
Abstract
Separation of C2H4 from C2H4/C2H2/C2H6 mixture with high working capacity is still a challenging task. Herein, we deliberately design a Th-metal-organic framework (MOF) for highly efficient separation of C2H4 from a binary C2H6/C2H4 and ternary C2H4/C2H2/C2H6 mixture. The synthesized MOF Azole-Th-1 shows a UiO-66-type structure with fcu topology built on a Th6 secondary building unit and a tetrazole-based linker. Such noticeable structure, is connected by a N,O-donor ligand with high chemical stability. At 100 kPa and 298 K Azole-Th-1 performs excellent separation of C2H4 (purity > 99.9%) from not only a binary C2H6/C2H4 (1:9, v/v) mixture but also a ternary mixture of C2H6/C2H2/C2H4 (9:1:90, v/v/v), and the corresponding working capacity can reach up to 1.13 and 1.34 mmol g−1, respectively. The separation mechanism, as unveiled by the density functional theory calculation, is due to a stronger van der Waals interaction between ethane and the MOF skeleton.
Similar content being viewed by others

Introduction
Ethylene is one of the most widely used feedstock molecules for the production of polymers and high-value organic chemicals1,2. It is usually produced by the thermal cracking of hydrocarbons. The removal of ethane and acetylene by-products that inevitably arise during these processes is one of the most challenging chemical separations due to the similarity of the physicochemical properties of ethane (kinetic diameter 4.4 Å, boiling point 184.55 K), ethylene (kinetic diameter 4.2 Å, boiling point 169.42 K), and acetylene kinetic diameter 3.3 Å, boiling point 188.40 K)3,4,5,6,7.
At present, cryogenic distillation is the main technology used to separate ethane and ethylene with the requirement of high pressure (5–28 bar) and low temperature (180–258 K)8,9, which indicates that this process is expensive and comes with a high energy penalty. And partial hydrogenation of acetylene into ethylene over catalyst10 or solvent extraction of cracked olefins11 are also involved with the purification of ethylene from acetylene. Adsorptive separation by porous materials is an alternative technology, especially, some metal-organic frameworks (MOFs)12,13,14,15,16,17,18,19,20,21 with high volume, designable pore characteristics, and countless structural possibilities, can be employed into the gas separation processes, the adsorption selectivity and capacity are higher than the results of conventional adsorbents22,23,24 such as zeolites and carbon-based, especially the adsorption and separation for C2H6/C2H48,22,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39.
For the MOFs with open metal site, the ethylene can easily bind to it, leading to highly selective uptake of ethylene over ethane, due to the electrostatic interaction between the π-electron in ethylene and the positive charge in open metal sites36,40,41,42,43,44, such as, HKUST-144, particularly in the low pressure region, is preferential adsorption of ethylene, which is supported by some theoretical calculations45,46. In contract, for some special MOFs, when the coordination positions of metal reaches to saturate, they can enable favorable adsorption towards ethane over ethylene through their unique pore wall that affords stronger van der Waals (vdW) interaction between the H of C2H6 and the MOF skeleton8,22,25,26,27,28,29,32,34,35,47. For example, ZIF-7 presents the first example of a microporous solid displaying the selective adsorption of paraffins over olefins48. However, MOFs showing such uncommon adsorption phenomenon (C2H6 over C2H4) is still rare until now.
Recently, Lu and co-workers49 report that they use TJT-100 to obtain the selective adsorption of ethane and acetylene over ethylene from a ternary mixture of C2H2/C2H6/C2H4 (0.5:0.5:99, v/v/v) and achieve a C2H4 purity greater than 99.9% (working capacity of 0.69 mmol g−1) by a single-breakthrough operation. Zaworoko et al.38 use a synergistic sorbent separation method for the one-step production of polymer-grade C2H4 from ternary (C2H2/C2H6/C2H4, working capacity of 0.32 mmol g−1) or quaternary (CO2/C2H2/C2H6/C2H4) gas mixtures with a series of physisorbents. In this regard, constructing MOFs with high working capacity is still highly desirable from the viewpoint of practical application.
The high-valence metal ions are often used to construct stable MOFs, such as Cr(III) for MIL-10150 and Zr(VI) for UiO-6651. However, due to the easy-to-hydrolysis nature of both Cr(III) and Zr(IV), it is still difficult to synthesize Cr(III) and Zr(IV) MOFs with high crystallization. Alternatively, another high-valence metal ion of Th(IV) shows less hydrolytic nature, suggesting an optimal metal ion to generate stable MOFs52,53,54,55,56. Generally speaking, according to the hard and soft acid and bases (HSAB) principle, the Zr-based or Th-based MOFs are usually constructed by O-donor carboxylic ligands. For example, the typical UiO-66-type structure is connected by various linear O-donor carboxylic ligands, as shown in the left of Supplementary Fig. 151,56,57. As we know, the N,O-donor ligands such as azole series have been attested to be an excellent organic linkers to construct a great number of MOFs (more than 900 tetrazole-based MOFs and more than 5000 triazole-based MOF from CCDC data)58,59,60. However, there is no Zr-based or Th-based MOFs built on N,O-donor ligands. As shown in the right of Supplementary Fig. 1, the construction of azole-based Zr- or Th-based MOFs is possible, because of the comparable coordination direction for carboxylate and azole molecules, while the introduction of azole unit to bind with Zr or Th ions is also possibly a powerful tool to modulate the electronic structure of metal center and the environment of pore wall, consequently leading to unique physical properties.
