
Abstract
Loci under balancing selection, where multiple alleles are maintained, offer a relevant opportunity to investigate the role of natural selection in shaping genetic dominance: the high frequency of heterozygotes at these loci has been shown to enable the evolution of dominance among alleles. In the butterfly Heliconius numata, mimetic wing color variations are controlled by an inversion polymorphism of a circa 2 Mb genomic region (supergene P), with strong dominance between sympatric alleles. To test how differences in dominance observed on wing patterns correlate with variations in expression levels throughout the supergene region, we sequenced the complete transcriptome of heterozygotes at the prepupal stage and compared it to corresponding homozygotes. By defining dominance based on non-overlapping ranges of transcript expression between genotypes, we found contrasting patterns of dominance between the supergene and the rest of the genome; the patterns of transcript expression in the heterozygotes were more similar to the expression observed in the dominant homozygotes in the supergene region. Dominance also differed among the three subinversions of the supergene, suggesting possible epistatic interactions among their gene contents underlying dominance evolution. We found the expression pattern of the melanization gene cortex located in the P-region to predict wing pattern phenotype in the heterozygote. We also identify new candidate genes that are potentially involved in mimetic color pattern variations highlighting the relevance of transcriptomic analyses in heterozygotes to pinpoint candidate genes in non-recombining regions.
Similar content being viewed by others


Introduction
Dominance between alleles determines the phenotype of heterozygotes and plays a key role in the evolutionary fate of alleles. Indeed, new adaptive variants are often dominant, because emerging alleles, initially at rare frequency within population, are more prone to be picked up by positive selection when expressed at heterozygous state (Haldane 1956). On the contrary, deleterious variants are generally recessive, because they escape purging if they are scarcely expressed (Charlesworth and Charlesworth 1999; Charlesworth and Willis 2009; Connallon and Hall 2018). In loci under balancing selection, where multiple alleles are maintained in sympatry, heterozygotes are frequent and natural selection thus frequently acts on heterozygotes, promoting the evolution of dominance (Otto and Bourguet 1999; Llaurens et al. 2009). Dominance can arise as an inherent property of the encoded protein, but can also be tuned by expression modifiers, especially in polymorphic loci (Wilkie 1994). At the translational level, one of the two protein copies in the heterozygote could be dis- or nonfunctional due to non-synonymous or missense mutations in the coding DNA sequence, such as the recessive O allele in the ABO blood group system (Yamamoto et al. 1990). At the transcriptional level, recessivity can arise from differences of overall level of expression of a gene between homozygotes, skewing the phenotype of the heterozygotes toward that of the allele with the higher expression. This is most clearly seen in cases of regulatory mutations, which can affect the expression of a gene in a particular tissue. For example, mutations at a regulatory element located 5-kb upstream of the tan gene in Drosophila erecta suppresses dark pigmentation in the abdomen of homozygous light females, but heterozygotes have pigmentation similar to homozygous dark females (Yassin et al. 2016). The regulatory mutations can also be part of the RNA transcripts, leading to allele-specific differential expression in the heterozygotes, such as in the case of the self-incompatibility locus in Arabidopsis halleri and A. lyrata where small RNAs specifically repress some alleles at the SCR gene (Durand et al. 2014). Remarkably, all these diverse molecular mechanisms are maintained by balancing selection, allowing selection to act on the phenotype of the heterozygotes, and therefore shaping the evolution of dominance (see Billiard et al. 2021 for a review). Both levels of gene and allele expression can trigger dominance in heterozygotes and be submitted to selection in polymorphic loci where heterozygotes have a high frequency.
The neo-tropical butterfly Heliconius numata is a fascinating example of adaptive polymorphism within populations, with multiple mimetic wing color patterns maintained in sympatry. These different wing patterns are submitted to strong natural selection exerted by predators favoring mimicry towards distinct toxic species (Joron et al. 1999). In this species, the discrete variations in wing color pattern are mainly controlled by the supergene P (Joron et al. 2006). The dissection of the supergene architecture is still in progress but, among the 129 genes located within the 2 Mb long supergene (Jay et al. 2018), the gene cortex is known to control variations in hindwing melanic patterns in H. numata (Nadeau et al. 2016; Livraghi et al. 2020). A number of other genes at the supergene are likely to control variations of other individual pattern elements (Saenko et al. 2019). Combinations of allelic variants at multiple linked genes will thus give rise to differentiated haplotypes at the supergene P, each governing distinct wing pattern features (Joron et al. 2006). By comparing the phenotypes of homozygotes and heterozygotes at the supergene, strong dominance between supergene alleles have been found in sympatry, whereas mosaics of dominance resulting in intermediate phenotypes were observed in heterozygotes obtained from crosses performed with individuals from different populations (Le Poul et al. 2014). Natural selection promoting mimicry favors coordinated dominance between supergene alleles found in sympatry, resulting in mimetic phenotypes expressed in heterozygotes (Le Poul et al. 2014; Arias et al. 2016). The mimicry in heterozygotes may stem from either the dominance of the derived mimetic alleles (i.e. Haldane’s sieve) or the modification of dominance of the ancestral alleles (Llaurens et al. 2015). Moreover, supergene alleles are characterized by different chromosomal inversions favoring their genetic differentiation (Joron et al. 2011), and alleles exhibiting the ancestral gene order are recessive to alleles exhibiting a rearranged gene order, the latter being introduced into H. numata via introgression from a closely-related species (Jay et al. 2018). We thus aim at understanding the molecular mechanisms involved in dominance between supergene alleles, which are likely shaped by natural selection through mimicry.
