the alteration of the quality and quantity of stem cells in human aging and the diseases of aging. Curr. Pharm. Des. 14, 2707–2718 (2008).
Janzen, V. et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 443, 421–426 (2006).
Wang, J. et al. Inhibition of activated pericentromeric SINE/Alu repeat transcription in senescent human adult stem cells reinstates self-renewal. Cell Cycle 10, 3016–3030 (2011).
Shyh-Chang, N., Daley, G. Q. & Cantley, L. C. Stem cell metabolism in tissue development and aging. Development 140, 2535–2547 (2013).
Di Mitri, D. et al. Tumour-infiltrating Gr-1 + myeloid cells antagonize senescence in cancer. Nature 515, 134–137 (2014).
Sousa-Victor, P. et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321 (2014).
Shomron, N. & Levy, C. MicroRNA-biogenesis and pre-mRNA splicing crosstalk. J. Biomed. Biotechnol. 2009, 594678 (2009).
Lewis, B. P., Burge, C. B. & Bartel, D. P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 (2005).
Winter, J. & Diederichs, S. Argonaute proteins regulate microRNA stability: increased microRNA abundance by Argonaute proteins is due to microRNA stabilization. RNA Biol. 8, 1149–1157 (2011).
Wang, B. et al. Distinct passenger strand and mRNA cleavage activities of human Argonaute proteins. Nat. Struct. Mol. Biol. 16, 1259–1266 (2009).
Osella, M., Bosia, C., Cora, D. & Caselle, M. The role of incoherent microRNA-mediated feedforward loops in noise buffering. PLoS Comput. Biol. 7, e1001101 (2011).
Siciliano, V. et al. MiRNAs confer phenotypic robustness to gene networks by suppressing biological noise. Nat. Commun. 4, 2364 (2013).
Hackl, M. et al. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging Cell 9, 291–296 (2010).
Rippe, C. et al. MicroRNA changes in human arterial endothelial cells with senescence: relation to apoptosis, eNOS and inflammation. Exp. Gerontol. 47, 45–51 (2012).
Singh, S. K., Pal Bhadra, M., Girschick, H. J. & Bhadra, U. MicroRNAs--micro in size but macro in function. FEBS J. 275, 4929–4944 (2008).
Rottiers, V. & Naar, A. M. MicroRNAs in metabolism and metabolic disorders. Nature reviews. Mol. Cell Biol. 13, 239–250 (2012).
Lanceta, J., Prough, R. A., Liang, R. & Wang, E. MicroRNA group disorganization in aging. Exp. Gerontol. 45, 269–278 (2010).
Chen, L. H., Chiou, G. Y., Chen, Y. W., Li, H. Y. & Chiou, S. H. MicroRNA and aging: a novel modulator in regulating the aging network. Ageing Res. Rev. 9 Suppl. 1, S59–S66 (2010).
Drummond, M. J. et al. Aging and microRNA expression in human skeletal muscle: a microarray and bioinformatics analysis. Physiol. Genomics 43, 595–603 (2011).
Thomson, D. W., Bracken, C. P. & Goodall, G. J. Experimental strategies for microRNA target identification. Nucleic Acids Res. 39, 6845–6853 (2011).
Hausser, J. & Zavolan, M. Identification and consequences of miRNA-target interactions--beyond repression of gene expression. Nat. Rev. Genet. 15, 599–612 (2014).
Zuk, P. A. et al. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 13, 4279–4295 (2002).
Rodriguez, A. M., Elabd, C., Amri, E. Z., Ailhaud, G. & Dani, C. The human adipose tissue is a source of multipotent stem cells. Biochimie 87, 125–128 (2005).
Gimble, J. M. & Guilak, F. Differentiation potential of adipose derived adult stem (ADAS) cells. Curr. Top. Dev. Biol. 58, 137–160 (2003).
Gruber, H. E. et al. Human adipose-derived mesenchymal stem cells: serial passaging, doubling time and cell senescence. Biotech. Histochem. 87, 303–311 (2012).
Jun, H. S., Dao, L. T., Pyun, J. C. & Cho, S. Effect of cell senescence on the impedance measurement of adipose tissue-derived stem cells. Enzyme Microb. Technol. 53, 302–306 (2013).
Shiloh, Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3, 155–168 (2003).
Aguilera, A. & Gomez-Gonzalez, B. Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 9, 204–217 (2008).
Lopez, M. F. et al. Proteomic signatures of serum albumin-bound proteins from stroke patients with and without endovascular closure of PFO are significantly different and suggest a novel mechanism for cholesterol efflux. Clin. Proteomics 12, 2 (2015).