In this work, we obtain a successful case via solvothermal reaction of Th(NO3)4 and 4-(1H-tetrazol-5-yl) benzoic acid (TBA). This compound shows a UiO-66-type structure, except for the replacement of secondary building unit such as Zr6 by Th6 and linkers such as O-donor ligand by N,O-donor ligand. This unique tetrazole-based structure allows it to perform high C2H6 uptake at room temperature and selective adsorption of C2H6 over C2H4, finally resulting in the promising application for C2H4 separation from the binary C2H6/C2H4 and ternary C2H4/C2H2/C2H6 mixture. Furthermore, both the grand canonical Monte Carlo (GCMC) simulations and density functional theory (DFT) calculations are carried out to disclose the separation mechanism.
Results
Synthesis, structure, and characterization of Azole-Th-1
The reaction between ligand TBA and Th(NO3)4 in N,N′-dimethylformamide (DMF) yields colorless octahedral crystals of Azole-Th-1 (Supplementary Fig. 2). The synthesis in detail is listed in Supplementary Information. The purity of the bulk samples was confirmed by powder X-ray diffraction (PXRD, Supplementary Fig. 3).
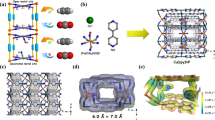
The PXRD discloses that the octahedral crystals of Azole-Th-1 crystallize in the cubic space group Fm3m, similar to UiO-66 (Fig. 1c). The length of a, b, c is 23.984(4) Å, longer than UiO-66-Zr (20.743 (5) Å)51 and UiO-66-Th (21.961(13) Å)56, mainly due to the longer linker of TBA (ca. 8.4 Å) in Azole-Th-1 than terephthalic acid (ca. 6.8 Å) in UiO-66. Six Th(IV) metal ions are combined together to give the Th6O4(OH)4(H2O)6 core (Fig. 1b), similar to the Zr6O4(OH)4(H2O)6 in UiO-66. The Th(IV) ion holds the nine-coordination surrounding with a monocapped square antiprismatic geometry. The Th-O bond length from O2−, OH−, and H2O is varied from 2.33 to 2.55 Å, slight shorter than the Th–N bond length of 2.74 Å. As observed in the literature61,62 for the TBA ligand usually showing highly disordered structure, similar trend is observed in Azole-Th-1 (Supplementary Fig. 4). The TBA ligand contacts with Th(IV) ions via both carboxylate and azole bridge, while two additional nitrogen atoms for each TBA ligand are free-standing without coordination. The 3D framework is formed by both the inorganic Th6O4(OH)4(H2O)6 core and TBA linkers, where each inorganic core connects to twelve identical Th6O4(OH)4(H2O)6 cores via twelve TBA linkers, finally constructing the UiO-66-like structure. Similarly, two different types of cages, viz. a super tetrahedron cage (Fig. 1d) and a super octahedron (Fig. 1e) with the largest cavity diameter of 1.1 nm and 1.2 nm, respectively, is observed in Azole-Th-1. This is larger the corresponding values of 0.88 nm in UiO-6651, due to the longer linkers of TBA in Azole-Th-1 than terephthalic acid in UiO-66. The solvent-accessible volume estimated by Platon program63 is 50.1% of the unit cell, suggesting high porosity of this MOF.

a Ligand TBA, including carboxylic acid donor (O-donor) and tetrazole donor (N-donor), b inner core Th6-cluster drawn alone for clarity (Th6O8), c unit cell structure in crystal Azole-Th-1, d, e two types of cages, including super tetrahedron cage d and super octahedron cage e. Where, Th-light blue ball b and polyhedron style c–e with purple color, O-red ball, C-gray ball, N-blue ball, and H-white ball.
The loss of trapped solvent molecules from Azole-Th-1, according to the thermogravimetric (TG) analysis, is before 75 °C (Supplementary Fig. 5). While the temperature increased to about 250 °C, the crystal structure begins to collapse, which is in agreement with the results of temperature dependent PXRD tests (Fig. 2a). As shown in Fig. 2a, the PXRD of Azole-Th-1 samples from room temperature to 200 °C are matching with the experimental and simulated results. While the temperature reaches to 250 °C, the peaks of PXRD disappear, which indicates that the crystal structure is destroyed by such high temperature. On the other hand, the chemical stability under water and different solvents environment, including seven organic solvents and a broad pH range from 1 to 12, were also traced by PXRD tests (Fig. 2b, c), where respective optical images of crystal were represented in Fig. 3. Note that the crystals of Azole-Th-1 render excellent stability in water and above solvents even after 30 days.

a Thermal stability from 100 to 400 °C, including simulated and experimental results, b soaking in water and seven different organic solvents 30 days, and c soaking in different pH solvents 30 days. Source data are provided as a Source Data file.

a after soaking in water and seven different organic solvents 30 days, b after soaking in different pH solvents 30 days.
Adsorption isotherm, selectivity, and breakthrough
The moderate thermal stability and high chemical stability of Azole-Th-1 prompts us to study the gas adsorption performances. To characterize the permanent porosity of the obtained material, the N2 adsorption isotherm at 77 K was measured. As shown in Fig. 4a, a fully reversible type I isotherm with a Brunauer Emmett Teller (BET) surface area of 983 m2 g−1 and a uniform pore size around 9.2 Å was exhibited. This pore size is comparable with that calculated LCD (the largest cavity diameter, 10.0 Å) by Zeo++ program64 (Supplementary Table 3).

a The N2 adsorption at 77 K with the insert of the distribution of pore size. b The adsorption isotherms of C2H6 and C2H4 at 298 K, including experiments and simulations. c Experimental adsorption isotherms of Azole-Th-1 for C2H6, C2H4, and C2H2 at 298 K from 0.01 to 1 bar with the insert of enlargement from 0.01 to 0.1 bar. d The adsorption heat enthalpy of C2H6 and C2H4, calculated from the single-component C2H6 and C2H4 adsorption data at 298 and 273 K. e Predicted mixture adsorption isotherms and selectivity of Azole-Th-1 by IAST method for a 50/50 C2H6/C2H4 mixture at 298 K. f A comparison in selectivity and C2H6 adsorption capacity at 298 K and 1 bar between the reported top-performing porous adsorbents for C2H6/C2H4 separation and our MOF. The purple triangle is our MOF. Source data are provided as a Source Data file.