In this paper, we thus compare the level of gene expression between individuals carrying either homozygous or heterozygous genotypes at the supergene P. We focus on two supergene alleles controlling the bicoloratus and tarapotensis phenotypes respectively. These two phenotypes are frequently observed in sympatry, and are mimetic to different communities of defended species, including the chemically-defended Ithominii species Melinaea mothone (bicoloratus mimicry ring) and Melinaea menophilus ssp. nov. (tarapotensis mimicry ring) (Joron et al. 1999). These two alleles are therefore under strong selection favouring mimicry to their respective communities (Chouteau et al. 2016). Heterozygotes Pbic/Ptar exhibit the bicoloratus phenotype so that the allele bicoloratus (referred to as Pbic hereafter) is dominant over the allele tarapotensis (Ptar hereafter) (Le Poul et al. 2014); Fig. 1A). Increased predation has been observed on intermediate phenotypes between tarapotensis and bicoloratus (Arias et al. 2016), therefore favouring dominance in heterozygotes Pbic/Ptar. Both alleles have derived gene orders, inferred from the discrepancy in synteny with closely-related Heliconius species (Joron et al. 2011). The bicoloratus allele (Pbic) displays a single inversion (P1) while the tarapotensis allele (Ptar) is thought to derive from a haplotype carrying a P1 inversion followed by two subsequent inversions P2 and P3 (Joron et al. 2011). As a result, the two alleles have the same gene order at the 400-kb long P1 inversion harboring the cortex gene and have different gene order at the circa 1.6-Mb long P2-P3 inversions (Fig. 1B). Because selection on phenotypic dominance between these two alleles is well documented, this stresses the need to investigate the molecular mechanisms underlying their genetic dominance.

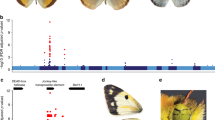
A Patterns of wing coloration in the bicoloratus (bic) and tarapotensis (tar) morphs of the species Heliconius numata and their co-mimics from the genus Melinaea. The diversity in wing pigmentation is associated with a dominance relationship between the two morphs: wing color pattern of the Pbic/Ptar heterozygotes show a striking similarity with the color pattern of Pbic/Pbic homozygotes except at two spots (blue arrows). B Three series of inversion at the supergene are associated with the two morphs bic and tar (Jay et al. 2021). The rearrangement P1 involves an event of inversion that spans over 400 kb and is associated with the phenotype bicoloratus. P2 and P3 involve two supplementary inversions of roughly 0.2 and 1.2 Mb respectively and are associated here with the phenotype tarapotensis.
Given that inversions limit recombination in the supergene region, the identification of candidate genes associated to wing pattern variations through widely used association analyses (e.g., GWAS, see Jay et al. 2022) or classical QTL mapping is difficult. Analysis of transcriptomic variation could overcome such difficulties because changes in expression levels among genes within the inversion could more likely associate with phenotypic differences than to their physical linkage. Using an RNAseq approach, we compared gene expression in the homozygotes (Pbic/Pbic and Ptar/Ptar) and heterozygotes (Pbic/Ptar) and specifically tested for departure from additivity. We focused our analyses only on transcripts where the range of expression levels did not overlap between Pbic/Pbic and Ptar/Ptar genotypes. By focusing on genes with contrasted levels of transcript expression in the homozygotes, we were thus able to compare patterns of dominance between the supergene and the rest of the genome.
