
Abstract
The progressive accumulation of dysfunctional mitochondria is implicated in aging and in common diseases of the elderly. To oppose this occurrence, organisms employ a variety of strategies, including the selective degradation of oxidatively damaged and misfolded mitochondrial proteins. Genetic studies in yeast indicate that the ATPase Associated with diverse cellular Activities (AAA+) family of mitochondrial proteases account for a substantial fraction of this protein degradation, but their metazoan counterparts have been little studied, despite the fact that mutations in the genes encoding these proteases cause a variety of human diseases. To begin to explore the biological roles of the metazoan mitochondrial AAA+ protease family, we have created a CRISPR/Cas9 allele of the Drosophila homolog of SPG7, which encodes an inner membrane-localized AAA+ protease known as paraplegin. Drosophila SPG7 mutants exhibited shortened lifespan, progressive locomotor defects, sensitivity to chemical and environmental stress, and muscular and neuronal degeneration. Ultrastructural examination of photoreceptor neurons indicated that the neurodegenerative phenotype of SPG7 mutants initiates at the synaptic terminal. A variety of mitochondrial defects accompanied the degenerative phenotypes of SPG7 mutants, including altered axonal transport of mitochondria, accumulation of electron-dense material in the matrix of flight muscle mitochondria, reduced activities of respiratory chain complexes I and II, and severely swollen and dysmorphic mitochondria in the synaptic terminals of photoreceptors. Drosophila SPG7 mutants recapitulate key features of human diseases caused by mutations in SPG7, and thus provide a foundation for the identification of Drosophila paraplegin substrates and strategies that could be used to ameliorate the symptoms of these diseases.
Similar content being viewed by others


Introduction
Mitochondria have essential cellular roles in ATP synthesis, calcium homeostasis, and metabolism, but these activities come at a cost. In particular, the production of ATP through oxidative phosphorylation leads to the generation of reactive oxygen species (ROS) that can damage mitochondrial proteins, lipids, and DNA1,2. Moreover, the respiratory chain complexes responsible for the production of ATP require the coordinated expression of mitochondrial and nuclear encoded subunits, and an imbalance in the stoichiometry of these subunits can result in protein misfolding and aggregation. The progressive accumulation of oxidatively damaged and misfolded mitochondrial proteins is strongly implicated in aging and common diseases of the elderly, including neurodegenerative diseases and cancer1,2,3. To oppose the accumulation of dysfunctional mitochondria, metazoans employ a variety of quality control strategies, including the selective degradation of dysfunctional mitochondria or their damaged components4,5. Although the recent identification of the mitophagy-promoting factors PTEN-induced putative kinase 1 (PINK1) and Parkin has led to rapid advancement in our understanding of mitochondrial degradation, comparatively less is known of the mechanisms by which damaged mitochondrial components are detected and degraded6,7,8.
Previous work suggests that the AAA+ family of mitochondrial proteases have a major role in mitochondrial quality control by degrading oxidatively damaged and misfolded proteins9,10. There are four major mitochondrial AAA+ proteases in metazoans including the inner membrane-localized proteases i-AAA and m-AAA, and the matrix-localized proteases LON and Clp9. The catalytic domains of the m-AAA and i-AAA proteases face the matrix and intermembrane space, respectively10. All four of these proteases form multimeric protein complexes that use energy derived from ATP hydrolysis to unfold and transport substrates to an internal proteolytic domain for degradation. Lon and i-AAA assemble as homo-oligomeric complexes, whereas Clp is composed of proteolytic (ClpP) and ATPase (ClpX) subunits. The m-AAA protease comes in two forms: hetero-oligomeric complexes of the paraplegin protein and the ATPase family gene 3-like 2 (AFG3L2) protein, and homo-oligomeric complexes of AFG3L29,10. The importance of these proteases is illustrated by the fact that mutations in the genes encoding them cause a variety of human diseases, including hereditary spastic paraplegia (HSP), spinocerebellar ataxia type 28, and perrault syndrome9,10,11,12,13. However, the substrates of these proteases, and the mechanisms by which mutations in the genes encoding them cause disease, are largely unknown.
To begin to explore the biological roles of the AAA+ mitochondrial protease family, we have created a CRISPR/Cas9 deletion allele of one of the Drosophila m-AAA family members, SPG7, which encodes a homolog of paraplegin. We found that SPG7 mutants display shortened lifespan, locomotor impairment, sensitivity to stressors, and degeneration of the indirect flight muscle (IFM) and the nervous system. These phenotypes were accompanied by mitochondrial trafficking defects in the nervous system, an accumulation of swollen mitochondria containing electron-dense aggregates, and significantly reduced activity of respiratory chain complexes I and II. Transmission electron microscopic analysis of photoreceptor neurons revealed severe architectural alterations restricted to the synaptic terminals, thus recapitulating the synaptopathy and axonopathy associated with HSP10. Our work provides a foundation to apply the powerful genetic tools of Drosophila to study the mechanisms underlying the neurological diseases associated with SPG7 mutations in humans10,14,15,16.
Results
Identification of a Drosophila paraplegin homolog
To identify a Drosophila paraplegin homolog, we used the human paraplegin protein sequence to conduct a Basic Local Alignment Search Tool (BLAST) search, and identified three Drosophila genes encoding proteins with 43–58% identity to human paraplegin. Two of these genes, CG6512 and CG3499, encode the previously characterized mitochondrial AAA+ protease family members Afg3l2 and dyme1l, respectively10,17. The third gene, CG2658, encodes a previously uncharacterized homolog of human paraplegin with 58% identity and 75% similarity at the protein level (Supplementary Figure S1A). Data from the Drosophila modENCODE and FlyAtlas data repositories indicate that the CG2658 gene is ubiquitously expressed in all tissues throughout development. The Pfam motif prediction algorithm predicts that the polypeptide encoded by CG2658 contains an AAA domain (amino acid residues 377–513) and an M41 metallopeptidase domain (amino acid residues 575–785) acting as a proteolytic center (Supplemental Figure S1B). The MitoProt algorithm predicts that the N-terminal region of paraplegin contains a mitochondrial targeting sequence, and a proteomic proximity ligation study detected peptides corresponding to CG2658 in the mitochondrial matrix18. We generated an antiserum to the CG2658 gene product and used this antiserum to confirm the mitochondrial localization of this protein (Fig. 1a–c). Given these findings, and the fact that CG2658 encodes the most closely related protein to paraplegin, we propose that this gene represents the Drosophila ortholog of SPG7.