This high porosity and desirable aperture encouraged us to further investigate C2H6/C2H4 separations in detail. Adsorption isotherms of single component C2H6 and C2H4 were collected at 298 K and 273 K, respectively, as presented in Fig. 4b and Supplementary Fig. 6. The adsorption isotherm of C2H6 is typically type I with a steep slope, which is a typical feature of strong adsorbates in microporous materials (Fig. 4b)8,22,25,27,28,29,30,32,33,34,35. And the adsorption amounts of C2H6 at both 273 and 298 K (121.7 and 100.2 cm3 g−1) were also higher than the corresponding C2H4 (111.3 and 80.7 cm3 g−1). Therefore, the Azole-Th-1 has a distinct preference for adsorbing ethane over ethylene. It is well-known that the magnitude of the adsorption enthalpies of porous materials reveals that the affinity of the pore surface toward adsorbents, determining the adsorptive selectivity39,65. This can be directly reflected on the adsorption heat enthalpy (Qst) of C2H6 and C2H4, giving 28.6 kJ mol−1 for C2H6 at the zero coverage, significantly higher than the values of 26.1 kJ mol−1 for C2H4 (Fig. 4d), strongly suggesting a higher affinity between host and guest for C2H6 than C2H4, where the detail virial-type analysis were provided in Supplementary Fig. 7.
At room temperature and 100 kPa, Azole-Th-1 affords ultrahigh adsorption capacity of C2H6 up to 100.2 cm3 g−1. This value exceeds most reported top-performing porous adsorbents for such use as shown in Table S2, including in Fe2(O2)(dobdc)35, MAF-4922, ZIF-834, ZIF-732, PCN-24526, MIL-142A28, Cu(Qc)237, Zn-atz-ipa38, etc., which are summarized in Fig. 4f. Correspondingly, adsorption capacity of C2H4 at the same conditions is 81.1 cm3 g−1, obviously, less than C2H6 about 20 cm3 g−1. Hence, selective adsorption of C2H6 over C2H4 is suggested. To estimate the adsorption selectivity, we employed the ideal adsorption solution theory (IAST)66 to analyze the experimental isotherm data, using composition of 50:50/10:90/1:15 C2H6/C2H4, as shown in Fig. 4e and Supplementary Fig. 8. The selectivity of C2H6 over C2H4 was up to 1.46 at room temperature and 100 kPa, which is slightly higher than that of other ratio mixture, both 1.44 for 10:90 and 1:15 C2H6/C2H4. To the best of our knowledge, Azole-Th-1 should be the first Th-MOF showing such abnormal adsorption behavior (C2H6 over C2H4).
Then, in order to check the actual separation ability of the gases mixture, the transient breakthrough simulations for C2H6/C2H4 (50/50, v/v) mixtures on Azole-Th-1 material was carried out at 298 K, as shown in Supplementary Fig. 9. This result demonstrates that the potential of producing pure product gas C2H4 during the time interval Δτ, also suggests excellent C2H6/C2H4 separation performance. And then, to further confirm the real practical separation ability, the actual dynamic adsorption breakthrough experiments for C2H6/C2H4 (10/90, 50/50, 1/15, v/v) binary mixtures on Azole-Th-1 material were also carried out (Fig. 5a, b, and Supplementary Fig. 10). The C2H4 broke through the adsorption bed and yield a high purity gas (>99.9%) at first, whereas after a certain time C2H6 slowly eluted and reached to the equilibrium (Fig. 5a, b, and Supplementary Fig. 10). During this period of time, polymer-grade (>99.9%) C2H4 can be generated at the outlet. The breakthrough time of ethane was later than that of ethylene for these three ratio mixtures, meaning that the Azole-Th-1 preferred to adsorb ethane over ethylene. The long breakthrough time interval between C2H4 and C2H6 suggests that the Azole-Th-1 is quite effective for C2H6/C2H4 separation. The experimental breakthrough results was well in consistent with the simulated breakthrough (Fig. 5b, Supplementary Fig. 10b, d), strongly suggesting its superior application for C2H4 purification. Furthermore, cycling breakthrough experiments on Azole-Th-1 were carried out under the same conditions. The breakthrough time interval for C2H6/C2H4 mixtures in five cycles (Fig. 5c) is comparable, showing that this material has a good regenerability. According to the polymer grade C2H4 produced during time interval at different C2H4/C2H6 ratio, 3 min (50/50), 5 min (90/10), and 3.5 min (15/1), the productivities of C2H4 (>99.9%) were 0.68, 1.13, and 0.79 mmol g−1, respectively. Hence, the polymer grade C2H4 with the max working capacity of 1.13 mmol g−1 with >99.9% purity was harvested from 90/10 gas mixture, which working capacity is nearly 1.3 times for Fe2(O2)(dobdc) (0.79 mmol g−1)35 and 3.6 times for MAF-49 (0.28 mmol g−1)22, the two best materials for C2H6/C2H4 separation. Some more detailed comparison with other MOF materials is shown in Supplementary Table 2. After the breakthrough experiments, the PXRD pattern of our sample was also consistent with the PXRD before the breakthrough (Supplementary Fig. 11), which further indicated that this material has a good regenerability and high stability.

a, b C2H6/C2H4 (10/90, v/v) binary mixture. c C2H6/C2H4 (50/50, v/v) binary mixture for five cycles. d C2H6/C2H4/C2H2(90/9/1, v/v/v) ternary mixture separation. Source data are provided as a Source Data file.