Lopez, M. F. et al. Heart-brain signaling in patent foramen ovale-related stroke: differential plasma proteomic expression patterns revealed with a 2-pass liquid chromatography-tandem mass spectrometry discovery workflow. J. Investig. Med. 60, 1122–1130 (2012).
Mendell, J. T. miRiad roles for the miR-17-92 cluster in development and disease. Cell 133, 217–222 (2008).
Chen, D. et al. miR-100 induces epithelial-mesenchymal transition but suppresses tumorigenesis, migration and invasion. PLoS Genet. 10, e1004177 (2014).
Ma, L., Li, G. Z., Wu, Z. S. & Meng, G. Prognostic significance of let-7b expression in breast cancer and correlation to its target gene of BSG expression. Med. oncol. 31, 773 (2014).
He, L. et al. A microRNA polycistron as a potential human oncogene. Nature 435, 828–833 (2005).
Nakanishi, H. et al. Loss of miR-125b-1 contributes to head and neck cancer development by dysregulating TACSTD2 and MAPK pathway. Oncogene 33, 702–712 (2014).
Kent, O. A. & Mendell, J. T. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene 25, 6188–6196 (2006).
Nishino, J., Kim, I., Chada, K. & Morrison, S. J. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell 135, 227–239 (2008).
Chen, F. et al. Let-7b inhibits human cancer phenotype by targeting cytochrome P450 epoxygenase 2J2. PLoS ONE 7, e39197 (2012).
Fu, T. Y. et al. Let-7b-mediated suppression of basigin expression and metastasis in mouse melanoma cells. Exp. Cell Res. 317, 445–451 (2011).
Khorshid, M., Hausser, J., Zavolan, M. & van Nimwegen, E. A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat. Methods 10, 253–255 (2013).
Stefani, G. & Slack, F. J. Small non-coding RNAs in animal development. Nature reviews. Mol. Cell Biol. 9, 219–230 (2008).
Winter, J. & Diederichs, S. Argonaute-3 activates the let-7a passenger strand microRNA. RNA Biol. 10, 1631–1643 (2013).
Sethupathy, P., Megraw, M. & Hatzigeorgiou, A. G. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat. Methods 3, 881–886 (2006).
Selbach, M. et al. Widespread changes in protein synthesis induced by microRNAs. Nature 455, 58–63 (2008).
Liu, J. Control of protein synthesis and mRNA degradation by microRNAs. Curr. Opin. Cell Biol. 20, 214–221 (2008).
Sun, D. et al. miR-99 family of MicroRNAs suppresses the expression of prostate-specific antigen and prostate cancer cell proliferation. Cancer Res. 71, 1313–1324 (2011).
Krek, A. et al. Combinatorial microRNA target predictions. Nature Genet. 37, 495–500 (2005).
Li, Z. et al. Foxa2 and H2A.Z mediate nucleosome depletion during embryonic stem cell differentiation. Cell 151, 1608–1616 (2012).
Wang, W. et al. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 10, 2117–2130 (1996).
Saccone, V. et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev. 28, 841–857 (2014).
Ye, Y. et al. TRE17/USP6 oncogene translocated in aneurysmal bone cyst induces matrix metalloproteinase production via activation of NF-kappaB. Oncogene 29, 3619–3629 (2010).
Pringle, L. M. et al. Atypical mechanism of NF-kappaB activation by TRE17/ubiquitin-specific protease 6 (USP6) oncogene and its requirement in tumorigenesis. Oncogene 31, 3525–3535 (2012).
Anbazhagan, A. N. et al. Translational repression of SLC26A3 by miR-494 in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 306, G123–G131 (2014).
Meseguer, S., Martinez-Zamora, A., Garcia-Arumi, E., Andreu, A. L. & Armengod, M. E. The ROS-sensitive microRNA-9/9* controls the expression of mitochondrial tRNA-modifying enzymes and is involved in the molecular mechanism of MELAS syndrome. Hum. Mol. Genet. 24, 167–184 (2015).
Grun, D., Wang, Y. L., Langenberger, D., Gunsalus, K. C. & Rajewsky, N. microRNA target predictions across seven Drosophila species and comparison to mammalian targets. PLoS Comput. Biol. 1, e13 (2005).
von Mering, C. et al. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 33, D433–D437 (2005).
Jeyapalan, J. C. & Sedivy, J. M. Cellular senescence and organismal aging. Mech. Ageing Dev. 129, 467–474 (2008).
Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 11, S27–S31 (2001).
Lin, A. W. et al. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 12, 3008–3019 (1998).
Kishi, S., Bayliss, P. E. & Hanai, J. A prospective epigenetic paradigm between cellular senescence and epithelial-mesenchymal transition in organismal development and aging. Transl. Res. 165, 241–249 (2015).
Lim, C. S. Cellular senescence, cancer, and organismal aging: a paradigm shift. Biochem. Biophys. Res. Commun. 344, 1–2 (2006).
Burd, C. E. et al. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 152, 340–351 (2013).
Paweletz, C. P. et al. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene 20, 1981–1989 (2001).
Liotta, L. A. & Kohn, E. C. The microenvironment of the tumour-host interface. Nature 411, 375–379 (2001).
Bartel, D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004).
Marson, A. et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 134, 521–533 (2008).
Shalgi, R., Lieber, D., Oren, M. & Pilpel, Y. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput. Biol. 3, e131 (2007).
Hornstein, E. & Shomron, N. Canalization of development by microRNAs. Nature Genet. 38, S20–S24 (2006).
Bilsland, A. E., Revie, J. & Keith, W. MicroRNA and senescence: the senectome, integration and distributed control. Crit. Rev. Oncog. 18, 373–390 (2013).
Perez-Mancera, P. A., Young, A. R. & Narita, M. Inside and out: the activities of senescence in cancer. Nat. Rev. Cancer 14, 547–558 (2014).
Tilkorn, D. J. et al. Sarcoma cells induce alteration in adipogenic differentiation. Anticancer Res. 32, 1167–1173 (2012).
Erhard, F. et al. Widespread context dependency of microRNA-mediated regulation. Genome Res. 24, 906–919 (2014).
Larsson, E., Sander, C. & Marks, D. mRNA turnover rate limits siRNA and microRNA efficacy. Mol. Syst. Biol. 6, 433 (2010).
van Kouwenhove, M., Kedde, M. & Agami, R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat. Rev. Cancer 11, 644–656 (2011).
Kedde, M. et al. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell 131, 1273–1286 (2007).
Mitchell, P. S. et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA. 105, 10513–10518 (2008).
Weber, J. A. et al. The microRNA spectrum in 12 body fluids. Clin. Chem. 56, 1733–1741 (2010).
Betel, D., Koppal, A., Agius, P., Sander, C. & Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 11, R90 (2010).
Acknowledgements
We thank Aelan Cell Technology members, particular James Tollervey and Alexandra Ortiz for help with manuscript preparation and discussions, and David Sarracino, Amol Prakash, Sirkka-Lisa Hostika, Denize Munoz and Glenn Geesman for technical assistance. This work was supported by Aelan Cell Technology, an NIA grant (R21 AGO43921) to V.V.L. and by the Alfred P. Sloan Foundation (BR-4839) to I.K.J. A.S. and A.N. are fellows of the Bolashag Foundation and their training was supported by a KazhMU sponsored program. M.F.L. and V.V.L. designed the experiments; P.N., M.G., David Sarracino, M.V., Y.Z. performed the experiments, L.W. developed and applied the bioinformatics method for data analysis; A.S., A.N. help with technical assistance; M.F.L., I.K.J. and V.V.L. discussed the results and wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
V.V.L., M.G. are full-time employees at Aelan Cell Technologies, Inc, a company that is commercializing biomarkers discovery. V.V.L. and M.G. all own stock in Aelan CT. M.F.L., B.K., M.V. are full-time employees at Nuclear Biotechnology, Inc. M.F.L., B.K., M.V. all own stock in Nuclea Bio. I.K.J. and L.W. are consultants to Aelan CT. This work was supported by NucleaBio and Aelan CT. The remaining authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lopez, M.F., Niu, P., Wang, L. et al. Opposing activities of oncogenic MIR17HG and tumor suppressive MIR100HG clusters and their gene targets regulate replicative senescence in human adult stem cells. npj Aging Mech Dis 3, 7 (2017). https://doi.org/10.1038/s41514-017-0006-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41514-017-0006-y
This article is cited by
-
DNA methylation changes related to nutritional deprivation: a genome-wide analysis of population and in vitro data
Clinical Epigenetics (2019)
-
MiR-152 Regulates Apoptosis and Triglyceride Production in MECs via Targeting ACAA2 and HSD17B12 Genes
Scientific Reports (2018)
-
LncRNA MIR100HG promotes cell proliferation in triple-negative breast cancer through triplex formation with p27 loci
Cell Death & Disease (2018)
