
Abstract
The application of genome-wide cytonuclear molecular data to identify management and adaptive units at various spatio-temporal levels is particularly important for overharvested large predatory organisms, often characterized by smaller, localized populations. Despite being “near threatened”, current understanding of habitat use and population structure of Carcharhinus galapagensis is limited to specific areas within its distribution. We evaluated population structure and connectivity across the Pacific Ocean using genome-wide single-nucleotide polymorphisms (~7200 SNPs) and mitochondrial control region sequences (945 bp) for 229 individuals. Neutral SNPs defined at least two genetically discrete geographic groups: an East Tropical Pacific (Mexico, east and west Galapagos Islands), and another central-west Pacific (Lord Howe Island, Middleton Reef, Norfolk Island, Elizabeth Reef, Kermadec, Hawaii and Southern Africa). More fine-grade population structure was suggested using outlier SNPs: west Pacific, Hawaii, Mexico, and Galapagos. Consistently, mtDNA pairwise ΦST defined three regional stocks: east, central and west Pacific. Compared to neutral SNPs (FST = 0.023–0.035), mtDNA exhibited more divergence (ΦST = 0.258–0.539) and high overall genetic diversity (h = 0.794 ± 0.014; π = 0.004 ± 0.000), consistent with the longstanding eastern Pacific barrier between the east and central–west Pacific. Hawaiian and Southern African populations group within the west Pacific cluster. Effective population sizes were moderate/high for east/west populations (738 and 3421, respectively). Insights into the biology, connectivity, genetic diversity, and population demographics informs for improved conservation of this species, by delineating three to four conservation units across their Pacific distribution. Implementing such conservation management may be challenging, but is necessary to achieve long-term population resilience at basin and regional scales.
Similar content being viewed by others


Introduction
The transition from conservation genetics to conservation genomics has lead to the development and increasing use of genome-wide genetic data capable of responding to complex ecological and evolutionary questions (Narum et al. 2013; Savolainen et al. 2013; Pujolar et al. 2014). Identifying conservation units (CU), including evolutionary significant units (ESU) and management units (MU) is essential for improved conservation of wild populations and to guarantee their evolutionary potential and long-term persistence (Funk et al. 2012; Savolainen et al. 2013). Importantly, the tremendous increase in number of markers facilitated through genotyping-by-sequencing methods has enabled reliable assessments of genetic variation, relatedness, effective population size, and has increased the statistical power and resolution of population adaptation and phylogenetic structure analyses (Portnoy and Heist 2012; Larson et al. 2014; Portnoy et al. 2015; Benestan et al. 2016). Accordingly, effective delimitations of ESUs and MUs enabling improved and informed management practices, especially for non-model organisms, is now possible (Ouborg et al. 2010; Willette et al. 2014; Shafer et al. 2015; Hamon et al. 2017). Despite delimitation of CUs being a standard practice in conservation genetics/genomics, debate remains regarding the importance of correct identification of adaptive loci and their use to inform conservation (de Guia and Saitoh 2007; Shafer et al. 2015; Garner et al. 2016), with a growing trend towards investigating statistical outlier loci (putatively adaptive) and local adaptation (Vincent et al. 2013; Steane et al. 2014; Candy et al. 2015).
Documenting genetic differences in marine environments is challenging due to limited evident gene flow barriers (Waples 1998; Selkoe et al. 2008), especially for highly migratory species such as sharks (Portnoy and Gold 2012; Portnoy et al. 2014). Barriers such as ocean currents, geographic distance, habitat discontinuity, or differential dispersal ability can be responsible for population structure in marine organisms (Dawson et al. 2002; Baums et al. 2012). A good example of this within the Pacific Ocean is the eastern Pacific barrier (EPB)—a 4000–7000 km stretch of ocean lacking intermediate islands—that separates the eastern from the central and west Pacific (Briggs 1974; Lessios and Robertson 2006; Gaither et al. 2016). While the advance of next generation sequencing techniques has increased access to these cost-effective genome-wide markers (Allendorf et al. 2010; Willette et al. 2014), they have not yet been widely used for Chondrichthyan studies (Portnoy et al. 2015). Conservation genetics studies of globally distributed sharks have traditionally used a combination of mitochondrial DNA (mtDNA) sequences and nuclear microsatellites to investigate population structure (Keeney and Heist 2006; Portnoy et al. 2010; Karl et al. 2010; Daly-Engel et al. 2012). The combination of both marker types has permitted identification of historic and current population demographic patterns, genetic diversity, and connectivity at the intra-specific level. These studies also have provided evidence for differential dispersal patterns between sexes (Portnoy et al. 2010; Daly-Engel et al. 2012). However, despite being widely used, conventional nuclear markers such as microsatellites present limitations (including homoplasy, null alleles, and shifts in allele size caused by mutations in flanking regions) to population structure investigations (Balloux et al. 2000; Portnoy and Heist 2012).
Chondrichthyans have experienced increasingly intensive fishing and habitat degradation pressure over recent decades. It is estimated that a hundred million sharks are killed annually and over-fishing has resulted in the loss of over 90% of sharks and large predatory fishes across all ocean basins (Myers and Worm 2003; Polidoro et al. 2012; Worm et al. 2013; Dulvy et al. 2014). Currently, the IUCN Red List for Threatened Species estimates one quarter of all shark and ray species are at risk of extinction (Dulvy et al. 2014). Common biological characteristics of chondrichthyans such as slow growth, late maturation and low fecundity (Hoenig and Gruber 1990) limit their recovery from anthropogenic pressure and lead to low resilience. These characteristics make it challenging to define a single conservation strategy for sharks, and proper management requires individual species assessments (Clarke et al. 2015). Understanding aspects of the biology, habitat use and population demographics is the first step towards improved conservation and management of sharks and rays, yet 50% are IUCN listed as data deficient (Dulvy et al. 2014), highlighting the need for more shark population structure and monitoring data for most species. Information on distribution patterns and population connectivity is crucial to avoid local depletion when a species is composed of more than one breeding unit (Shivji 2010; Clarke et al. 2015).
The Galapagos shark (Carcharhinus galapagensis, Snodgrass and Heller 1905) is a circumtropically distributed species with preference for isolated oceanic islands and seamounts in tropical and warm temperate waters (Compagno 1984; Wetherbee et al. 1996). However, there have been reports of individuals in open ocean habitats (Kohler et al. 1998). Studies of Galapagos shark behavior associate different depth preferences with different life history stages: adults preferring deeper—and juveniles shallower habitats (Lowe et al. 2006; Meyer et al. 2010). Others have shown reverse diel vertical movements by juveniles, which prefer deeper waters at night and shallower waters during daytime; seasonal changes in horizontal and vertical movements (Papastamatiou et al. 2015). Previous studies of Galapagos shark movements in Hawaii, based on acoustic telemetry, have indicated that individuals remain within a range of approximately 30 km for periods of up to 4 years (Lowe et al. 2006; Meyer et al. 2010; Papastamatiou et al. 2015). Most acoustic tagging from Hawaii and mark-recapture data from the Atlantic are congruent, indicating considerable site attachment in the species. However, occasionally some individuals migrate long distances >2000 km (Kohler et al. 1998); C. Meyer unpublished data). A recent Galapagos shark population genetic assessment in the southern Galapagos Islands identified two MU separated by only 50–60 km using mtDNA and SNPs (Pazmiño et al. 2017).