a Confocal images of indirect flight muscles using anti-paraplegin (upper panel) and anti-cytochrome c (middle panel) antisera. The merged image (lower panel) indicates that paraplegin and cytochrome c colocalize. Scale bar is 1 μm. b Mitochondrial and cytosolic fractions were isolated by differential centrifugation from adult flies and immunoblotted using antisera against paraplegin, Ndufs3 (mitochondria), Actin (cytosol), and Calnexin (endoplasmic reticulum) showing that paraplegin localizes to the mitochondrial fraction. c Purified mitochondrial and endoplasmic reticulum (ER) fractions produced from sucrose gradient sedimentation of a crude mitochondrial fraction were subjected to western blotting using antisera to paraplegin, Ndufs3 (mitochondria) and Calnexin (endoplasmic reticulum). Results indicate that paraplegin localizes to mitochondria, not ER. d Western blot analysis of w1118 and SPG7del mutants using an anti-paraplegin antiserum and an antiserum to actin as a loading control. Note that the band corresponding in size to the mature paraplegin protein is absent in SPG7del mutants
An SPG7 null mutant exhibits shortened lifespan and behavioral abnormalities
To explore the biological role of SPG7, we used the CRISPR/CAS9 system to replace the entire SPG7 coding sequence with the DsRed marker (Supplementary Figure S2)19. Flies with DsRed expression were subjected to whole-genome sequencing to verify the successful creation of an SPG7 deletion (SPG7del), and males hemizygous for this deletion were subjected to western blot analysis to confirm that SPG7del represents a null allele (Fig. 1d).These flies were then backcrossed six times to an isogenic w1118 stock to allow direct phenotypic comparison with a control stock.
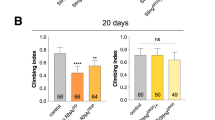
SPG7del mutants were viable and fertile, with no detectable anatomical or behavioral phenotypes at a young age. However, they were significantly shorter-lived than wild-type flies, with a median survival of 34 days compared to 73 days for control flies (Fig. 2a). Because premature aging in Drosophila is frequently associated with defective locomotor performance, we tested whether the shortened lifespan of SPG7del mutants was accompanied by climbing or flight defects. To test climbing ability, we tapped flies to the bottom of a vial and measured the distance climbed in a given interval of time. Flight ability was measured by releasing flies into the top of a graduated cylinder and noting where flies alight20. Strong fliers land near the top of the cylinder, whereas weaker fliers land lower in the cylinder. The climbing assay detected a severe climbing defect in SPG7del mutants at 4 weeks of age (Fig. 2b). SPG7del mutants also displayed a progressive biphasic flight defect: at 4 weeks of age, most flies landed near the top of the cylinder, but a small but significant fraction fell all the way to the bottom of the cylinder (Fig. 2c). The lifespan and locomotor phenotypes of SPG7del mutants were rescued by autosomes containing short duplications spanning the SPG7 gene (Supplemental Figure S3A), providing evidence that these phenotypes are a consequence of the SPG7 deletion.

a Kaplan–Meier survival curves of SPG7del flies (SPG7del, N = 671, 50% survival 34 days) and controls (w1118, N = 752, 50% survival 74 days). Significance was determined using a log-rank test (p < 0.0005). b SPG7del mutants exhibit an age-dependent climbing defect. Error bars represent s.e.m. (N = 200 for each genotype, ****p < 0.0005 by Student’s t-test). c SPG7del mutants exhibit a significant increase in the fraction of flies that are incapable of flight. The histograms represent the fraction of non-fliers of the indicated genotypes, and error bars represent the s.e.m. (n = 6 independent groups of 10–15 animals, *p < 0.05 from Student’s t-test). d SPG7del mutants are sensitive to heat-induced paralysis. The histograms depict the percentage of flies sensitive to heat shock. Error bars represent s.e.m. (N = 55 for w1118 and N = 83 for SPG7del flies, ****p < 0.0005 by Mann–Whitney U-test). e SPG7del mutants exhibit mechanical stress-induced paralysis. Recovery time from mechanical stress (bang sensitivity) is shown for flies of the indicated genotype and age. Error bars represent s.e.m. (N = 91 for each genotype, ****p < 0.0005 from Student’s t-test). f SPG7del flies are more sensitive to exposure to the oxidative stress-inducing agent paraquat (n = 200, ****p < 0.0005 by log-rank test)
Many Drosophila mutants that bear defects in genes encoding mitochondrial proteins exhibit stress sensitivity21. Thus, we tested the sensitivity of SPG7del mutants to three commonly used stressors: high temperature, mechanical stimulation (bang sensitivity), and exposure to an oxidative stress agent. The first two of these stressors can lead to paralysis in sensitive backgrounds, whereas the third can exacerbate the shortened lifespan of a mitochondrial mutant17. Exposing SPG7del mutants to 39 °C for 6 min resulted in a progressive age-dependent heat intolerance compared to wild-type controls (Fig. 2d). The SPG7del mutants also exhibited a progressive and severe bang-sensitive phenotype (Fig. 2e), and 4-week-old SPG7del mutants displayed seizure-like behavior with regular rhythmic wing and leg jerking even in the absence of mechanical stress (data not shown). Finally, SPG7del flies exhibited enhanced sensitivity to the oxidative stress-inducing agent paraquat (Fig. 2f). Together, these findings establish that SPG7del mutants are sensitive to a wide range of environmental stresses.
SPG7 del mutants exhibit muscular and neuronal degeneration
Mutations in human SPG7 cause the neurological disorder hereditary spastic paraplegia, a distal axonopathy characterized by the loss of corticospinal motor neurons, and deletion of the SPG7 gene in mice also results in axonal and synaptic alterations22,23. Thus, we tested whether the behavioral phenotypes of SPG7del mutants arise from similar neuronal defects. To evaluate whether SPG7del influences synapse formation or maintenance, we quantified the number of type Ib synaptic boutons in larval body wall muscle 4 segments A2 and A3 using presynaptic (anti-HRP) and postsynaptic (anti-discs large 1) markers24,25. We also used the motor neuron-specific driver D42-Gal4 to label motor neurons in adult fly legs with GFP, and analyzed axonal morphology in young (1 week) and old animals (4 week) using confocal microscopy26. Finally, we examined the integrity of photoreceptor neurons in 4-week-old animals using transmission electron microscopy (TEM)27. We did not detect alteration in the number of synaptic boutons in SPG7del third instar larvae (Fig. 3a), axonal loss in motor neurons in the adult leg (Fig. 3b), or reduction in the number of photoreceptor neurons in the visual system (Fig. 3c, d). However, in contrast to the well-organized photoreceptor synaptic terminals in wild-type flies, the synaptic terminals of SPG7del mutants were disorganized and showed accumulation of swollen and morphologically abnormal mitochondria (Fig. 3e, f)27,28. Together, these studies indicate that paraplegin influences synapse integrity in a cell type-dependent manner.

