
Abstract
Estrogen receptor α (ERα) is the crucial factor in ERα-positive breast cancer progression. Endocrine therapies targeting ERα signaling is one of the widely used therapeutic strategies for breast cancer. However, a large number of the patients become refractory to therapy. Abnormal expression of ERα co-regulator facilitates breast cancer development and tendency of endocrine resistance. Thus, it is necessary to discover the novel co-regulators modulating ERα action. Here, we demonstrate that histone deubiquitinase USP22 is highly expressed in breast cancer samples compared with that in the benign tissue, and high expression of USP22 was significantly associated with poorer overall survival in BCa samples. Moreover, USP22 associates with ERα to be involved in maintenance of ERα stability. USP22 enhances ERα-induced transactivation. We further provide the evidence that USP22 is recruited together with ERα to cis-regulatory elements of ERα target gene. USP22 promotes cell growth even under hypoxia condition and with the treatment of ERα antagonist in breast cancer cells. Importantly, the deubiquitination activity of USP22 is required for its functions on maintenance of ERα stability, thereby enhancing ERα action and conferring endocrine resistance in breast cancer.
Similar content being viewed by others


Introduction
Estrogen receptor α (ERα) induces target gene transcription to exert its physiological and pathological function in a ligand-dependent or -independent manner. Abnormal regulation of the liganded ERα signaling pathway in ERα-positive (ERα+) breast cancer (BCa) potentiates tumor progression and could facilitate resistance to endocrine therapy [1, 2]. ERα mediates the transcription of hypoxia-inducible factor 1 and participates in endocrine response in BCa [3]. On the other hand, inflammatory cytokines activates unliganded ERα through kinase-dependent phosphorylation, leading to endocrine resistance and invasiveness of BCa cells [4]. ERα-targeted therapy is therefore widely used in ERα+ BCa to inhibit ERα function, although therapy resistance occurs in the substantial fraction of patients [5, 6]. Therefore, a well understanding of molecular mechanisms modulating ERα action would be helpful to detect potential therapeutic targets for ERα+ BCa.
Upon activation of ERα, co-regulators are recruited by ERα to participate in modulating ERα-mediated transactivation via alteration of chromatin structure, posttranscriptional modification, or modulation of ERα protein stability [7,8,9]. A series of ERα co-regulators are involved in tumor proliferation, invasion, or endocrine resistance of BCa. HOXB7 acting as an ERα co-regulator enhances the transactivation of ERα target genes and HER2 leads to endocrine resistance in BCa [10]. FOXA1 has been demonstrated as a key factor necessary for ERα–chromatin interaction in BCa under different ligand conditions, including tamoxifen treatment [11]. The histone demethylase enzyme KMD3A is required for ERα action and cell growth in BCa [12]. JmjC-domain-containing protein JMJD6 controls the transactivation of ERα-bound enhancers and its downstream target genes and is a determinant of ERα+ BCa cell growth and tumorigenesis [13]. Thus, developing therapies to target regulation of ERα function may therefore provide effective means of ERα+ BCa treatment.
The deubiquitylating enzyme USP22 as a component of the deubiquitination module is involved in gene transcriptional regulation. We have previously demonstrated that USP22 regulates androgen receptor-induced transactivation [14]. In addition, USP22 exerts biological function through deubiquitinating nonhistone proteins. USP22 is involved in telomere maintenance by deubiquitinating telomeric-repeat-binding factor 1 [15]. USP22 stabilizes the NAD-dependent histone deacetylase SIRT1 by removing polyubiquitin chains conjugated onto SIRT1, subsequently leading to suppression of p53 function [16]. USP22 promotes cell proliferation by deubiquitinating the transcriptional regulator, far upstream element-binding protein 1 [17]. The deubiquitination of the cyclin D1 (CCND1) by USP22 is essential for regulating CCND1 stability and cell cycle progression in cancer cells [18].
USP22 has been reported as a member of 11 “Death-from-Cancer” genes [19, 20]. The recent studies have shown that high expression of USP22 is positively correlated with poor prognosis in numerous cancers [21,22,23,24,25,26,27,28,29,30,31]. However, the underlying molecular mechanism of USP22 in tumor progression remains poorly understood. In this study, our results demonstrate that USP22 is highly expressed in BCa samples compared with that in the benign tissue, and high expression of USP22 was significantly associated with poorer overall survival in BCa samples. We provide the evidence to show that USP22 maintains ERα stabilization through its deubiquitination activity. In addition, we have shown that USP22 enhances ERα-induced transactivation. USP22 is recruited together with ERα to cis-regulatory elements of ERα target gene. USP22 promotes cell growth in ERα+ BCa-derived cell lines. Importantly, the catalytic activity of USP22 is crucial for its co-regulatory function and promotion of cell growth even under the hypoxia condition in BCa. Taken together, these studies identify USP22 as an ERα co-regulator participates in promotion of cell growth and endocrine resistance progression by virtue of regulatory function on ERα signaling.