The Galapagos shark is “near threatened” according to the IUCN Red List of Threatened species (Bennett et al. 2003). However, information about population structure and connectivity across most of its distribution range is still lacking, and current knowledge of habitat use and population structure is limited to specific areas (Kohler et al. 1998; Meyer et al. 2010; Papastamatiou et al. 2015; Pazmiño et al. 2017). We performed a large-scale genetic assessment of Galapagos sharks across the Pacific Ocean using both nuclear genome-wide SNPs and mtDNA control region sequences. We aimed to: (1) assess the phylogeographic patterns and potential sex-biased dispersal signals of Galapagos sharks across the Indo-Pacific, (2) estimate the level of divergence within regionally defined populations using statistical outliers, and (3) estimate the effective population sizes (Ne) for each defined genetic population (CU) to inform conservation and management of the Galapagos shark.
Materials and methods
Tissue collection and DNA extraction
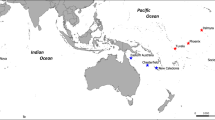
This study examined samples from nine locations across the Pacific Ocean and also included a few individuals from the southwestern Indian Ocean. Five southwest Pacific locations were sampled: Elizabeth (ELZ) and Middleton (MID) Reefs, Lord Howe (LHW), and Norfolk Islands (NOR) from Australia; Raoul Island (Kermadec Islands, KER) from New Zealand. One central Pacific location, Hawaii (HAW) and three east Pacific locations were sampled: the Revillagigedo Islands, Mexico (MEX) and two sites in the Galapagos Islands, Ecuador (EGAL and WGAL). Additionally, a token sample (three individuals) representing the southwest Indian Ocean was obtained from an isolated shallow seamount (Walters Shoals), 600 km east of South Africa, (SAF) (Fig. 1a). Given the small sample, SAF could not be included in a range of population genetic analyses. Genomic DNA was extracted from fin clips using a modified salting out protocol (Sunnucks and Hales 1996), DNA concentrations were spectrophotometrically (NanoDrop 1000, Thermo Scientific) estimated and DNA integrity was electrophoretically verified using 0.8% agarose in 1× Tris/Borate/EDTA (TBE) buffer. Undiluted and diluted aliquots of extracted DNA were stored at −20 °C.

a Sampling locations of Galapagos sharks across the Pacific and Indian Ocean as follows: west Pacific locations—Elizabeth (ELZ) and Middleton (MID) Reefs, Lord Howe Island (LHW), Norfolk Island (NOR), Kermadec Island (KER); east Pacific locations—Revillagigedo Islands in Mexico (MEX), east and west Galapagos Islands (EGAL and WGAL); central Pacific—Hawaii (HAW); and west Indian Ocean—Walters Shoal, South Africa (SAF). b Haplotype network of 229 mt control region sequences of Galapagos sharks. Sizes of circles are proportional to haplotype frequencies. Each dash crossing a branch represents one mutation between haplotype. c Population network of 200 individuals and 7274 neutral SNPs using the NetViewP pipeline. The network reconstruction is based on an Identity by Similarity (IBS) distance matrix and visualized at a maximum number of nearest neighbor (k-NN) threshold of 38
Nuclear SNP marker development using Genotype by Sequencing
Following DNA extractions, a quality control step involving a test restriction digest was performed at 37 °C for 3 h in a volume of 22 µl containing 5 µl undiluted DNA, 2 µl NE Buffer, 0.2 µl EcoRI enzyme and 14.8 µl DNase/RNase-free distilled water. Digestion controls contained all reagents as above, except EcoRI. Digestion was terminated by incubation at 65 °C for 20 min. Finally, 12.5 µl of digested, undigested and undiluted DNA were electrophoresed through an 0.8% agarose gel in 1× TBE for 45 min at 100 V and visualized using Biotium Gel-Green. Only high quality DNA obtained from this trial was sent for library preparation and sequencing at Diversity Arrays Technology (DArT PL) in Canberra, Australia.
Following a second DNA quality evaluation by DArT, double digestion was performed using methylation-sensitive restriction enzymes to digest 150–200 ng of gDNA. The resultant fragments were ligated to barcoded adapters and amplified using polymerase chain reaction (PCR). PCR products were then standardized in concentration and pooled for sequencing on a single HiSeq 2500 (Illumina) lane to yield approximately 2.5 million reads per sample. Preparation and sequencing of libraries was performed by DArT as per Sansaloni et al. 2010 and Kilian et al. 2012. SNPs were jointly developed and genotyped following standard procedures applied by DArT. For a comprehensive description of SNP calling and DArT quality filtering processes, we refer to Pazmiño et al. 2017. The initial data set consisted of 57,341 polymorphic SNP loci and downstream SNP quality control steps were performed before further analysis in order to reduce low-quality and uninformative data (Larson et al. 2014). Only loci with a call rate >85% were retained; the threshold for Minor Allele Frequencies (MAF) was determined at 2%. Linkage disequilibrium was tested using PLINK v1.09 (Purcell et al. 2007) by calculating the correlation coefficient of alleles at two loci, independent of allele frequency. Finally, we tested Hardy-Weinberg equilibrium (HWE) using GENODIVE v2.0 (Meirmans and Van Tienderen 2004). Loci displaying significant deviation from HWE expectations in all populations (p < 0.01) were removed.
Output files obtained from the afore-mentioned procedures were converted manually into a GENEPOP format file prior to being transformed into various other formats, using PGDSpider v2.0.6.0 (Lischer and Excoffier 2012). In order to identify statistical outlier loci we used two simulation approaches for the whole data set, the first approach was implemented in LOSITAN Selection Detection Workbench (Antao et al. 2008), and the second in ARLEQUIN. For analyses at the “within region level”, where hierarchical genetic structure is no longer required, the ARLEQUIN approach was replaced by an approach implemented in PCADAPT R package (Luu et al. 2016). LOSITAN uses a coalescent-based simulation approach to identify loci with unusually high or low pairwise FST values compared with pairwise FST values expected under neutrality to assess the relationship between FST and expected heterozygosity (He). Three independent runs were computed within a 95% confidence interval; an infinite alleles model was used with 100,000 iterations evaluating false discovery rate (FDR). ARLEQUIN performs coalescent simulations examining the joint null distribution of hierarchical FST and He and estimates p-values for each locus, while considering the hierarchical genetic structure of the data using a hierarchical island model (Slatkin and Voelm 1991). Hierarchical genetic structure was determined based on neutral variation and p-values were corrected using the Benjamini and Hochberg (1995) FDR method. The statistical method implemented using the PCADAPT R package detects outlier loci based on principal component analysis (PCA) by assuming that markers excessively related with population structure are candidates for local adaptation. p-values were adjusted with an FDR correction as implemented in the QVALUE R package (Storey 2015). To define the Neutral data set, all detected putative outliers were removed from the data. Loci were then divided into two data sets: one including neutral SNPs only, the other including outlier SNPs only.
Pairwise FST and expected (He) heterozygosity values were calculated for each locus of both data sets using ARLEQUIN, and were also independently evaluated using GENETIX v4.05 (Belkhir et al. 2004). Significance of pairwise FST values were assessed by running 10,000 permutations. Discriminant analysis of principal components (DAPC; Jombart et al. 2010) was also performed as an initial analysis of population structure for neutral and outlier loci independently, using the ADEGENET package in R Studio v0.98.977 (R Development Core Team 2008; Jombart and Ahmed 2011). Each individual was assigned to a predefined population (based on geographic location) for this analysis and an α-score optimization was used to determine the number of principal components to retain.