a Immunostaining of the neuromuscular junction from muscle 4 of third instar larvae of the indicated genotypes using anti-discs large 1 (upper panel) and anti-HRP (lower panel) antisera as postsynaptic and presynaptic markers, respectively. Right panel shows quantification of bouton numbers in the two genotypes (n = 10). Significance was determined using Student’s t-test. b Motor neurons were visualized in the femur region (highlighted in yellow box) from adult fly legs using the neuronal marker mCD8-GFP expressed using the D42-GAL4 driver from 1-week-old and 4-week-old w1118 control and SPG7del mutants (n = 20). Scale bar is 10 μm. c Representative transmission electron micrograph (TEM) images of photoreceptors from 4-week-old w1118 controls and SPG7del mutants. A single ommatidium is shown in d from w1118 control and SPG7del mutants. e TEM micrograph of lamina cartridge from 4-week-old w1118 control. f TEM micrographs of single lamina cartridges from 4-week-old SPG7del mutant. Note the presence of swollen mitochondria with aberrant cristae structure (red asterisks), and the disorganized appearance of photoreceptor terminals in SPG7del mutants, relative to controls. Scale bar for c 10 μm, for d 2 μm and for e, f 1 μm
Although the alterations in photoreceptor terminals demonstrate that paraplegin is critical for synaptic integrity, a synaptic defect confined to the visual system cannot explain the behavioral phenotypes of SPG7del mutants. To further explore the origin of these deficits, we performed histological analysis of paraffin-embedded brain and thorax sections. Brain sections from old SPG7del mutants revealed a significantly increased number of vacuoles relative to age-matched controls (Fig. 4a, b). The vacuoles were present throughout the brain, more frequently in the central neuropil region than in the optic lobes, indicating that loss of paraplegin causes progressive deterioration of brain tissue in Drosophila (Fig. 4a). Transverse thoracic sections also revealed a progressive loss of the integrity of IFMs in SPG7del mutants, consistent with the essential metabolic role of mitochondria in flight muscle maintenance (Fig. 4c). However, this phenotype was only partially penetrant (data not shown), and we therefore used a second assay to verify the findings. Phalloidin staining of indirect flight muscles confirmed the muscle degeneration phenotype of old SPG7del mutants (Fig. 4d). Together, our findings demonstrate an essential role for SPG7del in neuronal and IFM integrity.

a Brain integrity of w1118 control and SPG7del mutants was analyzed by hematoxylin- and eosin-stained, paraffin-embedded brain sections at 1 week and 4 weeks of age. The black arrowhead indicates a vacuole in SPG7del mutants. Scale bar is 100 μm. b Quantification of brain vacuole number (upper panel) and area (bottom panel) in 4-week-old flies of the indicated genotype (n = 6, **p < 0.005 from Student’s t-test). c Representative images of thoracic cross sections from w1118 control and SPG7del mutants at 1 week and 4 weeks age. Degenerating muscle tissues are indicated by the black arrowhead. Scale bar is 50 μm. d Confocal imaging of indirect flight muscles from 4-week-old w1118 control and SPG7del mutants stained using the actin-binding compound phalloidin. Degenerating muscle fibers are highlighted by white arrowhead. Scale bar is 10 μm
SPG7 del mutants accumulate morphologically and functionally abnormal mitochondria
The mitochondrial morphological defects in the photoreceptor terminals of SPG7del mutants, and the known role of paraplegin in mitochondrial protein degradation, suggest that the neuronal and muscular degeneration phenotypes of SPG7del mutants could derive from mitochondrial dysfunction. To explore this matter more fully, we performed several additional experiments. First, we investigated the ultrastructure of mitochondria in the IFMs using TEM in cross sections of thoraces6. Strikingly, IFMs from old SPG7del mutants (4 week) showed large, swollen, and loosely packed mitochondria with disorganized cristae (Fig. 5). These abnormal mitochondria frequently contained electron-dense material that appeared to reside in the matrix (Fig. 5b, c). This finding is consistent with previously published reports on other AAA+ protease mutants17,29,30.

a–c Representative TEM images from indirect flight muscles of 4-week-old w1118 control (a) and SPG7del (b, c) mutants. Note the presence of aberrant cristae and dense inclusions in mitochondria from SPG7del mutants relative to controls (arrowheads in c). Scale bar is 2 μm in a and b, and 0.5 μm in c
To test whether the mitochondrial morphological alterations detected in our TEM studies were also associated with compromised respiratory chain (RC) function, we used established assays to quantify RC activity31. RC complex activity was normal in young (1 week) SPG7del mutants (Fig. 6a, left panel), but we observed significant decreases in the activities of complexes I and II in 4-week-old SPG7del mutants relative to controls. However, the activities of complexes III and IV remained unaffected even in old SPG7del mutants (Fig. 6a, right panel). To explore the mechanisms underlying the reduced complex I and II activity in old SPG7del mutants, we analyzed the abundance of these complexes, as well as selected subunits of these complexes by immunoblotting. Blue native gels revealed an age-dependent decline in the abundance of assembled complex I in SPG7del mutants (Supplemental Figure S4), but the abundance of all tested subunits of complex I was normal in both young and old SPG7del mutants (Fig. 6b; Supplemental Figure S5). We also failed to detect a difference in the abundance of complex II subunits between SPG7del mutants and controls (Fig. 6b; Supplemental Figure S5), but we were unable to detect fully assembled complex II upon blue native gel analysis, possibly because the epitope detected by the antiserum we used is inaccessible in the fully assembled complex. Our findings suggest that the decrease in the activity of complex I reflects an age-dependent defect in the assembly of complex I. The molecular basis of reduced complex II activity will require further analysis.