Results
USP22 is highly expressed in breast cancer samples
We have previously identified USP22 as a subunit of deubiquitination module counteracts heterochromatin silencing and participates in histone H2A/H2B deubiquitination [14]. It has been reported that USP22 plays a crucial role in pathological processes of multiple cancers. However, the molecular mechanisms of USP22 in tumor progression and therapeutic resistance in BCa are largely unknown. Immunohistochemistry (IHC) experiments were thus performed to detect USP22 expression in a collection of breast benign hyperplasia (n = 37) and malignant BCa samples (n = 199). The results showed that USP22 was significantly highly expressed in BCa samples compared with that in breast benign tissues (Fig. S1a, b and Supplementary Table S1).
We then analyzed the correlation between the expression of USP22 and clinical prognosis in BCa samples using tissue array (n = 124) with the survival information of BCa patients and GEPIA (http://gepia.cancer-pku.cn/). The results demonstrated that high expression of USP22 was significantly associated with poorer overall survival in BCa samples (Fig. 1a, b). Unexpectedly, we found that the expression of USP22 was positively correlated with that of ERα in BCa samples, providing an evidence for the possibility of positive regulation between USP22 and ERα (Fig. 1c–f). In order to explore the clinical relevance of the expression of USP22 and ERα, we analyzed the expression correlation of USP22 and ESR1 encoding ERα from 825 BCa patients in publicly available gene expression profiling data in TCGA comprehensive molecular portraits of human breast tumors and cBio Cancer Genomics Portal (http://www.cbioportal.org/). Interestingly, USP22 expression correlates with 4934 genes, and 11,948 genes were correlated with ESR1 expression in clinical BCa samples (Pearson Score > 0.1 or Pearson Score < −0.1). Among them, the expression of 2721 genes was positively or negatively correlated with that of USP22 and ESR1, indicating that the 2721 genes may be involved in ERα-mediated transactivation regulated by USP22 to participate in progression of BCa (Fig. 1g and Supplementary Table S2).

a Kaplan–Meier estimates of overall survival of the subjects (n = 124). Comparison was made of groups between high USP22 expression and low USP22 expression. Marks on graph lines represent censored samples. P value refers to two-sided log-rank tests. b The correlation between USP22 expression and overall survival in BCa patients was assessed by GEPIA survival analysis. c USP22 and ERα protein levels were determined in 26 breast cancer and matched adjacent benign breast tissues by western blotting. GAPDH was used as a loading control. d, e Pearson correlation test was used as statistical significance between USP22 and ERα by quantification of the western bands. f USP22 and ERα immunostaining expression in breast cancer. g Venn diagram representing the overlap of genes co-expressed with USP22 or ESR1 identified in clinical breast cancer samples (Pearson Score > 0.1 or Pearson Score < −0.1). Data set obtained from the TCGA RNA-sequencing database and analyzed by cBio Cancer Genomics Portal (http://www.cbioportal.org/public-portal/). The information for 2721 genes co-expressed with USP22 and ESR1 was shown in Supplementary Table S2.
USP22 interacts with ERα in mammalian cells
We thus turn to examine whether USP22 associates with ERα in cells. Co-immunoprecipitation (Co-IP) experiments were performed in MCF-7 cells and T47D cells. The endogenous interaction was determined reciprocally by using anti-ERα or anti-USP22 antibody, demonstrating that the endogenous USP22 interacts with ERα (Figs. 2b, c and S4). GST Pull-down experiment was further performed with GST-tagged ERαAF1 or ERαAF2 expression vector. The results suggested that binding domain in ERα with USP22 is mainly in ERαAF2 region (Fig. 2d). Co-IP experiments were performed with generated USP22 mutant expression plasmid (USP22 HH/AA) [14] carrying loss-of-function mutation in deubiquitinase activity of USP22 as shown in Fig. 2a. Our results demonstrated that ERα was precipitated with USP22 or USP22 HH/AA, indicating that deubiquitinase activity of USP22 is not required for the interaction of USP22 with ERα in BCa cells (Fig. 2e).