Materials and methods
Samples, library sequencing, and de novo transcriptome assembly
The H. numata butterflies used in this study come from controlled crosses performed with individuals sampled around Tarapoto (Peru). Crosses were carried out among individuals with different genotypes at the color pattern supergene P (and therefore different wing color patterns). Individuals studied here all belong to the F2 generation of a single initial cross between a female with genotype Pbic/Parc and a male with genotype Ptar/Paur. The alternative alleles Parc and Paur control for the arcuella and aurora phenotypes that visually differ from bicoloratus and tarapotensis. These two alternative alleles have the same gene order than Ptar (i.e. they all have the three subinversions). The genotypes of the offspring were established using allele-specific primers followed by Sanger sequencing of the amplified fragments, as described in Saenko et al. 2019, therefore checking for recombinant haplotypes. We selected individuals with different combinations of Pbic and Ptar alleles from different crosses performed between F1 offspring with the relevant genotypes. The Pbic allele is associated with the bicoloratus form being dominant to the Ptar allele associated with tarapotensis, as described in Saenko et al. (2019). Forewing and hindwing wing discs were dissected from prepupae, a stage described as the key timing of expression of genes involved in color pattern development in Heliconius (Martin et al. 2012). Nevertheless, our experiment does not cover the whole timeframe of wing development, so that we cannot rule out that other genes involved in color pattern variations located in the P-supergene would be differentially expressed at other developmental stages not studied here. Ten individuals were selected, including 4 individuals with the genotypes Pbic/Pbic, 3 with Pbic/Ptar and 3 with Ptar/Ptar.
The reference transcriptome used in this study was assembled by Saenko et al. (2019) based on transcripts expressed in the wing discs at the 24 h and prepupal stages of 24 H. numata samples. Total RNA was extracted and cDNA libraries prepared and sequenced as described in Saenko et al. 2019. As reported in this paper, a mean of 42 ± 15 million passing Illumina quality filter reads was obtained for each sample. Raw data were filtered for low-quality reads (<Q30) with Prinseq v0.19.5 (Schmieder and Edwards 2011), for adapter sequences with Cutadapt v1.16 (Martin 2011) and for ribosomal RNA-like sequences with riboPicker v1.0.0 (Schmieder et al. 2012). The filtered reads were finally combined to generate a de novo reference transcriptome using Trinity r20140717 (Haas et al. 2013) with the following parameters: SS_lib_type = F, kmer_size = 25, max_pct_stdev = 100, minimum contig length = 200 bp. Of the 53719 transcripts of this reference transcriptome (obtained from all transcripts of the 24 afore-mentioned H. numata samples), 52525 are expressed in the prepupal stage. Of these, 50075, 51181 and 49808 were expressed in the Pbic/Pbic homozygotes, Pbic/Ptar heterozygotes and Ptar/Ptar homozygotes, respectively.
Estimating levels of dominance of the bic allele in heterozygotes using read counts (D)
The supergene allele Pbic is strongly dominant over the supergene allele Ptar, because Pbic/Ptar heterozygotes almost have the same wing color pattern as the Pbic/Pbic homozygotes (Fig. 1A). Therefore, in the heterozygotes Pbic/Ptar we generally expect the level of expression of the genes involved in determining wing patterns to be similar to the expression in the Pbic/Pbic homozygotes. Each wing pattern allele is associated with chromosomal rearrangements at the supergene involving one inversion (rearrangement P1 associated with the form bicoloratus) or two inversions (rearrangements P1, P2 and P3 in tarapotensis; Fig. 1B). The ancestral chromosomal arrangement, without any inversion, is absent from our crosses. To test whether the expression at the supergene in heterozygotes Pbic/Ptar matches the expression observed in the Pbic/Pbic homozygotes, we compared patterns of expression between Pbic/Ptar heterozygotes and the two corresponding homozygotes in terms of gene level of expression (D) along the genome.