a The activity of respiratory chain complexes in w1118 control and SPG7del mutants at 1 week (left panel) and at 4 weeks (right panel) of age. Aged SPG7del mutants exhibit reduced complex I and II activity (n = 3, *p < 0.05 from Student’s t-test). b Results of western blot analysis to quantify the abundance of the indicated respiratory chain complex subunits in young (1 week; left panel) and old (4 week; right panel) w1118 control and SPG7del mutants are shown. c Western blot analysis of UPRmt markers in young (1 week) and old (4 week) w1118 control and SPG7del mutants are shown
A failure to assemble or degrade misfolded mitochondrial proteins has been shown to activate the mitochondrial unfolded protein response (UPRmt) pathway, a nuclear response to mitochondrial stress that involves, among other things, induction of mitochondrial chaperones32. To test whether the electron-dense accumulations detected in IFM mitochondria activate the UPRmt, we examined the abundance of several different markers of this stress pathway, including Hsp60, mtHsp70, and Lon protease. We also examined the abundance of phosphorylated eIF2α, which serves to attenuate cytosolic translation in response to UPRmt33. Although we detected a trend towards increased abundance of phosphorylated eIF2α in both young and old SPG7del flies (Fig. 6c), the differences were not statistically significant. In addition, none of the tested markers of UPRmt showed significant induction (Fig. 6c). These findings indicate that, despite the electron-dense deposits in the matrix of muscle mitochondria of SPG7del mutants, any accumulation of misfolded proteins is insufficient to trigger induction of the UPRmt. Further work will be required to explain the origin of the electron-dense matrix accumulations in SPG7del mutants.
Loss of SPG7 causes mitochondrial trafficking defects in larval segmental nerves
Many loci associated with HSP encode factors that potentially influence mitochondrial trafficking. These findings, coupled with the mitochondrial phenotypes of SPG7del mutants, led us to explore the influence of SPG7 on axonal mitochondrial transport. We used the Drosophila larval segmental nerve to compare a variety of mitochondrial trafficking parameters in SPG7del mutants (Supplementary Movie 2) and w1118 controls (Supplementary Movie 1). Loss of SPG7 resulted in a significant increase in the fraction of mitochondria moving in the retrograde direction and an anterograde to retrograde transport ratio below 1 (Fig. 7a, b). We also found that the velocity of mitochondria moving in the anterograde direction was significantly decreased and there was a trend towards increased velocity of mitochondria moving in the retrograde direction in SPG7del mutants (Fig. 7c). However, the overall levels of activity of mitochondrial motors were unchanged as determined by the fraction of time a moving mitochondria remain stationary (Fig. 7d). Together our findings indicate that loss of SPG7 results in a net increase in the movement of mitochondria in the retrograde direction.

a Representative kymographs of axonal mitochondrial transport in larval segmental nerves of wild-type and SPG7del larvae. Anterograde movement is left to right and retrograde movement is right to left in the images. Vertical lines in the kymographs indicate stationary mitochondria. b Loss of SPG7 enhances net retrograde transport of mitochondria and decreases the anterograde/retrograde ratio (*p < 0.05 from Mann–Whitney U-test). c Loss of SPG7 results in reduced anterograde mitochondrial velocity (*p < 0.05 from Student’s t-test). d SPG7 does not influence the fraction of time mitochondria remain paused. Numbers in parenthesis in b and c indicate total number of mitochondria used for quantification. Significance was determined using Student’s t-test
Discussion
Mitochondrial dysfunction is a central hallmark of aging and a frequent occurrence in neurodegenerative disorders including Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease2,3,4. Mitochondrial proteases represent one of the first lines of defense against mitochondrial dysfunction by promoting the degradation of damaged and misfolded mitochondrial proteins. The importance of mitochondrial proteases is illustrated by the fact that mutations in the genes encoding these proteases are the cause of multiple human diseases. In particular, mutations in SPG7 have been implicated in cerebellar ataxia and progressive external ophthalmoplegia (PEO), and are responsible for 5–10% of HSP34. HSP is characterized by the degeneration of motor axons of the corticospinal tracts and of sensory axons of the fasciculus gracilis with preservation of the cell bodies. An SPG7 knockout mouse model recapitulates many characteristics of HSP;22 however, the molecular mechanisms underlying diseases associated with mutations in SPG7 remain largely unknown. To complement some of the deficiencies of vertebrate disease models, we created a Drosophila model of HSP by using CRISPR/CAS9 gene targeting to delete the Drosophila SPG7 homolog. We found that SPG7del mutants are short-lived and exhibit a variety of behavioral defects. Progressive neuron and muscle degeneration accompany these behavioral defects and dysmorphic mitochondria with electron-dense aggregates were detected in degenerating tissues, indicating that the phenotypes of SPG7del mutants are caused by mitochondrial dysfunction. Transmission electron microscopy of photoreceptor neurons revealed that the neurodegenerative phenotype of SPG7del mutants initiates at the synaptic terminal, thus recapitulating one of the hallmark characteristics of HSP. Our fly model provides a foundation for detailed exploration of the molecular mechanisms underlying HSP.
One of the most intriguing phenotypic features of SPG7del mutant flies is the appearance of abnormally large and dysfunctional mitochondria in the synaptic terminals of photoreceptor cells. These findings also corroborate with the specific pattern of degeneration referred to as “dying-back axonopathy” in HSP, which begins distally at the synaptic terminal and proceeds proximally towards the cell body35. Our current findings, coupled with previously published work on SPG7−/− mice, lead us to hypothesize that neuropathology in SPG7del mutants arises from mitochondrial dysfunction that initiates at the synapse and progresses to an axonal trafficking defect, axonal swelling, and ultimately neurodegeneration. Our finding that SPG7del mutants display increased retrograde mitochondrial transport in segmental nerves offers potential support for this model. Increased retrograde mitochondrial transport was also observed following treatment of cultured neurons with the mitochondrial uncoupling agent Antimycin A1, suggesting that this may represent an early stress response aimed at facilitating the turnover or repair of damaged synaptic mitochondria in the soma36. Our model would also potentially explain other genetic forms of HSP that are caused by mutations in genes that would be predicted to impact axonal trafficking, including SPG10, which encodes a kinesin heavy chain isoform involved in anterograde transport in axons; SPG4, which encodes Spastin, a protein involved in catalyzing microtubule severing; and SPG20, which encodes Spartin, a protein involved in endosomal trafficking and microtubule dynamics37,38,39.
Knockdown of SPG7 in Caenorhabditis elegans results in the accumulation of unfolded proteins and the induction of mitochondrial Hsp60, mtHsp70, and other factors through the UPRmt32. However, despite the accumulation of electron-dense material in the mitochondrial matrix of SPG7del mutants, we failed to detect evidence of UPRmt induction. One possible explanation for these discordant findings is that the UPRmt pathway may not be conserved between Drosophila and C. elegans. For example, the transcription factor Atfs-1 has a central role in the UPRmt, but this factor has only been conclusively identified in worms and no homolog of Atfs-1 has been reported in Drosophila. It is also possible that a different constellation of chaperones is induced during UPRmt in Drosophila to cope with the increased load of misfolded protein aggregates40,41. Alternatively, the accumulation of damaged and misfolded proteins in Drosophila may activate alternative quality control pathways, including the destruction of mitochondria through PINK1/Parkin-mediated mitophagy, or the degradation of damaged mitochondrial components through a PINK1-Parkin-mediated vesicular pathway5,42,43,44. Further work will be required to distinguish between these models.