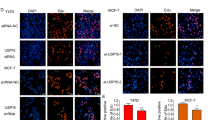
a Diagram representation of USP22, USP22 wt, and USP22 (HH/AA) (loss of function of USP22 deubiquitinase activity mutation). b, c Endogenous ERα and USP22 are associated with each other in MCF-7 cells. Reciprocal Co-IP and immunoblotting were performed with antibodies as indicated. A 5% fraction of the input cell lysate before immunoprecipitation was loaded as a control. d USP22 directly binds GST-ERαAF2 in vitro. GST, GST-ERαAF1, and ERαAF2 fusion proteins were incubated with 35S-USP22 in vitro. Bound proteins were detected with autoradiography. Black stars indicate GST and GST-fusion proteins. e FLAG-USP22 wt or USP22 (HH/AA) associates with ERα in HEK293 cells. HEK293 cells cotransfected with FLAG-USP22 or USP22 (HH/AA) and ERα expression plasmids with or without E2 treatment, the cell lysates were immunoprecipitated using anti-USP22 antibody and IgG as a control. Precipitates were analyzed by western blotting using indicated antibodies. f Subcellular localization of USP22 and ERα by immunofluorescence. HEK293 cells were cotransfected with FLAG-USP22 and ERα. Cells were stained with anti-ERα antibody (green) or anti-USP22 (red). DAPI was used to visualize the nucleus (blue). Merged images were illustrated as indicated. Scale bar, 20 μM. g Subcellular distribution of the endogenous USP22 and ERα in MCF-7 cells. Scale bar, 15 μM. h Subcellular distribution of the endogenous USP22 and ERα in T47D cells. Scale bar, 15 μM.
To study the subcellular distribution of USP22 and ERα in cells, HEK293 cells were cotransfected with ERα and FLAG-USP22 expression plasmids. We observed that USP22 is distributed in the nucleus regardless of estrogen (E2, 10−8 M) treatment (Fig. 2f, red). ERα is mainly located in cytoplasm without E2 (Fig. 2f, green). In the presence of E2, ERα is distributed in nucleus and co-located with USP22 (Fig. 2f). In consistent with the above results, we observed that the endogenous USP22 co-localizes with ERα in the nucleus in the presence of E2 in MCF-7 cells and T47D cells (Fig. 2g, h). Taken together, these data demonstrated that USP22 physically associates with ERα in BCa cells. These results, in concert with our analysis concerning the associated partners of USP22 with database STRING (http://www.bork.embl-heidelberg.de/STRING/) (Supplementary Fig. S2), demonstrated that USP22 associates with ERα encoded by ESR1.
USP22 maintains ERα stability by triggering deubiquitination of ERα
Having shown that USP22 is a core component of the deubiquitination module and USP22 associates with ERα in cells, we thus turn to investigate whether USP22 is involved in the deubiquitination of ERα and maintenance of ERα stability. Our data showed that upon inhibition of protein synthesis by cycloheximide, USP22 depletion accelerated ERα degradation, suggesting that USP22 maintains the stability of ERα (Fig. 3a). Meanwhile, USP22 HH/AA decreased ERα protein stability, compared with the effect of USP22 on ERα protein (Fig. 3b). Immunoprecipitation (IP)-based ubiquitination assays was further performed to provide the evidence that the influence of USP22 on ERα ubiquitination. Ubiquitinated proteins were enriched using ubiquitin (Ub) antibody from MCF-7 cell extracts with overexpression of ERα together with USP22. The results demonstrated that ectopic expression of USP22 significantly reduced the level of ERα ubiquitination (Fig. 3c). USP22 depletion or USP22 deubiquitinase inactive mutant (USP22 HH/AA) increased ubiquitination of ERα (Fig. 3d, e), indicating that deubiquitinase activity of USP22 is required for the function of USP22 on ERα deubiquitination and maintenance of ERα stability. Taken together, these findings indicate that USP22 stabilizes ERα by triggering ERα deubiquitination.

a Depletion of USP22 decreases endogenous ERα protein stability in MCF-7 cells. Cells transfected with siUSP22 or siCtrl were treated with 100 mmol/l cycloheximide (CHX) as indicated. The expression levels of ERα and USP22 were determined by western blot analysis. b MCF-7 cells were transfected with FLAG-USP22 wt or USP22 HH/AA expression plasmid. The expression of ERα was detected by western blot. GAPDH was used as internal control. c Immunoprecipitation of ubiquitinated proteins from MCF-7 cell extracts upon overexpression of USP22. Protein was harvested after MG132 (10 μM) treatment for 3 h and ubiquitinated ERα species were detected by western blots with anti-His. d Detection of ubiquitinated ERα upon USP22 depletion. Immunoprecipitation of ubiquitinated ERα from MCF-7 cell extracts upon shUSP22 were detected by western blot with anti-His. e HEK293 cells were transfected with expression plasmid encoding ERα and His-ub together with FLAG-USP22 wt or USP22 HH/AA. Protein was harvested after MG132 (10 μM) treatment for 3 h and immunoprecipitated with anti-ERα and immunoblotted with anti-His. f, g HEK293 cells were transfected with expression plasmid encoding ERα and FLAG-USP22 or USP22 HH/AA together with HA-tagged ubiquitin mutants, including K0 (lysineless), K48 (only K48-linked Ub) and K63 (only K63-linked Ub) as indicated. Cells were immunoprecipitated with ERα and immunoblotted with anti-HA.