The partitioning into putative genetically distinct populations was performed using the clustering approach implemented in STRUCTURE v2.3.4 (Pritchard et al. 2000), which investigates the likelihood that a sample belongs to K populations (K representing any number) based on allele frequencies at each locus. Data was analyzed for both neutral and outlier data sets using K values ranging from 1 to 10, with ten independent iterations, one million Markov chain Monte Carlo repetitions and an independent allele frequency burn-in of 100000. The most likely number of populations (K) was defined according to the DeltaK statistic as calculated using STRUCTURE HARVESTER webv0.6.93 (Earl and vonHoldt 2011). This was validated by hand in order to test for K = 1 specifically, since this is not otherwise evaluated (Pritchard and Wen 2003; Evanno et al. 2005) and was followed by population network analysis using NETVIEWP (Neuditschko et al. 2012; Steinig et al. 2016) in order to reveal fine and large scale genomic structure between and within populations. After performing an identity by similarity distance matrix reconstruction using PLINK, the NETVIEWP implementation calculates a minimum spanning tree based on the matrix, and finally the nearest neighbor network is constructed for different thresholds of the maximum numbers of nearest neighbors (k-NN) ranging from 10 to 100.
A phylogenetic analysis was performed to examine any underlying phylogenetic partitions in the data. This was done using the maximum likelihood (ML) criterion and required SNP data to be formatted into a hapmap file using a customized R script, which was analyzed using SNPHYLO (Lee et al. 2014), a pipeline specifically developed for large SNP data sets. The tree reconstruction was performed on a subset of 15 individuals from each sampling location (if available) in order to reduce computational time. ELZ and MID Reefs were considered a single location due to proximity and genetic similarity observed based on FST values in the present and previous analyses (van Herwerden et al. 2008). The Galapagos Islands population was split into two: east (EGAL) and west (WGAL) Galapagos according to Pazmiño et al. (2017). Three samples of the sister species, Carcharhinus obscurus, were used as outgroup in the analysis. A total of 1000 bootstrap replicates were performed to gauge support for identified phylogenetic structure.
Finally, contemporary effective population size was calculated based on the Linkage disequilibrium method (NeLD) for each population using NEESTIMATOR v.2.01 (Do et al. 2014), following an initial power assessment using NEOGEN software (Blower et al. 2016). This software incorporates life-history characteristics specific to the Galapagos shark to estimate the appropriate number of loci and individuals in order to accurately calculate Ne. Alleles with frequencies below critical values (PCrit of 0.02 or 0.05) were removed. Each population was previously filtered for linked loci in PLINK. Based on a standard measure of linkage disequilibrium (r2), we selected two thresholds (r2 = 0.10 and 0.20) and all loci above those thresholds were removed from the data to prevent LD bias in the Ne calculation, considering the number of genome wide SNPs used.
mtDNA sequencing and analyses
The control region (mtDNA) was amplified using PCR and GoTaq Flexi DNA polymerase (Promega). PCR primers were selected from Pardini et al. (2001): light strand ProL2 (5′-CTG CCC TTG GCT CCC AAA GC-3′, and (Keeney et al. 2003): heavy strand 282 H (5′-AAG GCT AGG ACC AAA CCT-3′. These primers have been successfully tested on Galapagos sharks (van Herwerden et al. 2008; Pazmiño et al. 2017). Reactions, PCR conditions and visualization were carried out following Pazmiño et al. (2017). Cleaned-up products were sent to Georgia Genomics Facility (http://dna.uga.edu, USA) for sequencing in forward and reverse directions. Forward and reverse sequences were assembled into contigs, trimmed to 945 bp, edited and aligned in GENEIOUS v5.4.7 (http://www.geneious.com, (Kearse et al. 2012).
Genetic diversity of the mtDNA control region was assessed as number of haplotypes, haplotype (h), and nucleotide (π) diversity within each locality using ARLEQUIN v.3.5.1.2 (Excoffier and Lischer 2010). An analysis of molecular variance (AMOVA) was also performed in ARLEQUIN. Pairwise ΦST was estimated after 10,000 permutations in order to detect population genetic partitioning between locations using ARLEQUIN. Correction for multiple testing was performed following the FDR procedure (Benjamini and Hochberg 1995). Additionally, we tested for demographic population expansion and reduction by calculating Tajima’s D (Tajima 1989) in DNASP v4.10 (Rozas et al. 2003). All positions containing missing data were eliminated for this purpose. MtDNA control region sequences were used for phylogenetic reconstruction under the ML method using default settings of the software MEGA 6.06 (Tamura et al. 2007). The model of sequence evolution was estimated using Partition Finder v1.1.0 (Lanfear et al. 2012) and posterior parameter distributions were examined using Tracer v.1.6 (Rambaut et al. 2014). A total of 1000 bootstrap replicates were performed. Finally, a haplotype network was calculated and drawn using NETWORK v4.2.0.1 (Bandelt et al. 1999) with a Median-joining algorithm and based on Maximum Parsimony.
Results
Neutral and outlier SNPs variation
A total of 208 individuals, including two C. obscurus were successfully genotyped for SNPs. After the first quality check step, including call rate and Minor Allele Frequency filters, the number of loci was reduced from the initial 57,341 to 8368 SNPs for 206 C. galapagensis. A total of 26 SNPs failed to conform to HWE across all populations and were removed from the data set. Ten pairs of loci were identified as linked (r2 > 0.2); subsequently one locus from each pair was randomly selected and deleted.
The number of outliers identified by different approaches varied and differed between data subsets. The whole Indo-Pacific outlier data set contained 31 and 559 loci using ARLEQUIN and LOSITAN, respectively. All loci detected by ARLEQUIN were common between these approaches. All outliers detected by both methods were removed to ensure a purely neutral Pacific-wide data set of 7274 SNPs in the first instance. At the regional scale, the central-west region (consisting of HAW, ELZ, MID, NOR, LHW, KER and SAF) contained 27 outliers common to both methods (LOSITAN and PCADAPT). All outliers, common and LOSITAN/PCADAPT-specific, were removed from the central-west Pacific data, with a total of 6476 neutral SNPs remaining. Finally, the east Pacific group (consisting of MEX, EGAL and WGAL) contained 13 common outliers (LOSITAN and PCADAPT). The east Pacific neutral data set contained a total of 6852 loci after removing these outliers. To define non-neutral data sets, loci were only considered as statistical outliers if detected by both analyses (Supplementary Table S1).
Heterozygosity values for neutral SNPs varied from 0.194 (±0.110) in South Africa (SAF) to 0.237 (±0.118) in Mexico (MEX) (Table 1). The ML tree from neutral SNP data (Fig. 2) showed geographic structure and supported differentiation between east and central–west Pacific (including SAF) populations with strong support (99%) for a monophyletic clade containing all samples from SAF, HAW and the rest of west Pacific populations. East Pacific samples (WGAL, EGAL and MEX) did not form a single monophyletic sister clade to the SAF–central–west Pacific clade, but were distributed across several highly supported sister clades. Fine-scale structure of the global population using neutral SNPs, examined using NETVIEWP analysis at k-NN = 38 (Fig. 1c), identified two distinct genetic clusters, as before: a SAF, HAW and west Pacific cluster and an east Pacific (MEX, EGAL and WGAL) admixed cluster. The overall clustering pattern was consistent at various k-NN thresholds ranging from 10 to 100. Broad-scale population structure of neutral SNPs was further tested independently using DAPC with prior group membership defined by locality, and this revealed a similar pattern seen based on both NETVIEWP and pairwise FST estimations (Fig. 3a). Galapagos shark population subdivision was also strongly supported by STRUCTURE analyses, which tests for the presence of distinct populations assuming a number of subpopulations (K) (between two and ten, Fig. 3b). The strongest and most likely substructure pattern corresponded to K = 2 based on DeltaK statistics computed in STRUCTURE HARVESTER (Earl and vonHoldt 2011). All results consistently highlighted an east vs. central-west Pacific genetic break for Galapagos shark. Neutral loci for the central-west Pacific within region cluster failed to identify further population structure (Fig. 3c), but SAF and HAW were differentiated from the west Pacific (Australian and New Zealand) populations using outlier SNPs (Fig. 3d). Similarly, within the eastern Pacific (where neutral SNPs failed to differentiate between the three sampling locations, Fig. 3e) outlier SNPs identified using either LOSITAN (n = 234, Fig. 3f) or PCADAPT (n = 346, data not shown) independently indicated differentiation between MEX and the Galapagos Islands (there were insufficient common loci detected by these methods (due to different assumptions of each method—Finite island model vs. no assumptions about population demographics, respectively).