Our finding that SPG7del mutants have reduced complex I abundance and activity agrees with previous work showing that fibroblasts from HSP individuals manifest a complex I assembly defect that results in reduced complex I activity45. However, in contrast to findings from HSP fibroblasts, we also detected a significant reduction in the activity of complex II in SPG7del mutants. Complex II has recently emerged as a regulator of cell death through increased ROS production46,47,48. We propose three alternative hypotheses to explain the decrease in the activity of complex II in SPG7del mutants. One possibility is that paraplegin is directly involved in the assembly of complex II. Another possibility is that paraplegin promotes the turnover or processing of a factor that influences electron transfer between succinate and ubiquinone-binding subunits. Finally decreased complex II activity in SPG7del mutants could result from mitochondrial calcium overload. Recent work indicates that Afg3l2 and paraplegin promote the degradation of the essential mitochondrial calcium uniporter regulator (EMRE)49,50. Loss of m-AAA protease activity results in the constitutive activation of mitochondrial calcium uniporter channel activity and increased Ca2+ influx into mitochondria. Mitochondrial Ca2+ overload has been implicated in ROS mediated cell death induction by disintegration of complex II46. Genetic and pharmacological manipulations designed to offset an increase in cytosolic calcium should enable a direct test of the role of calcium in the phenotypes of SPG7del mutants.
In summary our fruit fly model of HSP faithfully recapitulates key features of the human disease and provides a genetically tractable system to explore the molecular mechanisms underlying HSP. In previous work, we used a proteomic methodology to identify mitochondrial proteins with altered turnover kinetics in Drosophila mutants defective in key mitophagy-promoting and autophagy-promoting genes51. The application of this methodology to SPG7del mutants, along with the genetic tools available for gene manipulation in Drosophila, should enable rapid identification of paraplegin substrates and evaluation of their roles in pathogenesis. This work should provide important insight into the mechanisms responsible for diseases associated with mutations in SPG7, and, ultimately, the development of therapies for these disorders.
Materials and methods
Fly stocks and maintenance
All fly stocks and genetic crosses were maintained on standard cornmeal-molasses food at 25 °C, on a 12 h:12 h light-dark cycle. The w1118, UAS-CD8-GFP, UAS-MITO-GFP, Act-gal4, D42-gal4, CCAP-gal4, Da-gal4, y1 M{vas-Cas9.RFP-}ZH-2A w1118, and X chromosome duplication stocks Dp(1;3)DC048 and Dp(1;3)DC406 were obtained from the Bloomington Stock Center (Bloomington, IN, USA). The SPG7del allele was created using CRISPR-CAS9-mediated gene editing in accordance with a published procedure19. Briefly, our procedure involved replacing the SPG7 coding sequence with Ds-red through homology-mediated repair. The following primer sequences were used to design guide RNAs targeting the 5′ and 3′ UTR regions of SPG7:
5′-Guide RNA
Sense oligo—CTTCGTCGCAGCCGGTCCGCGATT
Antisense oligo—AAACAATCGCGGACCGGCTGCGAC
3′-Guide RNA
Sense oligo—CTTCGCTAATAAGACGCGTCGCGG
Antisense oligo—AAACCCGCGACGCGTCTTATTAGC
To facilitate homology-directed repair, the pHD-DsRed-attP vector containing the eye-specific 3xP3 promoter fused with dsRed was appended with sequences flanking the paraplegin coding sequences. The SPG7 flanking sequences were amplified from genomic DNA using the following primer sequences in PCR:
5′-Homology arm
Forward—CCGGCACCTGCGGCCTCGCATGCGGGTCTCACTCACCTTCACCC
Reverse—CCGGCACCTGCGGCCCTACCGCGGACCGGCTGCGACTACGTAACTCC
3′-Homology arm
Forward—GGCCGCTCTTCATATCGGCGGGCAAGGCGACTTTTAACAACC
Reverse—CCGGGCTCTTCTGACACTGACGGAATCCAGCCAGAAGTCGGG
The 5′ and 3′ homology arms were then cloned into the pHD-DsRed-attP vector using AarI and SapI restriction enzymes, respectively. Flies bearing the DsRed marker were identified by screening the injected adults for expression of red fluorescence in the compound eye. The absence of the paraplegin coding sequence was verified by whole-genome sequencing.
For all analyses involving adult flies, age refers to the number of days following eclosion.
Behavioral and lifespan analyses
All behavioral analyses were performed using male flies. Longevity assays involved 10–15 flies per vial. Food was changed every second day and the number of dead flies was counted. Kaplan–Meier lifespan curves were generated using Microsoft Office Excel, and log-rank test was used to determine statistical significance. For all remaining analyses, flies were anesthetized by briefly exposing them to CO2 and allowing them to recover for at least 24 h before the experiment.
Climbing behavior was assessed using the Rapid Iterative Negative Geotaxis (RING) assay at 1 week and 4 weeks of age, according to a previously published protocol52. Briefly, flies were transferred into plastic vials and loaded onto the RING apparatus (10–12 flies per vial). The apparatus was gently tapped down 3–4 times to initiate the climbing response and the height climbed by each fly after 4 s was recorded. The climbing assay was repeated three times and the average height climbed in the three trials was calculated.
To conduct the bang sensitivity test, groups of 5–6 flies were transferred into empty plastic vials and vortexed for 10 s at the maximum setting. The time required for flies to recover from paralysis was then recorded. Experiments were repeated with a different cohort of flies each time, and the mean recovery time was calculated.
To analyze heat sensitivity, groups of 5–6 flies were placed in watertight glass vial and vials were then submerged in a water bath preheated to 39 °C for 6 min. The number of paralyzed flies was then recorded following this heat challenge.
Flight assays were performed according to a previously published protocol20,21. Briefly, an acetate sheet coated with grease was inserted into a 2-liter graduated cylinder. Flies were tapped into a funnel at the top of the cylinder and the number of flies that fell to the bottom of the cylinder was counted to calculate the fraction of non-fliers after each trial.