Mechanism of gas adsorption by theoretical calculations
Theoretically, the determination of gas adsorption sites in the MOFs is of great significance for the design of some gas storage and separation materials based on MOFs31,67. Herein, the ultrahigh C2H6 storage capacity prompts us to explore the adsorption sites within this Azole-Th-1. Theoretical simulation is a powerful tool enabling us to unveil the adsorption mechanisms and provide the adsorption sites. Therefore, the C2H6/C2H4 binding affinity in Azole-Th-1 was firstly investigated by single-component sorption isotherms at 298 K and pressures up to 100 kPa. The GCMC simulations were performed for understanding the interactions and adsorption behaviors of C2H6 and C2H4 in Azole-Th-1 at the molecular level25,26,33,34,39. As shown in isotherm of C2H6/C2H4 adsorption (Fig. 4b), the maximum C2H6 and C2H4 uptake for Azole-Th-1 are 106.6 and 80.2 cm3 g−1 at 100 kPa and 298 K, respectively, both adsorbed tendency and the adsorption quantity are consistent with the experimental results (100.2 and 80.7 cm3 g−1), where the simulated details are listed in Supplementary Information.
The further investigations on the interaction between C2H6/C2H4 and MOF material can help us to understand the mechanism of gas adsorption, which could analysis the discrepancies the interactions between C2H6 and C2H4 with our material, respectively. According to the density distribution of C2H6 on Azole-Th-1 at 298 K and 100 kPa (Fig. 6), there are three main adsorbed areas in our material, including benzene region (I-region), the tetrazol heterocycle region (II-region), and carboxylate region (III-region), respectively. Then, the adsorptions under different pressure were analyzed (Supplementary Fig. 12). Due to the higher polarizability of C2H6 (44.7 × 10−25 cm3) compared with C2H4 (42.5 × 10−25 cm3)7, at the beginning of the adsorption, the C2H6 molecules are preferentially filled in region I at 2.6 kPa (Supplementary Fig. 12a), but the C2H4 molecules are almost not adsorbed until 10.3 kPa (Supplementary Fig. 12e). While the pressure is 5.1 kPa, the region II begins to be filled by C2H6 molecules (Supplementary Fig. 12b), the total capacity of adsorption reaches to 18.5 cm3 g−1, which corresponds to the adsorption capacity of C2H4 at 12.8 kPa (18.9 cm3 g−1), at that moment the region II is empty until the pressure increases to 17.9 kPa (Supplementary Fig. 12f). With the increase of pressure (to 48.7 kPa), region I and region II are almost filled saturated by C2H6 molecules (Supplementary Fig. 12c), and region III begins to be filled by C2H6 already, the total capacity of adsorption reaches to 73.2 cm3 g−1. The vdW interactions between C2H6 with aromatic rings (regions I and II) and carboxylate (region III) are more remarkable than the interactions between C2H4 with them. While the pressure reaches to 100 kPa, these three regions are saturated generally by C2H6 (Supplementary Fig. 12d), whereas, the C2H4 molecules only continue to be filled into regions I and II (Supplementary Fig. 12g), the C2H6 uptake (106.2 cm3 g−1) is significantly greater than the maximum C2H4 uptake (80.2 cm3 g−1). Hence, according to the GCMC simulations, the Azole-Th-1 prefers to adsorbing the C2H6 from 0.001 to 100 kPa at 298 K, and the C2H6 adsorption capacity is far greater than C2H4, which could achieve the separation of C2H6 and C2H4.

a The density distribution of C2H6 on Azole-Th-1 at 100 kPa and 298 K. b, c The structures of adsorptions for C2H6 and C2H4 at M2 model. Where, Th-light blue, O-red, C-gray, N-blue, H-white, C2H6-yellow molecule, and C2H4-green molecule, and the unit of distance is Å.
And then, according to the obtained adsorption regions, some DFT theoretical calculations about the mechanism of selective C2H6/C2H4 in Azole-Th-1 were investigated by the Dmol3 program package68 in the MS, the detail calculations were also presented in Supplementary Information. The determination of the adsorption point can quantify the interaction between the gas C2H6/C2H4 and Azole-Th-1 and analyze the mechanism of the gas adsorption. Because the calculations using the whole unit cell is too large, we used the fragmented cluster models cleaved from the unit cell for modeling the structures and energies to investigate the interaction points of C2H6/C2H4 adsorption. Due to the highly disordered structure of TBA, four fragment models were constructed. Accordingly, the fragment M1 to M4 were intercepted (Supplementary Fig. 13). Based on the distribution of density of C2H6/C2H4 through GCMC simulations, the geometries of fragmented models bound to C2H6/C2H4 were also obtained, as shown in Supplementary Fig. 14. By the calculations of binding energy for adsorbed geometries (Supplementary Table 5), obviously, the interactions between M1 to M4 with C2H6 (−43.09 kJ mol−1) were stronger than the interactions between that of fragmented models with C2H4 (−33.52 kJ mol−1), which results were in agreement with the heat of adsorption Qst. These can be attributed to the vdW interaction between the C–H in C2H6/C2H4 and conjugated π-systems in Azole-Th-1, especially, the conjugated region II (tetrazol).
In our opinion, there are three factors for the stabilities of adsorbed structures, the number of H atoms, the distances of vdW interaction, and the polarizability, where the summary about previous two factors were listed in Supplementary Tab. 6. As shown in Supplementary Fig. 14, C2H6 and C2H4 molecules are bound to several aromatic rings of ligands at three directions within a pore through vdW interactions. A single C2H6 or C2H4 molecule can form six or four pairs of C–H—π interactions with conjugated regions at least, where the vdW interaction between C–H and benzene ring of ligand (region I) are in the majority. The greater the number of H atoms, the stronger the C–H—π interaction between gas with different models, and finally the greater the adsorption capacity of C2H6 compared with C2H4.