All reads were aligned to the de novo transcriptome using Bowtie2 v2.1.0 with default parameters (Langmead and Salzberg 2012) and read counts were calculated with samtools idxstats (Li et al. 2009). To avoid any bias due to alternative splicing, all transcripts from a single gene were annotated and the read counts estimated separately. When they existed, orthologs were identified in the closely-related species H. melpomene by aligning the transcripts to the Hmel2_cds and HMEL2_scaffolds databases (downloaded from Lepbase v4 (Challi et al. 2016)) with NCBI BLASTn. This, however, has revealed that the number of assembled transcripts exceeded the number of transcripts in H. melpomene CDS. The higher number of transcripts in our assembly may be due to variations in alternative splicing between the two species and/or an incomplete annotation for H. melpomene (Saenko et al. 2019). Consequently, several numata transcripts mapped to melpomene intergenic regions. Read counts were normalized using the mean and standard deviation of all reads in the transcriptome per sample. Because of the small sample size in this study, reliance on mean expression level among samples belonging to each genotype (as in Saenko et al. 2019) could strongly be biased by outliers. To avoid this problem, only transcripts with no overlap in the levels of expression between the samples of each homozygous genotype were considered for further analyses. We thus selected transcripts for which i.e. the maximum read count among replicates of one homozygous genotype was inferior to the minimum read count of the alternative homozygous genotype. For those selected transcripts, dominance in heterozygotes was identified by comparing the number of counts in homozygotes and heterozygotes and then associating a score D ranging from −3 to 3. This dominance score D crucially depends on the direction of the difference in the level of expression between the two homozygous genotypes (Fig. 2): either the transcript is more highly expressed in Pbic/Pbic than in Ptar/Ptar homozygotes, or the other way around. We named Ni the read count of the transcripts in the different replicates of the genotype i and define the dominance score D depending on the distribution of the reads counts in the homozygotes where the transcript is highly vs. lowly expressed (Nhigh and Nlow respectively). In transcripts where the minimum counts observed in Pbic/Pbic homozygotes replicates is strictly above the maximum count among Ptar/Ptar homozygotes, we define Nhigh = NPbic/Pbic and Nlow = NPtar/Ptar (category 1). Symmetrically, in transcripts where the minimum count of a transcript among Ptar/Ptar homozygotes is strictly above its maximum count among Pbic/Pbic homozygotes, we define Nhigh = NPtar/Ptar and Nlow = NPbic/Pbic (category 2). The dominance score D is then defined as follows for both categories:

Two categories of transcripts were considered, depending on the levels of expression in the Pbic/Pbic and Ptar/Ptar homozygotes (category 1: when Min(NPbic/Pbic) > Max(NPtar/Ptar) and category 2 when Min(NPtar/Ptar) > Max(NPtbic/ Pbic). The dominance score was assessed depending on the minimum and maximum number of transcripts observed in the different replicates (represented by the transparent boxes) of the same homozygous (in red and blue) and heterozygous (in purple) genotypes (see Methods). A score ranging from −3 to 3 was then given to each transcript following its class in respect to the phenotypically-dominant Pbic/Pbic homozygous genotype.
D = 3 when overdominance of the bic allele is observed, i.e. when Min (NPbic/Ptar) > Max (Nhigh) in category 1 or when Max (NPbic/Ptar) < Min (Nlow) in category 2.
D = 2 when strict dominance of the bic allele is observed, i.e. when Min (NPbic/Ptar) > [(Min (Nhigh) + Max (Nlow)]/2 and Min (NPbic/Ptar) ≤ Max (Nhigh) in category 1 or when Max (NPbic/Ptar) < [(Min (Nhigh) + Max (Nlow)]/2 and Max (NPbic/Ptar) ≥ Min (Nlow) in category 2.
D = 1 when incomplete dominance of the bic allele is observed, i.e. when Max (NPbic/Ptar) ≤ Min (Nhigh) and Min (NPbic/Ptar) > [(Min (Nhigh) + Max (Nlow)]/2 in category 1 or when Min (NPbic/Ptar) ≥ Max (Nlow) and Max (NPbic/Ptar) < [(Min (Nhigh) + Max (Nlow)]/2 in category 2.
D = 0 when codominance of the two alleles is observed, i.e. when the range of expression levels observed in the heterozygotes overlap the midpoint of the range of expression observed in both homozygotes.
D = −1, when incomplete recessiveness of the bic allele is observed, i.e. when Min (NPbic/Ptar) ≥ Max (Nlow) and Max (NPbic/Ptar) < [(Min (Nhigh) + Max (Nlow)]/2 or when Max (NPbic/Ptar) ≤ Min (Nhigh) and Min (NPbic/Ptar) > [(Min (Nhigh) + Max (Nlow)]/2 in category 2.
D = −2, when strict recessivity of the bic allele is observed, i.e. when Max (NPbic/Ptar) < [(Min (Nhigh) + Max (Nlow)]/2 and Max (NPbic/Ptar) ≥ Min (Nlow) or when Min (NPbic/Ptar) > [(Min (Nhigh) + Max (Nlow)]/2 and Min (NPbic/Ptar) ≤ Max (Nhigh) in category 2.
D = −3, when underdominance of the bic allele is observed, i.e. when Max (NPbic/Ptar) < Min (Nlow) in category 1 or when Min (NPbic/Ptar) > Max (Nhigh) in category 2.
Non-parametric statistics (chi2-test) were then used to test whether frequencies of those categories differed (1) between genomic regions (supergene vs. rest of the genome) and (2) between transcripts where the level of expression was higher in Pbic/Pbic (category 1) vs. in Ptar/Ptar homozygotes (category 2).