Histological analysis
Histological analyses of brain and muscle tissues were performed as previously described53. Briefly, tissues were fixed in Carnoy’s fixation solution (10% acetic acid; 30% chloroform; 60% absolute ethanol) for 3.5 h and dehydrated in ethanol. Infiltration was performed using paraffin at 60 °C, and 4-μm sections were analyzed by hematoxylin and eosin staining. Images were collected on a Nikon Optiphot-2 using a ×10 objective. Brain vacuole size and area was quantified using ImageJ software.
Transmission electron microscopy
TEM was performed as previously described with some modifications54. Briefly, tissues from 28-day-old flies were dissected in fixative containing 2.5% glutaraldehyde, and 2% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.4, and incubated overnight at 4 °C. Fixed tissues were then postfixed in 1% OsO4, dehydrated in an ethanol series, and embedded using Epon. Samples were subjected to ultra-thin sectioning at 70 nm and stained with 6% uranyl acetate and a Reynolds lead citrate solution before TEM examination. Grids were viewed using a JEOL JEM 1400 transmission electron microscope.
Immunofluorescence and confocal microscopy
Thoraces from young (1 week) and old (4 week) flies were dissected in cold PBS buffer, then fixed in 4% paraformaldehyde for 1 h. Dissected tissues were then washed twice in PBS followed by staining with phalloidin-568 (Life Technologies) at a 1:250 dilution for 1 h and imaged using an Olympus FV-1000 confocal microscope with a 60x lens and a 1x digital zoom.
To examine the localization of paraplegin, fixed thoraces dissected from w1118 as described above were incubated overnight with rabbit anti-paraplegin (1:250) and mouse anti-cytochrome c (Cyto C) (1:1000, BD Biosciences) antibody. After washing with PBS (including 0.3% Triton X-100), tissues were incubated overnight with 1:500 anti-rabbit Alexa 488 secondary antiserum and 1:500 anti-mouse Alexa 568 secondary antiserum. Images were acquired sequentially with 488 nm and 561 nm lasers on an Olympus FV-1000 with a 60x lens.
To examine the morphology of neuromuscular junctions, third instar larvae were dissected in PBS buffer followed by fixation in 4% paraformaldehyde. Type 1b synaptic boutons of muscle 4 in abdominal segments 2 and 3- (A2–A3) were visualized using the presynaptic marker anti-HRP-Cy3 (1:200, Jackson Immunoresearch) and the postsynaptic marker anti-discs large 1 (1:50, DSHB).
To examine axonal morphology, legs from flies bearing the UAS-CD8-GFP and D42- GAL4 transgenes were dissected, and GFP fluorescence was visualized using confocal microscopy at 10x magnification. At least 20 legs from 10 different flies were imaged.
Mitochondrial respiratory chain activity assay
Mitochondrial fractions for respiratory chain activity assays were prepared using a published procedure with minor modifications55. Briefly, 800–900 flies were homogenized in 5 mM HEPES (pH 7.5), 210 mM mannitol, 70 mM sucrose, and 1 mM EGTA. The lysate was subjected to centrifugation at 3500 rpm for 5 min to remove cuticle and cellular debris. The supernatant was then subjected to centrifugation a second time at 15,000 rpm for 20 min to isolate the mitochondrial pellet. Mitochondria were resuspended in 250 mM sucrose, 2 mM EDTA, 100 mM Tris–HCl, pH 7.4, flash frozen in liquid nitrogen, and then stored at −80 °C.
Respiratory chain activity assays were performed as previously described with minor modifications55. Complex I activity was measured by monitoring the oxidation of NADH at 340 nm using ubiquinone-1 as an electron acceptor in a buffer containing 50 mM potassium phosphate (pH 7.5), 0.1 mM NADH, and 0.3 mM potassium cyanide. Background activity was determined using 10 μM rotenone, and used to calculate complex I-specific activity. Complex II activity was measured by monitoring the reduction of 2,6- dichlorophenolindophenol at 600 nm in a reaction mixture of 25 mM potassium phosphate (pH 7.5), 20 mM succinate, 80 μM 2,6-dichlorophenol-indophenol, 50 μM Decylubiquinone, and 0.3 mm potassium cyanide. Background activity was determined using 10 mM malonate, and used to calculate complex II-specific activity. Complex III activity was determined by monitoring the reduction of cytochrome c at 550 nm in a reaction mixture of 25 mM potassium phosphate (pH 7.5), 100 μM reduced decylubiquinone, 75 μM cytochrome c and 0.5 mM potassium cyanide. Background activity was calculated using antimycin A (10 μg ml−1), and used to calculate complex III-specific activity. Complex IV activity was performed by monitoring the oxidation of cytochrome c at 550 nm in a reaction mixture of 25 mM potassium phosphate (pH 7.5), and 0.05 mM reduced cytochrome c. Background activity was determined using 0.3 mM potassium cyanide, and used to calculate complex IV-specific activity. All activities were normalized against citrate synthase activity, which was determined by following the reduction of 5,5′-dithiobis(2-nitrobenzoic acid) at 412 nm in presence of acetyl-coenzyme A and oxaloacetate. Approximately 10–40 μg of mitochondria was used in all assays.
Blue native PAGE (BN-PAGE) analysis
BN-PAGE was performed according to a previously published protocol56. Briefly, 50 µg of mitochondria prepared from 1 week and 4 week old adult flies as solubilized in a buffer containing a digitonin:protein (mass:mass) ratio of 8 and subjected to centrifugation at 20,000×g for 10 min at 4 °C. The supernatent was treated with coomassie G-250 and used in Native PAGE analysis. Complex I assembly was analyzed using an anti-NDUFS3 antiserum.
Subcellular fractionation
Subcellular fractionation was performed as previously described57. Briefly, a crude mitochondrial pellet was obtained as described above and subsequently resuspended in buffer containing 0.27 M d-mannitol, 0.01 M Tris-base, and 0.1 mM EDTA and carefully overlaid on a sucrose gradient prepared by combining 1 ml of 1.7 M sucrose with 1.6 ml of 1 M sucrose. Samples were then subjected to ultracentrifugation at 40,000×g for 22 min and gradient fractions were recovered using a 1-ml syringe with a 20-G needle.
Paraquat sensitivity test
Two hundred flies were starved for 6 h in plastic vials containing filter paper soaked with water (10–15 flies per vial). Flies were then transferred to experimental vials containing filter paper soaked with 10 mM paraquat in 5% sucrose solution, or control vials containing filter paper soaked with 5% sucrose solution alone. Flies were transferred to new vials daily. The number of dead flies was counted daily over a period of 1 week.