Outgroup rooted Maximum Likelihood phylogram of C. galapagensis generated using SNPhylo software from Neutral SNPs and 1000 bootstrap replicates. Two C. obscurus individuals were used as out-group. Only bootstrap values >50% are shown

Population genetic structuring of Galapagos sharks from across the Indo Pacific based on either neutral or outlier SNP data. Panels A-C and E are derived from neutral SNPs and panels D and F are derived from outlier SNPs at different spatial scales as detailed below. a Discriminant analysis of principal components (DAPC) scatterplot of all locations sampled using 7274 neutral SNPs, drawn in the R package ADEGENET. Each dot represents an individual of C. galapagensis, and colors represent the population of origin: Elizabeth (ELZ, yellow) and Middleton (MID, red) Reefs, Lord Howe Island (LHW, dark blue), Norfolk Island (NOR, gray), Kermadec Island (KER, light green), Revillagigedo Islands in Mexico (MEX, pale blue), east and west Galapagos Islands (EGAL, pink and WGAL, purple), Hawaii (HAW, dark green), and Walters Shoals off Southern Africa (SAF, orange). Group membership was defined by sample locality and colors detailed here also apply to panels b–f. b Population assignment and clustering for K = 2 calculated for 7274 neutral SNPs from all locations sampled, using STRUCTURE software. c DAPC scatterplot at the within region level, using 6476 neutral SNPs for the central–west Pacific genetic cluster (HAW, SAF, KER, NOR, LHW, ELZ, and MID). d DAPC scatterplot for the within region level, using 27 outlier SNPs from the central–west Pacific genetic cluster (HAW, SAF, KER, NOR, LHW, ELZ, and MID); e DAPC scatterplot from 6852 neutral SNPs within the east Pacific cluster (MEX, WGAL and EGAL). f DAPC scatterplot from 234 outlier SNPs within the east Pacific cluster (MEX, WGAL and EGAL)
Effective population size (NeLD) estimates for the two populations were consistently recovered from all analyses (including STRUCTURE). Using an r2 = 0.20 threshold, the east Pacific population (n = 87) NeLD was estimated to be 820 (PCrit = 0.02) to 738 (PCrit = 0.05); while the central–west Pacific population (n = 110) NeLD ranged from 4618 (PCrit = 0.02) to 3421 (PCrit = 0.05). A more conservative threshold of r2 = 0.10 was also tested with no significant changes of estimated Ne for either population (Supplementary Table S2).
mtDNA genetic variation
A total of 229 C. galapagensis and three C. obscurus individuals were successfully sequenced for the mitochondrial control region (945 bp). Of the 945 base pairs (bp), 99 were polymorphic (10.4%), and 65% of these were parsimony informative. Summary statistics for mtDNA showed overall mtDNA haplotype (h) and nucleotide (π) diversity was 0.794 (±0.014) and 0.004 (0.000) respectively (Table 1). Hawaii had the highest overall haplotype and nucleotide diversity (h = 0.964 ± 0.077; π = 0.136 ± 0.078). A total of 30 different mtDNA haplotypes were identified. Three common haplotypes: Hap1, Hap2 and Hap20 represent 79.4% of the individuals (Fig. 1b). The remaining 20.5% individuals either shared haplotypes with nine (or fewer) individuals or contained unique haplotypes. Haplotype 1 occurred across the entire Pacific and in SAF. Haplotype 2 occurred in individuals from the Galapagos Islands (east Pacific) exclusively and haplotype 20 was restricted to Australia, New Zealand (southwest Pacific) and SAF. The AMOVA revealed significant differences between the east and central-west Pacific regions (Table 2). Additionally, variation among and within localities was also significant and explained 4.13 and 66.26% of the total variation, respectively. Estimates of population pairwise ΦST and FST indicated a pattern of broad scale phylogeographic structure (Table 3). The ΦST between the two Galapagos Islands populations (EGAL and WGAL) was low and non-significant. However, when comparing the Galapagos Islands with all remaining locations, both Galapagos populations were significantly different before and after FDR correction, with values ranging from 0.301 to 0.539 (p < 0.05). Additionally, significant differentiation was detected when comparing MEX and LHW (ΦST = 0.258), and MEX and MID (ΦST = 0.340). A similar pattern of significant differentiation between the east Pacific (MEX, WGAL and EGAL) and central-west Pacific (LHW, MID, NOR, ELZ, KER and HAW) populations was observed with FST ranging from 0.024 between MEX and HAW, to 0.035 between WGAL and KER. Mitochondrial data also revealed significant differences between HAW and Australian populations LHW, MID, NOR and ELZ.
Results from the ML analysis of mtDNA using the G + I (gamma distributed with invariant sites) evolutionary model, displayed a poorly supported phylogenetic tree (Supplementary Fig. S1). High Bootstrap values were observed only in two clades: the first one including samples from HAW, and a second clade represented by samples from ELZ, KER, NOR, LHW and MID. Neutrality tests for population expansion showed significantly negative values for Tajima’s D in the central-west Pacific populations (D = −2.114 p < 0.05), while non-significant Tajima’s D values were found for the east Pacific group members (MEX, EGAL and WGAL).
Discussion
Overall, our results are congruent with at least two Galapagos shark populations: one on either side of the Pacific Ocean and possibly three (east, central and west Pacific), four (Galapagos, Mexico, Hawaii and west Pacific) or more (the former and additional under- or un-sampled) Galapagos shark populations when taking more subtle marker-specific results into account. Specifically, we caution that the apparent lack of an Indian Ocean population is inconclusive and note that additional samples from southern Africa and elsewhere in the Indian Ocean are required to properly examine the Indo-Pacific wide population structure of Galapagos sharks. Geographic structure within the east and west Pacific C. galapagensis populations may also be further resolved with additional samples. Herein, neutral SNPs, outlier SNPs and mtDNA suggest a range of population structures within the Pacific. Similarly, in sandbar sharks (Carcharhinus plumbeus) mtDNA control region analysis identified strong divergence between Hawaii and the east coast of Australia (ΦST = 0.467), whereas nuclear microsatellites did not (FST = 0.062, Portnoy et al. 2010). Population structure and phylogeny based on neutral SNPs placed Hawaii (and Walters Shoals, southern Africa) within the west Pacific population; Mexico and Galapagos within the East Tropical Pacific population. In contrast, outlier SNPs differentiated Hawaii (and under sampled Walters Shoals) from the west Pacific, and Mexico from the Galapagos population. A similar pattern showing isolation of Hawaiian populations was reported for the coral Porites lobata (Baums et al. 2012) and the fish Acanthurus triostegus (Lessios and Robertson 2006). A strongly supported phylogenetic link (based on the SNP phylogeny) between east and west Pacific lineages via two Mexican animals suggests C. galapagensis entered the central-west Pacific from the east Pacific via Mexico.