Results
Patterns of expression between morphs differ within the supergene vs. the rest of the genome
Out of 53,224 genome-wide transcripts, we identified 5855 transcripts with non-overlapping expression level between the two homozygous genotypes (i.e. 11% of all transcripts) (Table 1). Within the P inversion, we identified 141 transcripts with such non-overlapping expression between homozygotes, out of the 669 transcripts mapped to the supergene (i.e. 21% of the transcripts located within the supergene). Consistent with previous studies based on average expression (Saenko et al. 2019; Jay et al. 2021), genes with non-overlapping expression levels are enriched in the P inversion relatively to the rest of the genome (Chi2 = 62.52, P < 0.001). At the genome-wide level, 2798 and 3057 transcripts are upregulated in Pbic/Pbic and Ptar/Ptar homozygotes respectively, indicating a slight deviation toward up-regulation in Ptar/Ptar homozygotes (Chi2 = 11.45, P < 0.001). However, within the P inversion, 56 and 39 transcripts are overexpressed in Pbic/Pbic and Ptar/Ptar homozygotes respectively. Interestingly, whereas differences in the P inversion does not deviate from parity (Chi2 = 3.08, P = 0.079), it contrasts with genome-wide pattern (Chi2 = 4.90, P = 0.027). A closer examination of the three subinversions P1, P2 and P3 revealed substantial differences. In the P1 inversion, which is present in both homozygotes, overexpression of transcripts is more frequent in Pbic/Pbic than in Ptar/Ptar (30 vs. 7, respectively; Chi2 = 14.30, P < 0.001)). An opposite pattern is found for P2, where 4 and 13 transcripts are overexpressed in Pbic/Pbic and Ptar/Ptar homozygotes respectively (Chi2 = 4.90, P = 0.029). For P3 differentially expressed transcripts, no deviation from parity was observed with 22 and 19 up-expressed transcripts in Pbic/Pbic and Ptar/Ptar homozygotes respectively (Chi2 = 0.22, P = 0.639). In fact, the difference in P2 is even larger than the genome-wide pattern which also indicates an overexpression in Ptar/Ptar homozygotes (Chi2 = 4.01, P = 0.045). In summary, the most important deviation from genome-wide pattern is biased towards Pbic/Pbic in P1 and Ptar/Ptar in P2.
A largely biased expression within the supergene toward P bic /P bic expression level in heterozygotes
When comparing genome-wide expression level in Pbic/Ptar heterozygotes to both homozygotes, we found that 997 and 739 transcripts showed expression level towards those of the Pbic/Pbic and Ptar/Ptar, respectively. This genome-wide pattern deviates from parity (Chi2 = 38.34, P < 0.001) towards the expression pattern observed in the Pbic/Pbic homozygotes. Out of those transcripts, 25 and 5 in the P inversion showed expression level towards those of Pbic/Pbic and Ptar/Ptar, respectively (Fig. 3). The pattern in the P inversion deviates from both parity (Chi2 = 13.33, P < 0.001), as well as from the whole-genome pattern reflecting a 5-fold enrichment in the inversion (Chi2 = 8.50, P = 3.55 × 10−3). Interestingly and in contrast to comparisons between homozygotes, the three subinversions did not show any difference in their dominance pattern. Nevertheless, the most important departure from genome-wide homozygotes-based expectations being at P2, with 6 and 1 heterozygous transcripts tending towards Pbic/Pbic and Ptar/Ptar, respectively (Chi2 = 53.69, P < 0.001 despite low number of observations).

Gray dots refer to transcripts with overlapping expression between the homozygous genotypes or between the homozygotes and the heterozygote. Transcripts with non-overlapping expression levels between the homozygous genotypes are indicated in violet and turquoise colors according to whether the expression was higher in the Pbic/Pbic (category 1) or Ptar/Ptar homozygous genotype (category 2), respectively. Vertical lines indicate borders of the three subinversions according the Hmel2 reference genome coordinates of chromosome 15.