During different types of poly-ubiquitination processes, lysine 48 (K48)- and K63-linked ubiquitination have been reported to be implicated in protein degradation and deliberately regulate protein fate [32, 33], we next examine the type of polyubiquitin chains on ERα by USP22. Our results demonstrated that ectopic expression of USP22 markedly reduced K63- as well as K48-linked ubiquitination of ERα (Fig. 3f). To further investigate whether deubiquitinase activity of USP22 is necessary for its function on reduction of polyubiquitin chains on ERα, the cells were transfected with USP22 wt or USP22 UCH mutant (USP22 HH/AA) for IP-based ubiquitination assays with HA-tagged Ub K48 or K63. Our results showed that USP22 wt cleaved the K48- and K63-linked polyubiquitin chains on ERα, whereas USP22 UCH mutant (USP22 HH/AA) did not (Fig. 3g). Taken together, these results suggest that the deubiquitinase activity of USP22 is essential for ERα deubiquitination through at least, if not all, K48- and K63-linkage.
The deubiquitinase activity of USP22 is required for its coactivation function on ERα-mediated transactivation
Having shown that USP22 associates with ERα to be involved in maintenance of ERα stability, we thus turn to ask whether USP22 regulates ERα-mediated transcriptional activity. The luciferase reporter assay was performed in MCF-7 cells. USP22 significantly enhanced ERα-mediated transcriptional activity (Fig. 4a). These results demonstrate that USP22 obviously enhances ERα action.

a USP22 enhances ERα-induced transactivation. MCF-7 cells were cotransfected with ERE-tk-luc and pRL-tk together with the expression plasmids as indicated with or without E2. b USP22 enhances the transcriptional activity induced by ERα or its two truncated mutants (ERαAF1 and ERαAF2) in HEK293 cells. c USP22 HH/AA carrying loss of function in deubiquitinase activity exhibits reduced coactivation function on ERα action. d The efficiency of USP22 depletion by siRNA against USP22 (siUSP22) detected by quantitative real-time PCR (qPCR) in MCF-7 cells. e The effects of USP22 on mRNA expression of endogenous ERα target genes. f, g The effects of USP22 on protein expression of endogenous ERα target genes as indicated. GAPDH was used as a control. h, i The influence of USP22 on HIF1α transcription under the hypoxic condition. MCF-7 cells were transfected with siUSP22 and siCtrl and treated with 10 nM E2 and 100 µM cocl2 for 24 h. mRNA expression (h) or protein expression for USP22, HIF1α (i). β-actin was used as a negative control.
ERα contains two functional domains including constitutive activation function 1 (AF1) at its N-terminus and ligand-dependent activation function 2 (AF2) at its C-terminal region. We thus turn to ensure which domains in ERα are regulated by USP22. Luciferase assay results showed that USP22 upregulated both ERαAF1 and ERαAF2 activity (Fig. 4b). Moreover, we want to ask whether the deubiquitinase activity is required for modulation function of USP22 on ERα action. The results showed that USP22 HH/AA had almost no effect on ERα-induced transactional activity, compared with the enhancement of ERα activity mediated by USP22 wt (Fig. 4c). These results indicate that USP22 potentiates ERα-induced transactivation, and the deubiquitinase activity of USP22 is indispensable for its co-regulation function on ERα action.
In order to examine whether USP22 is involved in modulation of the endogenous ERα target gene, quantitative PCR (qPCR) was perform. USP22 was knocked down by siRNA against USP22 (siUSP22) in MCF-7 or BT474 cell lines (Fig. 4d). USP22 depletion significantly decreased the mRNA expression of ERα target genes, including hTERT, c-Myc, E2F1, pS2, and GBEB1, compared with control (siCtrl) in the present of E2, while depletion of USP22 had no effect on ERα gene transcription level in MCF-7 and BT474 cells (Fig. 4e and Supplementary Fig. S5a). We also examined the influence of USP22 on protein expression of a series of ERα target genes. The results showed that USP22 depletion decreased, and ectopic expression of USP22 increased the protein levels of hTERT, c-Myc, and Cyclin D1 (Fig. 4f, g and Supplementary Fig. S5b). It is previously shown that HIF1α expression is directly modulated by ERα, and the expression of HIF1α or hypoxia meta gene signature is cooperated with a poor prognosis to endocrine treatment in ERα+ BCa. Thus, we turn to ask whether USP22 regulates ERα-induced HIF1α expression in hypoxia. The results demonstrated that USP22 depletion obviously reduced mRNA and protein expression of HIF1α under hypoxia (Fig. 4h, i and Supplementary Fig. S6). These results indicate that USP22 acts as a novel coactivator of ERα, and its deubiquitinase activity is required for regulation function of USP22 on ERα action.