Population structure analyses are self-referential, and consequently the geographic scale of analysis influences results, with subtle differences in structure more likely to be significant in regional than global analyses. For example, a regionally focused neutral SNP analysis of Galapagos shark population structure within the Galapagos archipelago identified two distinct populations (EGAL and WGAL; Pazmiño et al. 2017), whereas results from our current Pacific-wide analysis indicate that the Galapagos Islands all belong to a single regional genetic group (using neutral and outlier SNPs), which forms a separate cluster from Mexico (using outlier SNPs only). Only ~12% of polymorphisms are shared between the data set from Pazmiño et al. (2017) and our current data set, due to specific SNP filtering criteria. Hence, we highlight the importance of including both regional and global scale assessments to accurately inform conservation at different geographic scales. We did not find any regional population structure within the southwest Pacific (ELZ, MID, LHW, NOR, and KER) using either mtDNA, neutral or outlier SNPs (data not shown), despite local populations being separated by relatively large geographic distances.
Although neutral SNPs differentiate east from central and west Pacific Galapagos shark populations, mtDNA shows that (1) MEX is significantly different from LHW and MID but not from other central and west Pacific locations, hinting at connectivity between regions, possibly due to very few migrants per generation; (2) the HAW population is differentiated from most other locations, except KER and MEX, suggesting some level of connectivity between Mexico, Hawaii, and the Kermadec Islands (New Zealand); and (3) differentiation within the east Pacific, separating MEX from the Galapagos Islands (EGAL and WGAL, ΦST = 0.459–0.539). This third pattern could be due to secondary barriers between MEX and the Galapagos Islands, generating historical geographic isolation. Alternatively low sample sizes (MEX n = 6, HAW n = 8) could be affecting such patterns. Importantly, an acoustic telemetry study of Galapagos shark movements within the East Tropical Pacific (n = 76) indicated C. galapagensis is a highly resident species, with most migrations occurring within a range of 0–50 km (M. Hoyos-Padilla pers. Comm). In addition, no movement was recorded between Revillagigedo and the Galapagos Islands, despite including intermediate locations (potential stepping-stones). Bonnethead sharks (Sphyrna tiburo) show similar asymmetry between neutral SNPs and mtDNA in the west Atlantic region, possibly due to sex-biased dispersal (Portnoy et al. 2015), and also exhibit strong population structure indicative of a species complex rather than a single species (Fields et al. 2016).
Galapagos sharks showed high overall genetic diversity (a total of 30 mtDNA haplotypes), and mtDNA haplotype and nucleotide diversities (h = 0.794 ± 0.014; π = 0.004 ± 0.000) within the range of other oceanic shark species (h = 0.595 to 0.959; and π = 0.0013 to 0.013; Duncan et al. 2006; Keeney and Heist 2006; Chabot and Allen 2009; Portnoy et al. 2010; Clarke et al. 2015; Camargo et al. 2016). Among all populations, Hawaii had the highest haplotype and nucleotide diversity. The presence of divergent haplotypes with many mutations in this population suggests multiple colonization events from neighboring locations, further supporting Hawaii as an important location linking east and west Pacific populations, likely via Mexico in the east and New Zealand in the west. Additional sampling, including intermediate South Pacific Islands, is needed to better determine structure and patterns of movement and colonization in the central-south Pacific. Compared to nuclear SNPs, Galapagos shark mtDNA exhibited a greater (neutral) or smaller (outlier) magnitude of divergence, respectively. This phenomenon identifies differences in patterns of gene flow based on neutral vs. statistical outlier nuclear markers, and based on mitochondrial (maternal only) vs. nuclear (biparental) markers (Karl et al. 2010; Daly-Engel et al. 2012; Chabot 2015). Notably, other globally distributed live-bearing shark species purportedly displayed evidence of female philopatry and male-mediated gene flow (Chapman et al. 2015), based on tagging and genetic (mitochondrial and putatively neutral microsatellite markers) data (e.g., Carcharhinus limbatus, Hueter et al. 2004; Keeney et al. 2005; C. plumbeus, Portnoy et al. 2010). Additionally, mark-recapture and genome-wide SNP data have detected philopatry in bonnethead sharks (Sphyrna tiburo, Driggers III et al. 2014; Portnoy et al. 2015). However, the absence of genetic differentiation (mtDNA, neutral and outlier SNPs) within the west Pacific region suggests female Galapagos sharks are not philopatric, indicating that evidence for “natal philopatry” needs to be carefully examined prior to asserting sex-biased dispersal.
Galapagos sharks are capable of crossing extensive swathes of open ocean, evident from their broad geographic distribution (Compagno 1984), and also by empirical observations of tagged individuals swimming across up to 2000 km of ocean between remote Pacific islands (e.g., French Frigate Shoals, Hawaii and Palmyra Atoll, C. Meyer unpublished data). Despite this inherent capacity for long-distance movements and hence gene flow, we found clear evidence of at least two (east Pacific and central-west Pacific) and possibly four (west Pacific, Mexico, Galapagos Islands and Hawaii) Galapagos shark populations in the Pacific. Reliance on shelf habitats for crucial aspects of their ecology may ultimately explain the population structure seen in this potentially wide-ranging shark. Galapagos shark diet is composed largely of reef-associated organisms (Wetherbee et al. 1996), while juvenile Galapagos sharks form aggregations over reefs (Compagno 1984), suggesting that both foraging and natal ecology are tied to shelf habitats. Results based on outlier SNPs support the biogeographic provinces defined by Glynn and Ault (2000), which separate mainland Ecuador, Costa Rica, the Galapagos Archipelago and Cocos Island (Equatorial province) from mainland Mexico and the Revillagigedo Islands (Northern province) based on reef building coral species. This is consistent with empirical tracking studies showing Galapagos sharks to be highly reliant on oceanic islands, with most individuals showing long-term (up to 9 years, C. Meyer unpublished data) fidelity to a single, or several closely adjacent, islands, and repeated use of the same insular shelf habitats (0–200 m depth) (Papastamatiou et al. 2015). Thus oceanic islands apparently serve as important connecting steps for Galapagos shark dispersal within the Pacific Ocean. The EPB limits connectivity and gene flow for a range of tropical marine taxa (Rocha et al. 2007; Van Cise et al. 2016) including corals (Pocillopora damicornis, Combosch et al. 2011 and Porites lobata, Baums et al. 2012), fish (Myripristis berndti, Craig et al. 2007; Doryrhamphus excisus and Cirrhitichthys oxycephalus, Lessios and Robertson 2006), and lobsters (Panulirus penicillatus, Chow et al. 2011), and is a plausible explanation for the existence of at least two genetically distinct Galapagos shark populations within the Pacific. Furthermore, studies using mitochondrial control region and microsatellites also support the EPB as an important barrier defining phylogeographic structure for other globally distributed species such as tope sharks (Galeorhinus galeus, Chabot 2015; Chabot and Allen 2009) and silky sharks (Carcharhinus falciformis, Clarke et al. 2015). Notwithstanding, our results highlight the presence of migrants between the central-west and east Pacific regions. This pattern has been previously detected in sea urchins (Lessios et al. 2003) and fish (Lessios and Robertson 2006). The study by Lessios and Robertson (2006) reported examples of fish populations occurring on the two sides of the EPB with an extreme level of divergence (Doryrhamphus excisus and Cirrhitichthys oxycephalus), as well as transpacific species with populations that have recently, or continue to exchange genes (Myripristis berndti, Stethojulis bandanensis and Zanclus cornutus), demonstrating that the EPB is not completely impassable, but rather a permeable barrier for several marine species including Galapagos sharks.