Immunoblotting
Whole flies were homogenized in RIPA buffer and the supernatant was subjected to SDS-PAGE electrophoresis. Following electrophoresis, proteins were transferred to PVDF membrane, and the membrane was blocked with 5% milk, then incubated overnight with antisera. Antibodies were used at the following concentrations for immunoblots: COX IV (ab33985, Abcam), 1:5000; NDUFS3 (ab14711, Abcam), 1:1000; SDHB (ab14714, Abcam), 1:1000; phospho-EIF2α (ab32157, Abcam), 1:1000; EIF2α (ab26197, Abcam), 1:1000; Lon (NBP1-81734, Novus Biologicals), 1:5000; Hsp60 (4870S, Cell Signaling Technology), 1:1000; mtHsp70 (sc-13967, Santa Cruz Biotechnology), 1:5000; ATP Synthase β (A21351, Thermo Fisher Scientific), 1:1000; and Actin (MAB1501, Chemicon), 1:10,000. Chemiluminescence was used for antibody detection and western blot images were quantified using ImageJ software and normalized to Actin. Each experiment was performed at least three times.
Analysis of mitochondrial transport
Mitochondrial trafficking was measured in third instar larvae expressing UAS-MITOGFP driven by the CCAP-Gal4 driver58,59. Briefly, larvae were pinned with the dorsal surface facing upward and dissected in a buffer containing 128 mM NaCl, 5 mM EGTA, 4 mM MgCl2, 2 mM KCl, 5 mM HEPES and 36 mM sucrose, pH adjusted to 7.2. A small incision was made at the posterior end and continued along the dorsal midline. After removing internal organs, larvae were transferred to a chamber on a glass side constructed with the aid of double sided tape and imaged using an Olympus FV-1000 fluoview confocal microscope with a 60x water immersion lens. Images were captured at a rate of 1 frame per 3.25 s for 100 frames. One axon was analyzed per larvae and a total of 18 axons for control and 19 axons for SPG7del mutants were analyzed. Kymographs were constructed using the KymographBuilder plugin in ImageJ software, and analyzed as described previously58,59.
Statistics
All data is represented as mean ± s.e.m. Unless otherwise stated, statistical significance tests were calculated using an unpaired two-tailed Student’s t-test in GraphPad Prism 7.
References
Nunnari, J. & Suomalainen, A. Mitochondria: in sickness and in health. Cell 148, 1145–1159 (2012).
Sun, N., Youle, R. J. & Finkel, T. The mitochondrial basis of aging. Mol. Cell 61, 654–666 (2016).
Jaiswal, M., Sandoval, H., Zhang, K., Bayat, V. & Bellen, H. J. Probing mechanisms that underlie human neurodegenerative diseases in Drosophila. Annu. Rev. Genet. 46, 371–396 (2012).
Pickrell, A. M. & Youle, R. J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273 (2015).
Whitworth, A. J. & Pallanck, L. J. PINK1/Parkin mitophagy and neurodegeneration-what do we really know in vivo? Curr. Opin. Genet. Dev. 44, 47–53 (2017).
Greene, J. C. et al. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl Acad. Sci. USA 100, 4078–4083 (2003).
Clark, I. E. et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 (2006).
Narendra, D., Tanaka, A., Suen, D. F. & Youle, R. J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell. Biol. 183, 795–803 (2008).
Quiros, P. M., Langer, T. & Lopez-Otin, C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 16, 345–359 (2015).
Martinelli, P. & Rugarli, E. I. Emerging roles of mitochondrial proteases in neurodegeneration. Biochim. Biophys. Acta 1797, 1–10 (2010).
Jenkinson, E. M. et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am. J. Hum. Genet. 92, 605–613 (2013).
Strauss, K. A. et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 96, 121–135 (2015).
Hartmann, B. et al. Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. Elife 5. https://doi.org/10.7554/eLife.16078 (2016).
Pfeffer, G. et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 137, 1323–1336 (2014).
Pfeffer, G. et al. SPG7 mutations are a common cause of undiagnosed ataxia. Neurology 84, 1174–1176 (2015).
Choquet, K. et al. SPG7 mutations explain a significant proportion of French Canadian spastic ataxia cases. Eur. J. Hum. Genet. 24, 1016–1021 (2016).
Qi, Y., Liu, H., Daniels, M. P., Zhang, G. & Xu, H. Loss of Drosophila i-AAA protease, dYME1L, causes abnormal mitochondria and apoptotic degeneration. Cell Death Differ. 23, 291–302 (2016).
Chen, C. L. et al. Proteomic mapping in live Drosophila tissues using an engineered ascorbate peroxidase. Proc. Natl Acad. Sci. USA 112, 12093–12098 (2015).
Gratz, S. J. et al. Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics 196, 961–971 (2014).
Babcock, D. T. & Ganetzky, B. An improved method for accurate and rapid measurement of flight performance in Drosophila. J. Vis. Exp. e51223. https://doi.org/10.3791/51223 (2014).
Burman, J. L. et al. A Drosophila model of mitochondrial disease caused by a complex I mutation that uncouples proton pumping from electron transfer. Dis. Model. Mech. 7, 1165–1174 (2014).
Ferreirinha, F. et al. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J. Clin. Invest. 113, 231–242 (2004).
Pirozzi, M. et al. Intramuscular viral delivery of paraplegin rescues peripheral axonopathy in a model of hereditary spastic paraplegia. J. Clin. Invest. 116, 202–208 (2006).
Lloyd, T. E. & Taylor, J. P. Flightless flies: Drosophila models of neuromuscular disease. Ann. NY Acad. Sci. 1184, e1–e20 (2010).
Brent, J. R., Werner, K. M. & McCabe, B. D. Drosophila larval NMJ dissection. J. Vis. Exp. https://doi.org/10.3791/1107 (2009).
Sreedharan, J., Neukomm, L. J., Brown, R. H. Jr. & Freeman, M. R. Age-dependent TDP-43-mediated motor neuron degeneration requires GSK3, hat-trick, and xmas-2. Curr. Biol. 25, 2130–2136 (2015).
Borycz, J. A., Borycz, J., Kubow, A., Kostyleva, R. & Meinertzhagen, I. A. Histamine compartments of the Drosophila brain with an estimate of the quantum content at the photoreceptor synapse. J. Neurophysiol. 93, 1611–1619 (2005).
Zhai, R. G. et al. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 4, e416 (2006).
Bernstein, S. H. et al. The mitochondrial ATP-dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood 119, 3321–3329 (2012).