Further examination of dominance of gene expression in the P inversion revealed contrasting patterns between genes overexpressed in the two different homozygous genotypes (Fig. 3). Whereas the 25 bic-dominant transcripts were almost equally distributed along the three subinversions P1, P2 and P3, the 5 tar-dominant transcripts were all found at or near the inversion breakpoints. After rearranging the genomic regions according to the Ptar chromosome, all five transcripts located at the vicinity of the 200 kb long P2 subinversion. Those five transcripts belonged to three genes and two intergenic regions, namely HMEL000021 and HMEL000024 (Sur-8) on P1, an intergenic transcript close to HMEL032682 on P2, and HMEL011885 (orthologous to D. melanogaster CG11414) and an intergenic transcript close to HMEL011882 on P3 (Table 2). For the 25 bic-dominant transcripts, 11 belonged to intergenic regions whereas the remaining 14 transcripts belonged to 11 protein-coding genes. Two genes, namely HMEL000025 (Cortex) and HMEL000033 (Lmtk1, orthologous to D. melanogaster Ddr), had more than a single transcript. Both genes are on P1 and are overexpressed in Pbic/Pbic homozygotes. Those multiple transcripts are most likely the result of alternative splicing. Another evidence for alternative splicing comes from a partial overlap (~10 bp) between an intergenic transcript (comp38651_c0_seq1) that is overexpressed in Pbic/Pbic and shows a bic dominance in the heterozygote and the HMEL000021 transcript which is overexpressed in Ptar/Ptar and shows a tar dominance in the heterozygote. When correcting for the P1 inversion coordinates, these two overlapping transcripts with contrasting dominance patterns become adjacent to the P1/P2 breakpoint.
As indicated in the Methods above and Fig. 2, we assigned signs to dominance estimates according to the overlap between the minimal counts of the heterozygotes relative to the intermediate expression between the two homozygous genotypes. For example, for a gene with a higher expression of the Pbic/Pbic homozygotes, dominance will be positive if the heterozygous minimal counts will be higher than the intermediate expression level and negative if it is below this threshold. The opposite trend follows for a gene with a higher expression in the Ptar/Ptar homozygotes. We therefore investigated whether the dominance observed on the transcripts of the heterozygotes was caused by an overexpression of these transcripts in one out of the two homozygote genotypes. We thus compared the number of dominant transcripts where an overexpression was found in the Pbic/Pbic individuals (category 1) vs. the Ptar/Ptar homozygotes (category 2). At the genome-wide level, we indeed found an excess of transcripts for which expression levels in heterozygotes tend to follow the homozygous genotype with the highest expression level (Chi2 = 206.82, P < 0.001). A similar trend was also found for genes in the ancestral P1 inversion (Chi2 = 13.00, P < 0.001). However, for both P2 and P3 inversions, there was no deviation from parity (Chi2 = 0.06, P = 0.806) and the observed dominance patterns was thus not linked to an increased level of transcript expression in the Pbic/Pbic homozygous genotypes. This final observation indicates that nearly half of dominant transcripts in the two derived subinversions, P2 and P3, may be due to the downregulation of transcripts by the bic allele of genes that are overexpressed in the phenotypically recessive Ptar/Ptar homozygotes.
Discussion
Taken together our analyses of dominance of gene expression levels in Heliconius butterflies reveal that (1) expression levels of the transcripts in heterozygotes are more similar to the expression pattern observed in the dominant homozygous morph, (2) this dominance pattern is enriched but not restricted to the wing-pattern controlling P inversion, and (3) the most important deviation from homozygotes-based expectations is at the derived P2 subinversion.
Genome wide pattern of expression confirms the switch role of the P supergene
By comparing the genome-wide pattern of expression in pre-pupae with different genotypes at the supergene, we discovered important changes in transcript expressions. Genes in the supergene, such as the well-characterized gene cortex (Nadeau et al. 2016) located in the P1 subinversion, may regulate cascades of wing patterning and melanin synthesis genes found outside the P region, changing gene expression in multiple loci throughout the genome. Our study focused on the prepupal stage, where the pre-patterning of the wing starts to be expressed, and the P supergene probably behaves as a switch locus, acting upstream of the genetic pathways inducing variation in wing color pattern development. Consistent with this hypothesis, at the genome scale, the transcript expression patterns in the heterozygotes at the supergene is biased towards the expression observed in the dominant Pbic/Pbic rather than in the Ptar/Ptar. This biased expression pattern at the pre-pupal stage may indeed contributes to the development of the bicoloratus wing color pattern in Ptar/Pbic heterozygous imago. Many genes identified as causing color pattern variations are indeed transcriptional factors that may have a pleiotropic, upstream effect in the color pattern development pathway, rather than a downstream effect. For instance, the signaling gene WntA, that plays a major role in color pattern variation in butterflies, is thought to act as a major pre-patterning element, rather than a downstream gene determining the final identity of wing scales (Martin and Reed 2014).