Our findings have important implications for management and conservation of Pacific Galapagos sharks. We found strong divergence between two Pacific populations, but also identified connections between the east Pacific (via Mexico) and the central Pacific (via Hawaii) to New Zealand in the southwest Pacific, indicating that effective management requires protecting both demographically (albeit restricted) interconnected stocks, and the associated intermediate locations. The intra-regional Galapagos shark population structure identified by analysis of statistical outlier loci may indicate regional adaptive variation that may hinder or prevent effective replacement of extirpated sub-populations (Clarke et al. 2015). However, we acknowledge processes other than local adaptation may be responsible for significant structure based on outlier loci (Bierne et al. 2013; Cruickshank and Hahn 2014) and further investigation is required to confirm their functional nature.
Our genetic effective population size (Ne) estimates suggest Pacific Galapagos sharks are currently genetically healthy overall, with the central-west Pacific Galapagos shark stock having almost five-fold more breeding individuals than the east Pacific population. However, Ne estimates for the two Galapagos Islands sub-populations were low (Pazmiño et al. 2017), emphasizing the need for appropriate, regional management. Overall, our study highlights the importance and potential impacts of using genome-wide genetic data for applied Galapagos shark conservation. Using a dramatically increased number of variable genetic markers compared to previous studies (van Herwerden et al. 2008) has lead to a precise estimation of diversity and population demographic parameters, including effective population size, relevant for the species conservation.
We highlight the importance of using both neutral and outlier markers to better understand population structure and genetic diversity of the species at a global scale to efficiently delimitate CU, and ultimately to achieve effective conservation and management in the short and long term. Our understanding of population structure may be enhanced by studying additional material from under- (Mexico and Walters Shoals) and unsampled (e.g., the Indian and Atlantic Oceans) intermediate locations across the Galapagos shark distribution. In order to enhance long-term conservation efforts of Galapagos sharks we further recommend evaluating the stability of identified regional populations and regular monitoring of each identified stock in order to document temporal demographic changes within stocks. Informing for improved conservation management of this near threatened shark species across its Pacific Ocean distribution was relatively straightforward and necessary, but implementing such Pacific-wide management may be challenging.
Data accesibility
All mtDNA sequences were uploaded to GenBank [accession numbers MG241666-MG241897]. The raw SNPs data set were deposited on Dryad (https://doi.org/10.5061/dryad.h2kh3).
References
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11:697–709
Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G (2008) LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinforma 9:323
Balloux F, Brunner H, Lugon-Moulin N, Hausser J, Goudet J (2000) Microsatellites can be misleading: an empirical and simulation study. Evolution 54:1414–1422
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Baums IB, Boulay JN, Polato NR, Hellberg ME (2012) No gene flow across the eastern Pacific Barrier in the reef-building coral Porites lobata. Mol Ecol 21:5418–5433
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (1996–2004) GENETIX 4.05, logicielsous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5171 (http://www.genetix.univ-montp2.fr/genetix/intro.htm)
Benestan L, Quinn BK, Maaroufi H, Laporte M, Clark FK, Greenwood SJ et al. (2016) Seascape genomics provides evidence for thermal adaptation and current-mediated population structure in American lobster (Homarus americanus). Mol Ecol 25:5073–5092
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57:289–300
Bennett M., Gordon I, Kyne PM (2003). SSG Australia & Oceania regional rorkshop, March 2003. Carcharhinus galapagensis. The IUCN Red List of Threatened Species. Version 2014.1. http://www.iucnredlist.org
Bierne N, Roze D, Welch JJ (2013) Pervasive selection or is it…? Why are FST outliers sometimes so frequent? Mol Ecol 22:2061–2064
Blower DC, Riginos CR, Ovenden JR (2016) NeOGen: a tool to predict genetic effective population size(Ne) for low-fecundity, slowly-maturing, iteroparous species, and to assist empirical Ne study design. (http://www.molecularfisherieslaboratory.com.au/neogen-software/)
Briggs JC (1974) Marine zoogeography. McGraw-Hill, New York, NY, USA
Camargo SM, Coelho R, Chapman D, Howey-Jordan L, Brooks EJ, Fernando D et al. (2016) Structure and genetic variability of the oceanic whitetip shark, Carcharhinus longimanus, determined using mitochondrial DNA. PLoS One 11(5):1–11
Candy JR, Campbell NR, Grinnell MH, Beacham TD, Larson WA, Narum SR (2015) Population differentiation determined from putative neutral and divergent adaptive genetic markers in Eulachon (Thaleichthys pacificus, Osmeridae), an anadromous Pacific smelt. Mol Ecol Resour 15:1421–1434
Chabot CL (2015) Microsatellite loci confirm a lack of population connectivity among globally distributed populations of the tope shark Galeorhinus galeus (Triakidae). J Fish Biol 87:371–385
Chabot CL, Allen LG (2009) Global population structure of the tope (Galeorhinus galeus) inferred by mitochondrial control region sequence data. Mol Ecol 18:545–552
Chapman DD, Feldheim KA, Papastamatiou YP, Hueter RE (2015) There and back again: a review of residency and return migrations in sharks, with implications for population structure and management. Annu Rev Mar Sci 7:547–570
Chow S, Jeffs A, Miyake Y, Konishi K, Okasaki M et al. (2011) Genetic Isolation between the Western and eastern Pacific populations of pronghorn spiny lobster Panulirus penicillatus. PLoS One 6:e29280
Clarke CR, Karl SA, Horn RL, Bernard AM, Lea JS, Hazin FH et al. (2015) Global mitochondrial DNA phylogeography and population structure of the silky shark, Carcharhinus falciformis. Mar Biol 162:945–955
Combosch DJ, Vollmer SV (2011) Population genetics of an ecosystem-defining reef coral Pocillopora damicornis in the tropical eastern Pacific. PLoS One 6:e21200
Compagno LJ (1984) FAO species catalogue, Sharks of the world: an annotated and illustrated catalogue of shark species known to date. Part 2. Carcharhiniformes. FAO Fish Synop 125:251–655
Craig MT, Eble JA, Bowen BW, Robertson DR (2007) High genetic connectivity across the Indian and Pacific Oceans in the reef fish Myripristis berndti (Holocentridae). Mar Ecol Prog Ser 334:245–254
Cruickshank TE, Hahn MW (2014) Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol Ecol 23:3133–3157
Daly-Engel TS, Seraphin KD, Holland KN, Coffey JP, Nance HA, Toonen RJ et al. (2012) Global phylogeography with mixed-marker analysis reveals male-mediated dispersal in the endangered scalloped hammerhead shark (Sphyrna lewini). PLoS One 7:e29986
Dawson MN, Louie KD, Barlow M, Jacobs DK, Swift CC (2002) Comparative phylogeography of sympatric sister species, Clevelandia ios and Eucyclogobius newberryi (Teleostei, Gobiidae), across the California transition zone. Mol Ecol 11:1065–1075