USP22 and ERα were recruited to cis-regulatory element of ERα target gene
In order to determine whether USP22 or ERα is recruited to estrogen response elements, which are cis-regulatory elements of ERα target genes. Chromatin immunoprecipitation (ChIP) assays were performed with ectopic expression of USP22 or depletion of USP22 induced by lentivirus infection in MCF-7 cells. Our data showed that USP22 or ERα was recruited to cis-regulatory elements of c-Myc promoter. In addition, ectopic expression of USP22 remarkably increased the recruitment of ERα to cis-regulatory elements. Meanwhile, USP22 depletion led to the contrary effect on ERα recruitment (Fig. 5a, b and Supplementary Fig. S7). These results suggest that USP22 or ERα is recruited to cis-regulatory elements of c-Myc promoter. USP22 facilitates ERα recruitment to the same region of ERα target gene.

a, b The regulation function of USP22 on histone H2A/H2B ubiquitination. MCF-7 cells with ectopic expression of USP22 (a) or depletion of USP22 (b) in the presence or absence of E2 were performed for the ChIP assay with antibodies as indicated. The precipitated cis-regulatory elements of c-Myc were normalized to convert DNA signal into percentage. Data represent the mean values (±SD) of triplicate real-time PCR. IgG was used as a nonspecific control for ChIP. GAPDH promoter regions were used as negative control. c, d USP22 and ERα are recruited together to cis-regulatory elements of c-Myc. ChIP/re-ChIP experiments were performed using antibodies as indicated. DNA eluted from unprecipitated chromatin was used as input. Chromatin samples were analyzed by qPCR.
We further investigated whether USP22 is recruited to ERα target gene together with ERα. ChIP–re-ChIP assays were performed in MCF-7 cells. ChIP assays were first performed with antibodies against USP22 or ERα in the soluble chromatin derived from MCF-7 cells. Then, precipitates and supernatants from the first ChIP were respectively subjected to re-IP with antibodies against a second protein as indicated (Fig. 5c, d). The data suggest that USP22 and ERα act in a combinatorial pattern at cis-regulatory elements of ERα target gene.
USP22 promotes the cell growth in ERα-positive breast cancer
To further investigate the potential function of USP22 in BCa progression, we constructed the MCF-7 cell lines with stable knockdown of USP22 (shUSP22) or ectopic expression of USP22 induced by lentivirus infection, a scramble shRNA or vector as a control (shCtrl or Ctrl). The ectopic expression of USP22 tagged with FLAG or USP22 depletion by shUSP22 in MCF-7 cell line was confirmed by western blot (Fig. 6a). The cell cycle distribution was performed by flow cytometry in MCF-7 cells. The results showed that USP22 depletion significantly decreased the proportion of S phase in MCF-7 cells and T47D cells, thereby suppressing cell proliferation (Fig. 6b and Supplementary Fig. S9b). In addition, ectopic expression of ERα partially reversed the reduced proportion of S phase following USP22 depletion (Supplementary Fig. S8). These results indicate that USP22 is involved in the regulation of cell cycle distribution, and the function of USP22 is partially related to ERα. We further examined the influence of USP22 on cell growth in BCa cell line. The colony formation assay was performed to determine the cell proliferative capability in MCF-7 cell line. The data demonstrated that when USP22 was knocked down (shUSP22), the MCF-7 cells formed a lower number of colonies than that in control cells (shCtrl). Meanwhile ectopic expression of USP22 led to a significant increase in number of colonies (Fig. 6c). However, USP22 depletion did not show obvious change in cell proliferation in ERα-negative BCa cell line (MDA-MB-231) (Fig. 6d). It has been previously described that ERα regulates the HIF1α pathway to confer endocrine resistance. We then examined the proliferative capability in MCF-7 cell line with ectopic expression of USP22 under hypoxic condition. The colony formation assay demonstrated that when cocl2 was added, MCF-7 cell line with ectopic expression of USP22 formed a larger number of colonies than that in the control cells (Fig. 6e). USP22 knockdown also formed a lower number of colonies in T47D cells (Supplementary Fig. S9c, d). Conclusively, these results suggest that USP22 promotes cell proliferation even under the hypoxia condition.