Suzuki, C. K., Suda, K., Wang, N. & Schatz, G. Requirement for the yeast gene LON in intramitochondrial proteolysis and maintenance of respiration. Science 264, 891 (1994).
Spinazzi, M., Casarin, A., Pertegato, V., Salviati, L. & Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 7, 1235–1246 (2012).
Haynes, C. M., Fiorese, C. J. & Lin, Y. F. Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol. 23, 311–318 (2013).
Baker, B. M., Nargund, A. M., Sun, T. & Haynes, C. M. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 8, e1002760 (2012).
Crosby, A. H. & Proukakis, C. Is the transportation highway the right road for hereditary spastic paraplegia? Am. J. Hum. Genet. 71, 1009–1016 (2002).
Rugarli, E. I. & Langer, T. Translating m-AAA protease function in mitochondria to hereditary spastic paraplegia. Trends Mol. Med. 12, 262–269 (2006).
Lin, M. Y. et al. Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron 94, 595–610.e6 (2017).
Reid, E., Dearlove, A. M., Rhodes, M. & Rubinsztein, D. C. A new locus for autosomal dominant “pure” hereditary spastic paraplegia mapping to chromosome 12q13, and evidence for further genetic heterogeneity. Am. J. Hum. Genet. 65, 757–763 (1999).
Hazan, J. et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 23, 296–303 (1999).
Patel, H. et al. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat. Genet. 31, 347–348 (2002).
Morrow, G. et al. Changes in Drosophila mitochondrial proteins following chaperone-mediated lifespan extension confirm a role of Hsp22 in mitochondrial UPR and reveal a mitochondrial localization for cathepsin D. Mech. Ageing Dev. 155, 36–47 (2016).
Zhang, L. et al. TRAP1 rescues PINK1 loss-of-function phenotypes. Hum. Mol. Genet. 22, 2829–2841 (2013).
McLelland, G. L., Soubannier, V., Chen, C. X., McBride, H. M. & Fon, E. A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295 (2014).
Thomas, R. E., Andrews, L. A., Burman, J. L., Lin, W. Y. & Pallanck, L. J. PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genet. 10, e1004279 (2014).
Greene, A. W. et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 13, 378–385 (2012).
Atorino, L. et al. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J. Cell Biol. 163, 777–787 (2003).
Kluckova, K. et al. Ubiquinone-binding site mutagenesis reveals the role of mitochondrial complex II in cell death initiation. Cell Death Dis. 6, e1749 (2015).
Hwang, M. S. et al. Mitochondrial Ca(2+) influx targets cardiolipin to disintegrate respiratory chain complex II for cell death induction. Cell Death Differ. 21, 1733–1745 (2014).
Bezawork-Geleta, A., Rohlena, J., Dong, L., Pacak, K. & Neuzil, J. Mitochondrial complex II: at the crossroads. Trends Biochem. Sci. 42, 312–325 (2017).
Konig, T. et al. The m-AAA protease associated with neurodegeneration limits MCU activity in mitochondria. Mol. Cell 64, 148–162 (2016).
Tsai, C. W. et al. Proteolytic control of the mitochondrial calcium uniporter complex. Proc. Natl Acad. Sci. USA 114, 4388–4393 (2017).
Vincow, E. S. et al. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl Acad. Sci. USA 110, 6400–6405 (2013).
Gargano, J. W., Martin, I., Bhandari, P. & Grotewiel, M. S. Rapid iterative negative geotaxis (RING): a new method for assessing age-related locomotor decline in Drosophila. Exp. Gerontol. 40, 386–395 (2005).
Davis, M. Y. et al. Glucocerebrosidase deficiency in drosophila results in alpha-synuclein-independent protein aggregation and neurodegeneration. PLoS Genet. 12, e1005944 (2016).
Poole, A. C. et al. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl Acad. Sci. USA 105, 1638–1643 (2008).
Zhang, K. et al. The C8ORF38 homologue Sicily is a cytosolic chaperone for a mitochondrial complex I subunit. J. Cell Biol. 200, 807–820 (2013).
Jha, P., Wang, X. & Auwerx, J. Analysis of mitochondrial respiratory chain supercomplexes using blue native polyacrylamide gel electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol. 6, 1–14 (2016).
Williamson, C. D., Wong, D. S., Bozidis, P., Zhang, A. & Colberg-Poley, A. M. Isolation of endoplasmic reticulum, mitochondria, and mitochondria-associated membrane and detergent resistant membrane fractions from transfected cells and from human cytomegalovirus-infected primary fibroblasts. Curr. Protoc. Cell Biol. 68, 3.27.1–33 (2015).
Wang, X. & Schwarz, T. L. Imaging axonal transport of mitochondria. Methods Enzymol. 457, 319–333 (2009).
Devireddy, S., Sung, H., Liao, P. C., Garland-Kuntz, E. & Hollenbeck, P. J. Analysis of mitochondrial traffic in Drosophila. Methods Enzymol. 547, 131–150 (2014).
Acknowledgements
We thank Dr. Scott Kennedy (Department of Pathology, University of Washington) for assistance with whole-genome sequencing of the SPG7del mutant; Dr. Glen MacDonald (Microscopy and Imaging Facility, Virginia Merrill Bloedel Hearing Research Center, University of Washington) for technical assistance with confocal imaging and analysis; Dr. Ying-tzang Tien (Department of Pathology, University of Washington) for histological staining of Drosophila tissue sections; Dr. Bobbie Schneider (Fred Hutchinson Cancer Research Center) for assistance with electron microcopy and all members of the Pallanck lab (particularly, Evelyn Vincow) for critical review of this work and manuscript. This work was supported by a grant from the National Institute of Health to LP (R21NS094901).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pareek, G., Thomas, R.E. & Pallanck, L.J. Loss of the Drosophila m-AAA mitochondrial protease paraplegin results in mitochondrial dysfunction, shortened lifespan, and neuronal and muscular degeneration. Cell Death Dis 9, 304 (2018). https://doi.org/10.1038/s41419-018-0365-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-018-0365-8
This article is cited by
-
SPG7 mutations in amyotrophic lateral sclerosis: a genetic link to hereditary spastic paraplegia
Journal of Neurology (2020)
-
Lon protease inactivation in Drosophila causes unfolded protein stress and inhibition of mitochondrial translation
Cell Death Discovery (2018)
-
Inactivation of Lon protease reveals a link between mitochondrial unfolded protein stress and mitochondrial translation inhibition
Cell Death & Disease (2018)