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimatorv2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Driggers III WB, Frazier BS, Adams DH, Ulrich GF, Jones CM, Hoffmayer ER et al. (2014) Site fidelity of migratory bonnethead sharks Sphyrna tiburo (L. 1758) to specific estuaries in South Carolina, USA. J Exp Mar Biol Ecol 459:61–69
Dulvy NK, Fowler SL, Musick JA, Cavanagh RD, Kyne PM, Harrison LR et al. (2014) Extinction risk and conservation of the world’s sharks and rays. eLife 3:e00590
Duncan KM, Martin AP, Bowen BW, De Couet HG (2006) Global phylogeography of the scalloped hammerhead shark (Sphyrna lewini). Mol Ecol 15:2239–2251
Earl DA, vonHoldt BM (2011) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Fields AT, Feldheim KA, Gelsleichter J, Pfoertner C, Chapman DD (2016) Population structure and cryptic speciation in bonnethead sharks Sphyrna tiburo in the south-eastern U.S.A. and Caribbean. J Fish Biol 89:2219–2233
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol 27:489–496
Gaither MR, Bowen BW, Rocha LA, Briggs JC (2016) Fishes that rule the world: circumtropical distributions revisited. Fish Fish 17:664–679
Garner BA, Hand BK, Amish SJ, Bernatchez L, Foster JT, Miller KM et al. (2016) Genomics in conservation: case studies and bridging the gap between data and application. Trends Ecol Evol 31:81–83
de Guia APO, Saitoh T (2007) The gap between the concept and definitions in the evolutionarily significant unit: the need to integrate neutral genetic variation and adaptive variation. Ecol Res 22:604–612
Glynn PW, Ault JS (2000) A biogeographic analysis and review of the far eastern Pacific coral reef region. Coral Reefs 19:1–23
Hamon P, Grover CE, Davis AP, Rakotomalala J-J, Raharimalala NE, Albert VA et al. (2017) Genotyping-by-sequencing provides the first well-resolved phylogeny for coffee (Coffea) and insights into the evolution of caffeine content in its species: GBS coffee phylogeny and the evolution of caffeine content. Mol Phylogenet Evol 109:351–361
Hoenig JM, Gruber SH (1990) Life-history patterns in the elasmobranchs: implications for fisheries management. In: Pratt HL Jr, Gruber SH, Taniuchi T (eds) Elasmobranch as living resources: advances in the biology, ecology, systematics, and the status of the fisheries. NOAA/National Marine Fisheries Service, Hawaii, USA, 1–16
Hueter RE, Heupel MR, Heist EJ, Keeney DB (2004) Evidence of philopatry in sharks and implications for the management of shark fisheries. J North Atl Fish Sci 35:239–247
Jombart T, Ahmed I (2011) adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Karl SA, Castro ALF, Lopez JA, Charvet P, Burgess GH (2010) Phylogeography and conservation of the bull shark (Carcharhinus leucas) inferred from mitochondrial and microsatellite DNA. Conserv Genet 12:371–382
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S et al. (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Keeney DB, Heist EJ (2006) Worldwide phylogeography of the blacktip shark (Carcharhinus limbatus) inferred from mitochondrial DNA reveals isolation of western Atlantic populations coupled with recent Pacific dispersal. Mol Ecol 15:3669–3679
Keeney DB, Heupel M, Hueter RE, Heist EJ (2003) Genetic heterogeneity among blacktip shark, Carcharhinus limbatus, continental nurseries along the U.S. Atlantic and Gulf of Mexico. Mar Biol 143:1039–1046
Keeney DB, Heupel MR, Hueter RE, Heist EJ (2005) Microsatellite and mitochondrial DNA analyses of the genetic structure of blacktip shark (Carcharhinus limbatus) nurseries in the northwestern Atlantic, Gulf of Mexico, and Caribbean Sea. Mol Ecol 14:1911–1923
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H et al. (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. Pompanon F, Bonin A (eds) Data production and analysis in population genomics: methods and protocols. Methods Mol Biol, vol 888. Springer, New York, NY, p 67–89
Kohler NE, Casey JG, Turner PA (1998) NMFS cooperative shark tagging program, 1962–1993: an atlas of shark tag and recapture data. Mar Fish Rev 60:1–87
Lanfear R, Calcott B, Ho SYW, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701
Larson WA, Seeb LW, Everett MV, Waples RK, Templin WD, Seeb JE (2014) Genotyping by sequencing resolves shallow population structure to inform conservation of Chinook salmon (Oncorhynchus tshawytscha). Evol Appl 7:355–369
Lee T-H, Guo H, Wang X, Kim C, Paterson AH (2014) SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom 15:162
Lessios HA, Kane J, Robertson DR (2003) Phylogeography of the pantropical sea urchin Tripneustes: Contrasting patterns of population structure between oceans. Evolution 57:2026–2036
Lessios H, Robertson D (2006) Crossing the impassable: genetic connections in 20 reef fishes across the eastern Pacific barrier. Proc R Soc B Biol Sci 273:2201–2208
Lischer HEL, Excoffier L (2012) PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28:298–299
Lowe CG, Wetherbee BM, Meyer CG (2006) Using acoustic telemetry monitoring techniques to quantify movement patterns and site fidelity of sharks and giant trevally around French Frigate Shoals and Midway Atoll. Atoll Res Bull 543:281–303
Luu K, Bazin E, Blum MGB (2016) pcadapt: an R package to perform genome scans for selection based on principal component analysis. Mol Ecol Resour 17:67–77
Meirmans PG, Van Tienderen PH (2004) Genotype and genodive: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4:792–794
Meyer CG, Papastamatiou YP, Holland KN (2010) A multiple instrument approach to quantifying the movement patterns and habitat use of tiger (Galeocerdo cuvier) and Galapagos sharks (Carcharhinus galapagensis) at French Frigate Shoals, Hawaii. Mar Biol 157:1857–1868
Myers RA, Worm B (2003) Rapid worldwide depletion of predatory fish communities. Nature 423:280–3
Narum SR, Buerkle CA, Davey JW, Miller MR, Hohenlohe PA (2013) Genotyping-by-sequencing in ecological and conservation genomics. Mol Ecol 22:2841–2847
Neuditschko M, Khatkar MS, Raadsma HW (2012) NetView: A High-Definition Network-Visualization Approach to Detect Fine-Scale Population Structures from Genome-Wide Patterns of Variation. PLoS One 7:e48375
Ouborg NJ, Pertoldi C, Loeschcke V, Bijlsma RK, Hedrick PW (2010) Conservation genetics in transition to conservation genomics. Trends Genet 26:177–187
Pardini AT, Jones CS, Noble LR, Kreiser B, Malcolm H, Bruce BD et al. (2001) Sex-biased dispersal of great white sharks. Nature 412:139–140
Papastamatiou YP, Meyer CG, Kosaki RK, Wallsgrove NJ, Popp BN (2015) Movements and foraging of predators associated with mesophotic coral reefs and their potential for linking ecological habitats. Mar Ecol Prog Ser 521:155–170
Pazmiño DA, Maes GE, Simpfendorfer CA, van Herwerden L (2017) Genome-wide SNPs reveal small scale conservation units in the highly vagile Galápagos shark (Carcharhinus galapagensis). Conserv Genet 18:1151–1163
Polidoro BA, Brooks T, Carpenter KE, Edgar GJ, Henderson S, Sanciangco J et al. (2012) Patterns of extinction risk and threat for marine vertebrates and habitat-forming species in the tropical eastern Pacific. Mar Ecol Prog Ser 448:93–104
Portnoy DS, Gold JR (2012) Evidence of multiple vicariance in a marine suture-zone in the Gulf of Mexico. J Biogeogr 39:1499–1507
Portnoy DS, Heist EJ (2012) Molecular markers: progress and prospects for understanding reproductive ecology in elasmobranchs. J Fish Biol 80:1120–1140