Heterozygous patterns of expression in the P region suggests a combination of molecular mechanisms involved in coordinated phenotypic dominance
Our approach focused on the detection of non-additive patterns of transcript expression. Nevertheless, assuming a threshold above which a certain developmental pathway is triggered, intermediate expression of a transcript in heterozygotes might be sufficient to generate dominance (Gilchrist and Nijhout 2001). Here, we focused on the deviation from additivity, therefore identifying biased patterns of expression of transcript in the heterozygotes, which might represent only a fraction of the molecular mechanisms involved in phenotypic dominance. In the P1 subinversion, there is a strong overexpression of transcripts in the Pbic/Pbic homozygotes as compared to Ptar/Ptar homozygotes. This high expression level in the P1 region is also found in the Pbic/Ptar heterozygotes, suggesting that high level of P1 transcripts might trigger the development of the bicoloratus phenotype. The identification of upregulated transcripts in the cortex gene might play a major role in the developmental switch between tarapotensis and bicoloratus. Interestingly, the downregulation in heterozygotes of transcripts belonging to the P2 subinversion, that are otherwise overexpressed in the recessive tarapotensis morph might also contribute to the development of the bicoloratus phenotypes. Such downregulation might be promoted by selection acting on the dominance at the P supergene, contributing to the coordinated pattern of dominance observed in sympatric homozygotes (Le Poul et al. 2014). If such a hypothesis turns out to be correct, this would indicate that the evolution of complex dominance patterns in Heliconius may have involved a step-wise process, with (1) a Haldane’s sieve-like evolution of a dominant derived allele at P1, where the over-expression of the dominant transcripts drive phenotypic development towards the bicoloratus phenotype and (2) the evolution of repression in the genes located in the younger sub-inversions P2 and P3, preventing the expression of the transcripts triggering the development of the recessive tarapotensis phenotypes. This study thus represents a promising step towards the understanding of the evolution of dominance relationships in complex phenotypic traits. Investigating the pattern of different heterozygous combinations at the P supergene would certainly shed light on the different molecular mechanisms causing the coordinated dominance observed at the phenotypic scale, allowing to reconstruct the evolutionary history of dominance relationships.
Candidate genes for the development of color pattern with multiple elements
The bicoloratus and tarapotensis color pattern phenotypes differ in a number of melanic and yellow elements, that might be controlled by different genetic bases. The identification of contrasted patterns of expression in the different transcripts within the supergenes in heterozygotes may shed light on the genetic architecture controlling variations of such a complex color pattern. Out of the 669 transcripts of the 2 Mb-long P inversion, our analysis identified only 30 candidate transcripts, and therefore represents a significant step towards the identification of the molecular basis of dominance evolution in this important model of adaptive polymorphism. Although the functional role of these different genes on wing pattern development is not obvious, the genes for which heterozygotes have a biased expression toward the Pbic/Pbic expression level can be putative candidates for the developmental switch between tarapotensis and bicoloratus color patterns. Note that these genes did not display any signal of differential expression when we compared expression levels across homozygotes (Saenko et al. 2019). Nevertheless, a switch in developmental pathway can be triggered by variations in the levels of certain signaling proteins but can also be triggered by variations in the protein sequence. We thus hope our study will stimulate larger-scale investigation of expression patterns, allowing to refine our knowledge on the combinations of genetic elements involved in such major developmental switch.
Data availability
The data can be found in the NCBI SRA and TSA repositories under the number PRJNA555830.
References
Arias M, le Poul Y, Chouteau M, Boisseau R, Rosser N, Théry M et al. (2016) Crossing fitness valleys: empirical estimation of a fitness landscape associated with polymorphic mimicry. Proc R Soc B 283:20160391
Billiard S, Castric V, Llaurens V (2021) The integrative biology of genetic dominance. Biol Rev 96:2925–2942
Challi RJ, Kumar S, Dasmahapatra KK, Jiggins CD, Blaxter M (2016) Lepbase: the Lepidopteran genome database. bioRxiv https://www.biorxiv.org/content/10.1101/056994v1.