a Confirmation of MCF-7 cell lines with stable knockdown of USP22 (shUSP22) or ectopic expression of USP22 induced by lentivirus infection, a scramble shRNA or vector as a control (shCtrl or Ctrl). b Cell cycle analysis by flow cytometry for MCF-7 cell line with knockdown of USP22 (shUSP22). c, d Effects of USP22 on cell growth in MCF-7 cells or MDA-MB-231 cells as illustrated by the colony formation assay. All experiments were performed in both E2-free and 10−8 M E2 media and repeated at least three times. e Effects of USP22 on cell growth in MCF-7 cells as illustrated by the colony formation assay under hypoxic conditions. MCF-7 cells were treated with 10 nM E2 and 100 µM Cocl2 or vehicle for 24 h. f Representative images of tumor-bearing nude mice 28 days after injection as indicated. g Representative photographs of tumors in each group isolated from nude mice 4 weeks after injection. h The growth curves of tumor xenografts with ectopic expression of USP22 or control plotted from tumor volumes at points representing different time. i Average tumor weights from tumors as indicated after 28 days. j Average tumor volume from tumors as indicated after 28 days. The results represent mean ± SD, *P < 0.05, **P < 0.01, ***P < 0.001.
To determine the effect of USP22 on BCa cell growth in vivo, the MCF-7 cell line carrying ectopic expression of USP22 and control shRNA (Ctrl) were, respectively, injected into the left (USP22) or right (Ctrl) flank of the 4-week-old female BALB/C-null mice at per injection site (Fig. 6f). Subsequently, we measured the tumor size every week after hypodermic injection. As shown in Fig. 6g, the size of tumor from USP22-MCF-7 cells was mostly much larger in size than those from ctrl-MCF-7 cells. After 1 week of the cell injection, we measured long diameter and short diameter of tumor for every other week and calculated the tumor volume. As shown in Fig. 6h, tumors from USP22-MCF-7 cells were mostly much larger in size than those from ctrl-MCF-7 cells. After 4 weeks, tumors were harvested and weighed. The tumor weight of USP22-MCF-7 cells was obviously higher than those from Ctrl-MCF-7 cells (Fig. 6i). Furthermore, the tumor volume of USP22-MCF-7 cells exhibited markedly greater growth rate than those from Ctrl-MCF-7 cells (Fig. 6j). The results above indicate that USP22 promotes BCa cell growth in mice.
USP22 increases the resistance of breast cancer cells to ERα antagonists
Endocrine therapy against the ERα signaling pathway has been putatively considered as a potential method to deal with ERα+ BCa. An ERα antagonist, ICI 182,780 induces ERα degradation and blocks ERα-mediated transactivation. 4-Hydroxytamoxifen (tamoxifen) acting as an ERα antagonist inhibits estrogen binding to ERα, thereby suppressing ERα action. The effects of the ERα antagonists, ICI 182,780 and tamoxifen, on ERα protein stability and ERα activity were detected as shown in Fig. 7a, b. We thus turn to examine the influence of USP22 on cell growth/proliferation with the treatment of E2 and ERα antagonists in MCF-7 cells, T47D cells and tamoxifen resistant BT474 cells. As shown in Fig. 7d, our results demonstrated that USP22 knockdown suppressed E2-induced cell proliferation, meanwhile ectopic expression of USP22 increased E2-induced cell proliferation in MCF-7 cells (Supplementary Fig. S10b). Furthermore, knockdown of USP22 reduced the inhibition of cell proliferation by ICI 182,780 or tamoxifen (Fig. 7e, f). On the other hand, cell proliferation inhibition mediated by ICI 182,780 or tamoxifen was remitted by USP22 in time-response analysis in MCF-7 cells (Supplementary Fig. S10c, d). Consistently, the effect of USP22 on cell proliferation in T47D cells with the treatment of E2 or ERα antagonists was similar as that in MCF-7 cells (Supplementary Fig. S9e–g). In addition, knockdown of USP22 enhances the sensitivity of BT474 cells to tamoxifen (Fig. 7g, h), and when MCF-7 cells were transfected with USP22, the cells became more resistant to tamoxifen (Supplementary Fig. S10e). These data indicate that USP22 increases the resistance to antagonists in MCF-7, T47D, or BT474 cells.

Confirmation of the effects of ICI 182,780 (a) or tamoxifen (TAM) (b) on ERα stability or ERα action as indicated. c Confirmation of the efficiency of USP22 depletion in MCF-7 cell lines with stable knockdown of USP22 (shUSP22) induced by lentivirus infection, a scramble shRNA as a control (shCtrl). d Knockdown of USP22 inhibits cell proliferation in MCF-7 cells. e, f USP22 knockdown enhances the sensitivity of MCF-7 cells to ERα antagonists. MCF-7 cells carrying lentivirus-mediated USP22 knockdown (shUSP22) or the control cells (shCtrl) were treated with 0.1 µM of ICI 182,780 or 0.5 µM of TAM in the presence of E2 (10 nM) for the indicated days. Cell viability was measured using the MTS assay, and proliferation rates were calculated by values relative to the value of “0 day”. Points represent mean of three replicates. *P < 0.05, **P < 0.01, ***P < 0.001. g Confirmation of BT474 cell lines with stable knockdown of USP22 (shUSP22) induced by lentivirus infection, a scramble shRNA as a control (shCtrl). h Effect of USP22 depletion on cell growth in BT474 cells treated with TAM. i Schematic diagram illustrating the function of USP22 on modulation of ERα action and the role of USP22 in promotion of cell growth and endocrine resistance in breast cancer.