Because of the use of previously unreported ligand, the conjugated region tetrazole can enhance the binding with gas. As shown in Supplementary Fig. 14, C2H6@M1 (all O-donors of ligands coordinated on the Th(IV) inner cluster), there are two pairs of vdW interactions between C–H with carboxylic acid (region III). However, as to C2H6@M2 (Fig. 6, all N-donor of ligands coordinated on the Th(IV) inner cluster), because the gas molecule was near to the region II, which contributed to the increasing of the interaction probability between gas and tetrazol. The four pairs of C–H—π interactions with region II could make the adsorption structure more stable with −46.90 kJ mol−1 binding energy than the structure without the interaction between C2H6 with tetrazol (C2H6@M1, −25.87 kJ mol−1).
And then, the average distances between C2H6 with benzene region (I), tetrazol region (II), and carboxylic acid region (III) are 3.67, 3.65, and 3.08 Å, respectively. And the average distances between C2H4 with these three regions are 3.41 and 3.25 Å (no interaction with region III), which results agreed with the GCMC results (Supplementary Fig. 12g, C2H4 only filled into regions I and II). According to these distances of interaction, it is hard to analysis the stabilization of the adsorption structures. Besides the distance and number of C–H—π interactions, C2H6 as a more polarizable molecule can interact more strongly by induced dipole interactions with the framework compared to the less polarizable C2H4 molecule.
Separation of ternary mixture of C2H6/C2H2/C2H4
In traditional C2H4 production process, trace amounts of C2H2 (about 1%) also exist in the ethylene feed. So, this material was further investigated for the simultaneous capture of C2H2 and C2H6 from a ternary mixture of C2H6/C2H2/C2H4. As shown in Fig. 5d, highly efficient separation of C2H4 from a 90:1:9 (v/v/v) gas mixture of C2H6/C2H2/C2H4 was achieved by passing the mixture over a packed column of activated Azole-Th-1 material. It can be observed that C2H4 achieves a breakthrough first, with no evidence of C2H2 or C2H6 flow before its breakthrough, which indicates that this material can produce an high purity C2H4 (>99.9%) after only a single-breakthrough operation. The working capacity is up to 1.34 mmol g−1, far exceeding the top-performing materials reported by Lu et al. (0.69 mmol g−1)49 and Zaworoko et al. (0.32 mmol g−1)38, strongly suggesting its promising applications in this target. Note that, according to the adsorption isotherm of C2H2 (Fig. 4c), the maximum of capacities of C2H2 (81.1 cm3 g−1) and C2H4 (80.7 cm3 g−1) are almost equal to each other. In addition, the adsorption heat enthalpy of C2H2 is 25.4 kJ mol−1 at the zero coverage, which is lower slightly than Qst of C2H4 (26.1 kJ mol−1). And why the high purity of C2H4 (>99.9%) can be acquired. Therefore, in order to explore this problem, the C2H6, C2H4, and C2H2 at low pressure region (<0.1 bar) was checked (inserted into Fig. 4c). Before pressure of 0.05 bar, the adsorption of C2H2 is obviously bigger than both C2H6 and C2H4, giving a hierarchy of C2H2>C2H6>C2H4. This is well consistent with the separation conditions of 90:1:9 (v/v/v) gas mixture of C2H6/C2H2/C2H4.
Discussion
In a word, we reported a Th-azole framework (Azole-Th-1) by introducing an previously unreported ligand TBA showing a preferential adsorption of ethane over ethylene. Notably, Azole-Th-1 samples exhibited good stability for soaking in water, various organic solvents, and different pH (1–12) solvents about 30 days, respectively. Moreover, preferential adsorption of ethane over ethylene was confirmed by measuring the adsorption isotherms and breakthrough curves. Azole-Th-1 had relative high ethane and ethylene adsorption capacities, 4.5 and 3.6 mmol g−1 at 298 K and 100 kPa, respectively. The adsorption selectivity of binary mixture C2H6/C2H4(1:1, v/v) was ~1.46 at pressure below 100 kPa and 298 K. Five cycles of ethane adsorption–desorption cycle experiments revealed that Azole-Th-1 had a good regenerability. The polymer grade C2H4 with the max working capacities of 1.13 mmol g−1 with 99.9% purity was harvested from 10/90 gas mixture. Furthermore, Azole-Th-1 also can purify the C2H4 (purity >99.9%) from ternary mixture C2H6/C2H2/C2H4 (90:1:9, v/v/v) with working capacities of 1.34 mmol g−1. Some DFT calculations suggested that the greater vdW interaction between ethane and Azole-Th-1 than ethylene and material, −43.09 and −33.52 kJ mol−1, respectively, which were in agreement with the isosteric heat of ethane (28.6 kJ mol−1) and ethylene (26.1 kJ mol−1). In brief, these excellent properties, the perfect pH stability and high C2H4 purity (>99.9%) from a ternary 90:1:9 mixture of C2H6/C2H2/C2H4, etc., make Azole-Th-1 a promising candidate for efficient separation of ethane/ethylene. It will be much more challenging and difficult to separate more complex gas mixtures. Those small gas molecules such as N2 and CH4 will not affect the ternary C2H6/C2H2/C2H4 separation very much because of their very weak interactions with the framework, leading to very low uptakes of N2 and CH4 at the room temperature. However, some other gas molecules, particularly CO2, is expected to significantly affect the C2H6/C2H2/C2H4 separation, because the uptakes of CO2 are comparable to these C2 hydrocarbons. Until now, no suitable porous materials have been reported yet for the efficient C2H6/C2H2/C2H4/CO2 separation. Before any porous materials can be realized for this very challenging separation, step-by-step separations by different adsorbents will be necessary to get high purity C2 hydrocarbons.