Portnoy DS, Hollenbeck CM, Belcher CN, Driggers WB, Frazier BS, Gelsleichter J et al. (2014) Contemporary population structure and post-glacial genetic demography in a migratory marine species, the blacknose shark, Carcharhinus acronotus. Mol Ecol 23:5480–5495
Portnoy DS, Mcdowell JR, Heist EJ, Musick JA, Graves JE (2010) World phylogeography and male-mediated gene flow in the sandbar shark, Carcharhinus plumbeus. Mol Ecol 19:1994–2010
Portnoy DS, Puritz JB, Hollenbeck CM, Gelsleichter J, Chapman D, Gold JR (2015) Selection and sex-biased dispersal in a coastal shark: the influence of philopatry on adaptive variation. Mol Ecol 24:5877–5885
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pritchard JK, Wen W (2003) Documentation for structure software: Version 2. http://pritch.bsd.uchicago.edu.elibrary.jcu.edu.au
Pujolar JM, Jacobsen MW, Als TD, Frydenberg J, Munch K, Jónsson B et al. (2014) Genome-wide single-generation signatures of local selection in the panmictic European eel. Mol Ecol 23:2514–2528
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. (2007) PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Rambaut A, Suchard MA, Xie D, Drummond A (2014) Tracer v1.6. http://beast.community/tracer
R Development Core Team (2008) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna http://www.R-project.org/
Rocha LA, Craig MT, Bowen BW (2007) Phylogeography and the conservation of coral reef fishes. Coral Reefs 26:501–512
Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Sansaloni CP, Petroli CD, Carling J, Hudson CJ, Steane DA, Myburg AA et al. (2010) A high-density diversity arrays technology (DArT) microarray for genome-wide genotyping in Eucalyptus. Plant Methods 6:16
Savolainen O, Lascoux M, Merilä J (2013) Ecological genomics of local adaptation. Nat Rev Genet Lond 14:807–20
Selkoe KA, Henzler CM, Gaines SD (2008) Seascape genetics and the spatial ecology of marine populations. Fish Fish 9:363–377
Shafer ABA, Wolf JBW, Alves PC, Bergström L, Bruford MW, Brännström I et al. (2015) Genomics and the challenging translation into conservation practice. Trends Ecol Evol 30:78–87
Shivji MS (2010) DNA forensic applications in shark management and conservation. In: Carrier JC, Musick JA, Heithaus MR (eds) Sharks and their relatives II, Marine Biology. CRC Press, Florida, FL, 593–610
Slatkin M, Voelm L (1991) F(st) in a Hierarchical Island Model. Genetics 127:627
Snodgrass RE, Heller E (1905) Shore fishes of the Revillagigedo, Clipperton, Cocos and Galapagos islands. Proceedings of the Washington Academy of Sciences, Washington, DC, USA
Steane DA, Potts BM, McLean E, Prober SM, Stock WD, Vaillancourt RE et al. (2014) Genome-wide scans detect adaptation to aridity in a widespread forest tree species. Mol Ecol 23:2500–2513
Steinig EJ, Neuditschko M, Khatkar MS, Raadsma HW, Zenger KR (2016) netview p: a network visualization tool to unravel complex population structure using genome-wide SNPs. Mol Ecol Resour 16:216–227
Storey JD with contributions from Bass AJ, Dabney A and Robinson D (2015) qvalue: Q-value estimation for false discovery rate control. R package version 2.4.2 http://github.com/jdstorey/qvalue
Sunnucks P, Hales DF (1996) Numerous transposed sequences of mitochondrial cytochrome oxidase I-II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol Biol Evol 13:510–524
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Van Cise AM, Morin PA, Baird RW, Lang AR, Robertson KM, Chivers SJ et al. (2016) Redrawing the map: mtDNA provides new insight into the distribution and diversity of short-finned pilot whales in the Pacific Ocean. Mar Mammal Sci 32:1177–1199
van Herwerden L, Almojil D, Choat H (2008) Population genetic structure of Australian Galápagos reef sharks Carcharhinus galapagensis at Elizabeth and Middleton Reefs Marine National Nature Reserve and Lord Howe Island Marine Park. Final report to the Department of the Environment, water, heritage and the arts. Queensland. Molecular Ecology and Evolution Laboratory, School of Marine and Tropical Biology, James Cook University
Vincent B, Dionne M, Kent MP, Lien S, Bernatchez L (2013) Landscape genomics in Atlantic Salmon (Salmo salar): searching for gene–environment interactions driving local adaptation. Evolution 67:3469–3487
Waples RS (1998) Separating the wheat from the chaff: patterns of genetic differentiation in high gene flow species. J Hered 89:438–450
Wetherbee BM, Crow GL, Lowe CG (1996) Biology of the Galapagos shark, Carcharhinus galapagensis, in Hawai’i. Environ Biol Fishes 45:299–310
Willette D, Allendorf F, Barber P, Barshis D, Carpenter K, Crandall E et al. (2014) So, you want to use next-generation sequencing in marine systems? Insight from the Pan-Pacific Advanced Studies Institute. Bull Mar Sci 90:79–122
Worm B, Davis B, Kettemer L, Ward-Paige CA, Chapman D, Heithaus MR et al. (2013) Global catches, exploitation rates, and rebuilding options for sharks. Mar Policy 40:194–204
Acknowledgements
We thank the Ecuadorian Government, who financially supported this study through a National Secretary of Higher Education, Science and Technology (SENESCYT) scholarship; The Galápagos National Park for permission to collect and export samples under permit 065-2013PNG and 014-2014PNG; The Molecular Ecology and Evolutionary Laboratory (MEEL) staff for laboratory, analytical, and logistic support. We would also like to acknowledge DST/NRF African Coelacanth Ecosystem Programme (ACEP) for financial and logistical support; the New Zealand Department of Conservation and Pew Environment Group for the funding provided and the permit approval for sampling at the Kermadec Islands Marine Reserve, and funding provided from the Data Deficient Species Fund. Finally, we thank and acknowledge the effort of Mark Scott who collected samples from Norfolk Island.
Author contribution
D.A.P., L.vH., G.E.M., C.A.S., and M.E.G. designed the research project. D.A.P and L.vH. conducted fieldwork and collected samples from the Galápagos Islands for genetic analyses. M.H., C.A.J.D., C.G.M., S.E.K., collected and provided samples from the remaining locations. DAP had primary responsibility for conducting laboratory work, data analysis, and writing of the manuscript. All authors have reviewed and contributed to the current version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Pazmiño, D., Maes, G.E., Green, M.E. et al. Strong trans-Pacific break and local conservation units in the Galapagos shark (Carcharhinus galapagensis) revealed by genome-wide cytonuclear markers. Heredity 120, 407–421 (2018). https://doi.org/10.1038/s41437-017-0025-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-017-0025-2
This article is cited by
-
What Darwin could not see: island formation and historical sea levels shape genetic divergence and island biogeography in a coastal marine species
Heredity (2023)
-
Shallow seamounts represent speciation islands for circumglobal yellowtail Seriola lalandi
Scientific Reports (2021)
-
Sex assignment in a non-model organism in the absence of field records using Diversity Arrays Technology (DArT) data
Conservation Genetics Resources (2021)
-
Contrasting patterns of genetic connectivity in brooding and spawning corals across a remote atoll system in northwest Australia
Coral Reefs (2020)
-
Contrasting global, regional and local patterns of genetic structure in gray reef shark populations from the Indo-Pacific region
Scientific Reports (2019)