Discussion
It is known that ERα-induced nongenomic and genomic signaling pathways are crucial for ERα+ BCa progression and development of endocrine resistance. Identification of novel co-regulators of ERα would provide the potential therapeutic targets for endocrine resistance in ERα+ BCa. In this study, our findings demonstrate that high expression of USP22 was significantly associated with poor overall survival in BCa (Fig. 1a, b). We further show the high expression of USP22 is positively correlated with that of ERα in clinical BCa samples (Fig. 1c–f). Our data, which demonstrate the function of USP22 on modulation of ERα stability in BCa, further provide a mechanism for the clinical findings.
USP22 maintains the stability of ERα via triggering ERα deubiquitination
USP22 belonging to ubiquitin-specific proteases (USP) family participates in protein deubiquitination on histone or nonhistone proteins. Here, our data have demonstrated that USP22 maintains ERα stability by triggering the deubiquitination of K48- and K63-linked polyubiquitin chains on ERα (Fig. 3). It has been shown that several deubiquitinases (DUB), including USP5, CYLD, and UCH37, deubiquitinate both K48- and K63-linked polyubiquitinated proteins, here we provide the evidence that USP22 is a novel deubiquitinase reducing K48/K63 ubiquitination of ERα.
It has been demonstrated that a series of E3 Ub ligases trigger ERα ubiquitination to promote ubiquitin-mediated ERα proteolysis and regulate ERα-induced transcriptional activity, thereby to be essential for BCa progression. MDM2 as a Ub ligases interacts with ERα to promote ERα degradation and acts as an ERα co-activators in BCa cells [34]. Several lines of evidence demonstrate that although BRCA1 functions as an E3 ligase for ERα, the influences of BRCA1 on ERα action are controversial, it can act as a co-repressor or coactivator of ERα in deferent cellular contexts [35, 36]. The carboxyl terminus of Hsc70-interacting protein (CHIP) containing Ub ligase activity promotes ERα degradation and attenuates ERα-mediated transactivation [37]. The previous studies have demonstrated that ubiquitination-mediated ERα degradation links co-active or co-repressive regulation on ERα action, suggesting that the amount of ERα protein is not necessary for ERα activity. Our findings here identify USP22 as a DUB participates in maintenance of ERα stability enhances ERα-mediated transactivation.
USP22 co-activates ERα-induced transactivation
ERα is a crucial factor for ERα+ BCa progression and transforming to endocrine resistance. ERα exerts its oncogenic functions on BCa through the recruitment of a large number of co-regulators participating in modulation of ERα action. In this study, by analyzing TCGA data, we found that a series of genes regulated by USP22 were common to those induced by ESR1 in BCa clinical samples (Fig. 1g and Supplementary Table S2). These findings prompted the hypothesis that USP22 may function as a coactivator of ERα. The luciferase assay results demonstrated that USP22 obviously enhances ERα-induced transactivation, whereas USP22 with deubiquitinase activity mutant (USP22 HH/AA) had no co-active effect on ERα action. We further observed the upregulation of endogenous ERα target genes with ectopic expression of USP22 and downregulation of ERα target genes upon deletion of USP22 (Fig. 4). Co-IP experiments supported USP22 and ERα association (Fig. 2). ChIP assay analyses further strengthened the findings that USP22–ERα complex was recruited to cis-regulatory elements of ERα target gene. Our studies indicate that USP22 as a key ERα co-regulator enhances the transcription of ERα target genes.
USP22 acts as a therapeutic target for ERα-positive breast cancer
A growing body of findings have demonstrated that DUB-family members are implicated in multiple carcinomas. Thus, development of active site inhibitors would be crucial for tumor therapy [38, 39]. Proteasome-associated DUB (USP14 and UCHL5) inhibitors b-AP15 and platinum pyrithione enhance ubiquitin-mediated ERα degradation and inhibit ERα action, thereby inducing cell growth inhibition in BCa [40]. P5091 as a USP7 inhibitor has been reported to induce apoptosis in multiple myeloma cells and overcomes bortezomib resistance [41]. However, no agents that specifically target USP22 enzymatic activity are in experimental and clinical trials. A small molecule WP1130 acting as a deubiquitinase inhibitor has been found to induce rapid accumulation of K48/K63-linked polyubiquitinated proteins into juxtanuclear aggresomes [42]. Our results have shown that USP22 is capable to deubiquitinate K48- and K63-linked polyubiquitin chains of ERα. Thus, WP1130 might be a potential inhibitor for USP22, although the specific inhibitor for USP22 is still unknown.