Methods
The synthesis
A mixture of 4-(1H-Tetrazol-5-yl) benzoic acid (TBA, 0.019 g, 0.10 mmol), Th(NO3)4 (0.048 g, 0.10 mmol), N,N′-dimethylformamide (DMF, 3.0 mL), and ionic liquid of tetramethylguanidine chloride (0.015 mg, 0.1 mmol) were placed in a 20 mL screw-capped glass capped jar, then five drops of concentrated hydrochloric acid were added to the mixture. The mixture was sealed and heated at 110 °C for 3 days. The reaction system was cooled to 30 °C with about 6 °C per min cooling rate. After filtration and washed with excess of N,N′-dimethylacetamide (DMA), colorless block crystals were collected as a pure phase (see PXRD in Fig. 2).
Gas-adsorption and breakthrough experiments
The original sample about 100 mg was activated at 60 °C under high vacuum for 12 h in gas adsorption apparatus before the gas adsorption measurement. The BET of the MOFs were investigated by nitrogen adsorption and desorption at 77 K using a Belsorp-max. The single-component isotherms of C2H6, C2H4, and C2H2 were collected at 298 and 273 K on a Belsorp-max. The breakthrough separation apparatus consisted of two fixed-bed stainless steel column. One column was loaded with MOF powder (1.9810 g), while the other reactor was used as a blank control group to stabilize the gas flow. The horizontal reactors were placed in a temperature-controlled environment, maintained at 298 K. The flow rates of all gases mixtures were regulated by mass flow controllers, and the effluent gas stream from the column is monitored by a gas chromatography (TCD-Thermal Conductivity Detector, detection limit 0.1%). Prior to each breakthrough experiment, we regenerated the sample by flushing the adsorption bed with helium gas (100 mL per min) for 30 min at 298 K.
Data availability
The X-ray crystallographic coordinated for structure reported in this study has been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number 1969398. This data can be obtained free of charge from the CCDC via https://www.ccdc.cam.ac.uk/structures/. The source data underlying Figs. 2, 4, and 5 and Supplementary Figs. 5, 6, 8, 9, 10a, 10c, and 11 are provided as a Source Data file. And other data, if not included in the article or Supplementary Information or Source Data, are available from the authors on request.
References
Amghizar, I., Vandewalle, L. A., Van Geem, K. M. & Marin, G. B. New trends in olefin production. Engineering 3, 171–178 (2017).
He, Y. B., Rajamani, K. & Chen, B. L. Metal–organic frameworks with potential for energy-efficient adsorptive separation of light hydrocarbons. Energy Environ. Sci. 5, 9107–9120 (2012).
Li, B. et al. Introduction of pi-complexation into porous aromatic framework for highly selective adsorption of ethylene over ethane. J. Am. Chem. Soc. 136, 8654–8660 (2018).
Liao, P. Q., Zhu, A. X., Zhang, W. X., Zhang, J. P. & Chen, X. M. Self-catalysed aerobic oxidization of organic linker in porous crystal for on-demand regulation of sorption behaviours. Nat. Commun. 6, 6350 (2015).
Li, J. R., Sculley, J. & Zhou, H. C. Metal-organic frameworks for separations. Chem. Rev. 112, 869–932 (2012).
Lee, C. Y. et al. Kinetic separation of propene and propane in metal-organic frameworks: controlling diffusion rates in plate-shaped crystals via tuning of pore apertures and crystallite aspect ratios. J. Am. Chem. Soc. 133, 5228–5231 (2011).
Li, J. R., Kuppler, R. J. & Zhou, H. C. Selective gas adsorption and separation in metal-organic frameworks. Chem. Soc. Rev. 38, 1477–1504 (2009).
Qazvini, O. T., Babarao, R., Shi, Z. L., Zhang, Y. B. & Telfer, S. G. A robust ethane-trapping metal-organic framework with a high capacity for ethylene purification. J. Am. Chem. Soc. 141, 5014–5020 (2019).
Bux, H., Chmelik, C., Krishna, R. & Caro, J. Ethene/ethane separation by the MOF membrane ZIF-8: molecular correlation of permeation, adsorption, diffusion. J. Membr. Sci. 369, 284–289 (2011).
Bos, A. N. R. & Westerterp, K. R. Mechanism and kinetics of the selective hydrogenation of ethyne and ethene. Chem. Eng. Process.: Process Intensification 32, 1–7 (1993).
Weissermel, K. & Arpe, H. J. Industrial Organic Chemistry, 4th edn. (Wiley, New York, 2003).
Jin, J., Zhao, X., Feng, P. Y. & Bu, X. H. A cooperative pillar-template strategy as a generalized synthetic method for flexible homochiral porous frameworks. Angew. Chem. Int. Ed. 57, 3737–3741 (2018).
Li, H. et al. Microporous metal-organic framework with dual functionalities for efficient separation of acetylene from light hydrocarbon mixtures. ACS Sustain. Chem. Eng. 7, 4897–4902 (2019).
Bao, Z. B. et al. Potential of microporous metal-organic frameworks for separation of hydrocarbon mixtures. Energy Environ. Sci. 9, 3612–3641 (2016).
Yang, L. F. et al. A single-molecule propyne trap: highly efficient removal of propyne from propylene with anion-pillared ultramicroporous materials. Adv. Mater. 30, 8 (2018).
Huang, Y. L. et al. Tuning the C2/C1 hydrocarbon separation performance in a bioMOF by surface functionalization. Eur. J. Inorg. Chem. 2019, 4205–4210 (2019).
Nijem, N. et al. Tuning the gate opening pressure of metal-organic frameworks (MOFs) for the selective separation of hydrocarbons. J. Am. Chem. Soc. 134, 15201–15204 (2012).
Jin, G. X., Wang, J., Liu, J. Y., Ma, J. P. & Dong, Y. B. Visual recognition and removal of C2H2 from C2H4/C2H2 mixtures by a Cu-I-MOF. Inorg. Chem. 57, 6218–6221 (2018).