Charlesworth B, Charlesworth D (1999) The genetic basis of inbreeding depression. Genet Res 74:329–340
Charlesworth D, Willis JH (2009) The genetics of inbreeding depression. Nat Rev Genet 10:783–796
Chouteau M, Arias M, Joron M (2016) Warning signals are under positive frequency-dependent selection in nature. PNAS 113:2164–2169
Connallon T, Hall MD (2018) Genetic constraints on adaptation: a theoretical primer for the genomics era. Ann NY Acad Sci 1422:65–87
Durand E, Méheust R, Soucaze M, Goubet PM, Gallina S, Poux C et al. (2014) Dominance hierarchy arising from the evolution of a complex small RNA regulatory network. Science 346:1200–1205
Gilchrist MA, Nijhout HF (2001) Nonlinear developmental processes as sources of dominance. Genetics 159:423–432
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J et al. (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512
Haldane JBS (1956) The theory of selection for melanism in Lepidoptera. Proc R Soc Lond B 145:303–306
Jay P, Whibley A, Frézal L, Rodríguez de Cara MÁ, Nowell RW, Mallet J et al. (2018) Supergene evolution triggered by the introgression of a chromosomal inversion. Curr Biol 28:1839–1845
Jay P, Chouteau M, Whibley A, Bastide H, Parrinello H, Llaurens V et al. (2021) Mutation load at a mimicry supergene sheds new light on the evolution of inversion polymorphisms. Nat Genet 53:288–293
Jay P, Leroy M, Le Poul Y, Whibley A, Arias M, Chouteau M et al. (2022) Association mapping of colour variation in a butterfly provides evidence that a supergene locks together a cluster of adaptive loci. Philos Trans R Soc B 377:20210193
Joron M, Frezal L, Jones RT, Chamberlain NL, Lee SF, Haag CR et al. (2011) Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477:203–206
Joron M, Papa R, Beltrán M, Chamberlain N, Mavárez J, Baxter S et al. (2006) A conserved supergene locus controls colour pattern diversity in Heliconius butterflies. PLOS Biol 4:e303
Joron M, Wynne IR, Lamas G, Mallet J (1999) Variable selection and the coexistence of multiple mimetic forms of the butterfly Heliconius numata. Evol Ecol 13:721–754
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Method 9:357–359
Le Poul Y, Whibley A, Chouteau M, Prunier F, Llaurens V, Joron M (2014) Evolution of dominance mechanisms at a butterfly mimicry supergene. Nat Commun 5:1–8
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079
Livraghi L, Hanly JJ, Loh LS, Ren A, Warren IA, Concha C et al. (2020) The gene cortex controls scale colour identity in Heliconius. bioRxiv https://www.biorxiv.org/content/10.1101/2020.05.26.116533v2.full.
Llaurens V, Billiard S, Castric V, Vekemans X (2009) Evolution of dominance in sporophytic self-incompatibility systems: I. Genetic load and coevolution of levels of dominance in pollen and pistil. Evolution 63:2427–2437
Llaurens V, Joron M, Billiard S (2015) Molecular mechanisms of dominance evolution in Müllerian mimicry. Evolution 69:3097–3108
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12
Martin A, Reed RD (2014) Wnt signaling underlies evolution and development of the butterfly wing pattern symmetry systems. Dev Biol 395:367–378
Martin A, Papa R, Nadeau NJ, Hill RI, Counterman BA, Halder G et al. (2012) Diversification of complex butterfly wing patterns by repeated regulatory evolution of a Wnt ligand. PNAS 109:12632–12637
Nadeau NJ, Pardo-Diaz C, Whibley A, Supple MA, Saenko SV, Wallbank RWR et al. (2016) The gene cortex controls mimicry and crypsis in butterflies and moths. Nature 534:106–110
Otto SP, Bourguet D (1999) Balanced polymorphisms and the evolution of dominance. Am Nat 153:561–574
Saenko SV, Chouteau M, Piron-Prunier F, Blugeon C, Joron M, Llaurens V (2019) Unravelling the genes forming the wing pattern supergene in the polymorphic butterfly Heliconius numata. EvoDevo 10:16
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864
Schmieder R, Lim YW, Edwards R (2012) Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 28:433–435
Wilkie AO (1994) The molecular basis of genetic dominance. J Med Genet 31:89–98
Yamamoto F, Clausen H, White T, Marken J, Hakomori S (1990) Molecular genetic basis of the histo-blood group ABO system. Nature 345:229–233
Yassin A, Bastide H, Chung H, Veuille M, David JR, Pool JE (2016) Ancient balancing selection at tan underlies female colour dimorphism in Drosophila erecta. Nat Commun 7:10400
Acknowledgements
This study was funded by ANR JCJC DOMEVOL and the Emergence program of Paris City council to VL and by ANR SUPERGENE to MJ.
Author information
Authors and Affiliations
Contributions
HB designed and performed the dominance analyses on RNA-Seq data and wrote and edited the manuscript. SVS extracted the RNA, performed the RNA-Seq, assembled the transcriptome and wrote and edited the manuscript. MC designed the experimental scheme, bred the butterflies, performed the crosses and wrote and edited the manuscript. MJ designed the experimental scheme and wrote and edited the manuscript. VL designed the experimental scheme, supervised the project and wrote and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Rowan Barrett.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bastide, H., Saenko, S.V., Chouteau, M. et al. Dominance mechanisms in supergene alleles controlling butterfly wing pattern variation: insights from gene expression in Heliconius numata. Heredity 130, 92–98 (2023). https://doi.org/10.1038/s41437-022-00583-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00583-5