Luo, M. B. et al. The MOF+ technique: a significant synergic effect enables high performance chromate removal. Angew. Chem. Int. Ed. 56, 16376–16379 (2017).
Fan, C. B. et al. Significant enhancement of C2H2/C2H4 separation by a photochromic diarylethene unit: a temperature- and light-responsive separation switch. Angew. Chem. Int. Ed. 56, 7900–7906 (2017).
Luo, F. et al. UTSA-74: a MOF-74 isomer with two accessible binding sites per metal center for highly selective gas separation. J. Am. Chem. Soc. 138, 5678–5684 (2016).
Liao, P. Q., Zhang, W. X., Zhang, J. P. & Chen, X. M. Efficient purification of ethene by an ethane-trapping metal-organic framework. Nat. Commun. 6, 8697 (2015).
Narin, G. et al. Light olefins/paraffins separation with 13X zeolite binderless beads. Sep. Purif. Technol. 133, 452–475 (2014).
Eldridge, R. B. Olefin/paraffin separation technology: a review. Ind. Eng. Chem. Res. 32, 2208–2212 (1993).
Chen, Y. et al. An ethane-trapping MOF PCN-250 for highly selective adsorption of ethane over ethylene. Chem. Eng. Sci. 175, 110–117 (2018).
Lv, D. et al. Selective adsorption of ethane over ethylene in PCN-245: Impacts of interpenetrated adsorbent. ACS Appl. Mater. Interfaces 10, 8366–8373 (2018).
Pires, J., Pinto M. L. & Saini, V. K. Ethane selective IRMOF-8 and its significance in ethane–ethylene separation by adsorption. ACS Appl. Mater. Interfaces 6, 12093–12099 (2014).
Chen, Y. et al. Highly adsorptive separation of ethane/ethylene by an ethane-selective MOF MIL-142A. Ind. Eng. Chem. Res. 57, 4063–4069 (2018).
Liang, W. et al. Ethane selective adsorbent Ni(bdc)(ted)0.5 with high uptake and its significance in adsorption separation of ethane and ethylene. Chem. Eng. Sci. 148, 275–281 (2016).
Böhme, U. et al. Ethene/ethane and propene/propane separation via the olefin and paraffin selective metal–organic framework adsorbents CPO-27 and ZIF-8. Langmuir 29, 8592–8600 (2013).
Li, J. R. et al. Porous materials with pre-designed single-molecule traps for CO2 seletive adsorption. Nat. Commun. 4, 1538 (2013).
Chen, D. et al. A combined theoretical and experimental analysis on transient breakthroughs of C2H6/C2H4 in fixed beds packed with ZIF-7. Microporous Mesoporous Mater. 208, 55–65 (2015).
Pillai, R. S., Pinto, M. L., Pires, J., Jorge, M. & Gomes, J. R. B. Understanding gas adsorption selectivity in IRMOF-8 using molecular simulation. ACS Appl. Mater. Interfaces 7, 624–637 (2015).
Wu, Y., Chen, H., Liu, D., Qian, Y. & Xia, H. Adsorption and separation of ethane/ethylene on ZIFs with various topologies: Combining GCMC simulation with the ideal adsorbed solution theory (IAST). Chem. Eng. Sci. 124, 144–153 (2015).
Li, L. et al. Ethane/ethylene separation in a metal-organic framework with iron peroxo sites. Science 362, 443–446 (2018).
Lin, R. B. et al. Molecular sieving of ethylene from ethane using a rigid metal-organic framework. Nat. Mater. 17, 1128–1133 (2018).
Lin, R. B. et al. Boosting ethane/ethylene separation within isoreticular ultramicroporous metal-organic frameworks. J. Am. Chem. Soc. 140, 12940–12946 (2018).
Chen, K. J. et al. Synergistic sorbent separation for one-step ethylene purification from a four-component mixture. Science 366, 241–246 (2019).
Fan, W. et al. Fine-tuning the pore environment of the microporous Cu-MOF for high propylene storage and efficient separation of light hydrocarbons. ACS Cent. Sci. 5, 1261–1268 (2019).
Zhang, Y. et al. Highly selective adsorption of ethylene over ethane in a MOF featuring the combination of open metal site and pi-complexation. Chem. Commun. 51, 2714–2717 (2015).
Luna-Triguero, A., Vicent-Luna, J. M., Gómez-Álvarez, P. & Calero, S. Olefin/Paraffin separation in open metal site Cu-BTC metal–organic framework. J. Phys. Chem. C 121, 3126–3132 (2017).
Bloch, E. D. et al. Hydrocarbon separations in a metal-organic framework with open iron(II) coordination sites. Science 335, 1606–1610 (2012).
Martins, V. F. D. et al. Ethane/ethylene separation on a copper benzene-1,3,5-tricarboxylate MOF. Sep. Purif. Technol. 149, 445–456 (2015).
Wang, Q. M. et al. Metallo-organic molecular sieve for gas separation and purification. Microporous Mesoporous Mater. 55, 217–230 (2002).
Wang, S. Y., Yang, Q. Y. & Zhong, C. L. Adsorption and separation of binary mixtures in a metal-organic framework Cu-BTC: A computational study. Sep. Purif. Technol. 60, 30–35 (2008).
Nicholson, T. M. & Bhatia, S. K. Electrostatically mediated specific adsorption of small molecules in metallo-organic frameworks. J. Phys. Chem. B 110, 24834–24836 (2006).
Cai, J. et al. A doubly interpenetrated metal–organic framework with open metal sites and suitable pore sizes for highly selective separation of small hydrocarbons at room temperature. Cryst. Growth Des. 13, 2094–2097 (2013).
Gücüyener, C., van den Bergh, J., Gascon, J. & Kapteijn, F. Ethane/ethene separation turned on its head: selective ethane adsorption on the metal-organic framework ZIF-7 through a gate-opening mechanism. J. Am. Chem. Soc. 132, 17704–17706 (2010).
Hao, H. G. et al. Simultaneous trapping of C2H2 and C2H6 from a ternary mixture of C2H2/C2H4/C2H6 in a robust metal–organic framework for the purification of C2H4. Angew. Chem. Int. Ed. 57, 16067–16071 (2018).
Férey, G. et al. A chromium terephthalate-based solid with unusually large pore volumes and surface area. Science 309, 2040–2042 (2005).
Jasmina Hafizovic, Cavka et al. A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability. J. Am. Chem. Soc. 130, 13850–13851 (2008).
Dolgopolova, E. A., Rice, A. M. & Shustova, N. B. Actinide-based MOFs: a middle ground in solution and solid-state structural motifs. Chem. Commun. 54, 6472–6483 (2018).
Dolgopolova, E. A. et al. Multifaceted modularity: A key for stepwise building of hierarchical complexity in actinide metal–organic frameworks. J. Am. Chem. Soc. 139, 16852–16861 (2017).
Ok, K. M., Sung, J. Y., Hu, G., Jacobs, R. M. J. & O’Hare, D. TOF-2: A large 1D channel thorium organic framework. J. Am. Chem. Soc. 130, 3762–3763 (2008).
Wang, Y. et al. Employing an unsaturated Th(4+) site in a porous thorium-organic framework for Kr/Xe uptake and separation. Angew. Chem. Int. Ed. 57, 5783–5787 (2018).
Falaise, C., Charles, J. S., Volkringer, C. & Loiseau, T. Thorium terephthalates coordination polymers synthesized in solvothermal DMF/H2O system. Inorg. Chem. 54, 2235–2242 (2015).
Puchberger, M. et al. Can the clusters Zr6O4(OH)4(OOCR)12 and [Zr6O4(OH)4(OOCR)12]2 be converted into each other? Eur. J. Inorg. Chem. 2006, 3283–3293 (2006).
Burgun, A. et al. Mapping-out catalytic processes in a metal–organic framework with single-crystal X-ray crystallography. Angew. Chem. Int Ed. 56, 8412–8416 (2017).
Sun, W. Q. et al. Programmable self-assembly of heterometallic Palladium(II)–Copper(II) 1D grid-chain using dinuclear Palladium(II) corners with pyrazole–carboxylic acid ligands. Chem. Asian J. 13, 1108–1113 (2018).
Chen, D. M. et al. Microporous cobalt(II)–organic framework with open O-donor sites for effective C2H2 storage and C2H2/CO2 separation at room temperature. Inorg. Chem. 56, 14767–14770 (2017).
Wang, D., Zhao, T., Li, G., Huo, Q. & Liu, Y. A porous sodalite-type MOF based on tetrazolcarboxylate ligands and [Cu4Cl]7+ squares with open metal sites for gas sorption. Dalton Trans. 43, 2365–2368 (2014).
Pachfule, P., Chen, Y., Sahoo, S. C., Jiang, J. & Banerjee, R. Structural isomerism and effect of fluorination on gas adsorption in copper-tetrazolate based metal organic frameworks. Chem. Mater. 23, 2908–2916 (2011).
Spek, A. L. PLATON. Utrecht University (2001).
Willems, T. F., Rycroft, C. H., Kazi, M., Meza, J. C. & Haranczyk, M. Algorithms and tools for high-throughput geometry—based analysis of crystalline porous materials. Microporous Mesoporous Mater. 149, 134–141 (2012).
Yang, Y. Y., Lin, Z. J., Liu, T. T., Liang, J. & Cao, R. Synthesis structures and physical properties of mixed-ligand coordination polymers based on a V-shaped dicarboxylic ligand. Cryst. Eng. Commun. 17, 1381–1388 (2015).
Myers, A. & Prausnitz, J. M. Thermodynamics of mixed gas adsortion. AIChE J. 11, 121–127 (1965).
Kubota, Y. et al. Metastable sorption state of a metal-organic porous material determined by in situitu synchrotron powder diffraction. Angew. Chem. Int. Ed. 45, 4932–4936 (2006).
Delley, B. From molecules to solids with the Dmol3 approach. J. Chem. Phys. 113, 7756 (2000).
Acknowledgements
We thanks to the National Natural Science Foundations of China (21966002, 21871047, 21661001, and 21922810), the Natural Science Foundation of Jiangxi Province of China (20181ACB20003), and the Training Program for Academic and Technical Leaders of Major Disciplines in Jiangxi Province (20194BCJ22010).
Author information
Authors and Affiliations
Contributions
B.C. and F.L. conceived and designed the research, and gave valuable comments on the analysis; L.L. carried out the transient breakthrough experiments; R.K. calculated the selectivity and simulated the breakthrough; Z.X. carried out all theoretical calculations and interpretations, created all figures and wrote the draft; X.X., J.X., and Y.F. carried out experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Shane Telfer, Michael Zaworotko and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, Z., Xiong, X., Xiong, J. et al. A robust Th-azole framework for highly efficient purification of C2H4 from a C2H4/C2H2/C2H6 mixture. Nat Commun 11, 3163 (2020). https://doi.org/10.1038/s41467-020-16960-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-16960-9
This article is cited by
-
Hydrogen bond unlocking-driven pore structure control for shifting multi-component gas separation function
Nature Communications (2024)
-
Benchmark single-step ethylene purification from ternary mixtures by a customized fluorinated anion-embedded MOF
Nature Communications (2023)
-
Fine pore engineering in a series of isoreticular metal-organic frameworks for efficient C2H2/CO2 separation
Nature Communications (2022)
-
Rational tuning of thorium-organic frameworks by reticular chemistry for boosting radionuclide sequestration
Nano Research (2022)
-
One-step ethylene production from a four-component gas mixture by a single physisorbent
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