In summary, our study has demonstrated that USP22 as a modulator of ERα protein level associates with ERα to participate in deubiquitination of K48- and K63-linked Ub chains of ERα, thereby inhibiting ERα degradation. On the other hand, USP22 emerges as a novel coactivator of ER signaling, thereby promoting cell growth and proliferation in BCa. These results support the concept that USP22 is a major driver of BCa progression and a potential therapeutic target for BCa and endocrine resistance (Fig. 7i).
Materials and methods
Antibodies
The antibodies used in this study were: anti-FLAG (cat# PA1-984B, Sigma), anti-His (cat# 66005, Proteintech), anti-HA (cat# GNI4110 Shanghai genomics), anti-USP22 (cat# A78455, Sigma), anti-ERα (cat# 8644, Cell Signaling), anti-c-Myc (cat# 10828, Proteintech), anti-hTERT (cat# C2C3-2, Genetex), anti-Cyclin D1 (cat# 2926, Cell Signaling), anti-HIF1α (cat# 20960, Proteintech), anti-GAPDH (cat# AC036, ABclonal), and anti-β-actin(cat# 60008, Proteintech). The detailed description of cell culture, transfections, and luciferase dual-reporter assays has been included in Supplementary Data.
Cell culture, transfections, and luciferase dual-reporter assays
Human BCa cell line MCF-7, T47D, BT474, and MDA-MB-231 were obtained from Prof. Yujie Sun’s lab. HEK293 cell line were obtained from the American Type Culture Collection. The above cell lines were authenticated by STR profiling. The HEK293, T47D, and MDA-MB-231 cells were cultured in Dulbecco’s modified Eagle’s medium, the MCF-7 cell was cultured in MEM, the BT474 cell was cultured in RPMI1640 (GIBCO-BRL). All the culture media were contained 10% fetal bovine serum, 2 mM glutamine.
Cell lines were cotransfected with the listed constructs according to the manufacturer’s instructions by using jetPRIMETM DNA Transfection Reagent (Polyplus transfection). After 24 h of transfections, cells were harvested and lysed in 80 μl of passive lysis buffer (Promega). Luciferase activities were analyzed using a Promega dual-luciferase reporter assay system. Firefly luciferase activity was normalized to the activity of Renilla luciferase control. Relative luciferase activity was analyzed using the luminometer Lumat LB 9507 (Berthold Technologies, Germany). All the results represent the means ± SD based on at least three independent experiments.
Co-Immunoprecipitation, western blot analysis, and GST pull-down assays
For Co-IP, the whole cell lysates were extracted and equal protein amounts were performed to Co-IP with specific antibody. The crude extracts and immune complexes were analyzed by western blotting. The Co-IP, western blot, and GST pull-down assays used in this study have been described previously in detail [43].
Immunofluorescence
Cells were fixed in 4% paraformaldehyde at room temperature for 15 min, and then blocked in 1% donkey serum albumin. The cells were incubated with primary antibody overnight at 4 °C and subsequently secondary antibody conjugated to FITC or Cy 3 or Cy 5 (Jackson Immunoresearch Laboratories Inc). Cells were stained with DAPI (Roche) to visualize the nuclei.
RNA isolation, quantitative real-time PCR (qPCR), siRNA transfection, and lentiviral production and infection
These sections have been described in Supplementary Data.
Chromatin Immunoprecipitation (ChIP), and ChIP–re-ChIP
Transfected MCF-7 cells were cross-linked with 1% formaldehyde (final concentration), and then were lysed with lysis buffer and sonicated on ice. IP of sonicated chromatin solutions was conducted by overnight incubation at 4 °C with anti-USP22 or anti-ERα or Histone modification antibodies. Protein A-sepharose beads were added, and then washed sequentially with low salt buffer, high salt buffer, LiCl buffer, and TE buffer. The protein–DNA complexes were eluted and the crosslinking was reversed, the purified DNA was resuspended in TE buffer. DNA samples (1 μl) were then amplified by real-time PCR. Primer pairs for the c-Myc promoter were sense: 5′-AGGCGCGCGTAGTTAATTC-3′, antisense: 5′-CGCCCTCTGCTTTGGGA-3′. The ChIP–re-ChIP assay has been performed in MCF-7 cells as previously described [44].
Xenograft tumor growth
Animal work was carried out in compliance with the ethical regulations approved by the Animal Ethics Committee of China Medical University. Two groups (six each) of 4-week-old female BALB/C-null mice were injected subcutaneously into 5.0 × 106 MCF-7 cells with lentivirus-mediated stable expression of USP22 and control vector, respectively. MCF-7 cells were suspended in 100 µl medium with half Metrigel (BD Biosciences). Tumors was measured weekly using an electronic caliper and the volume was calculated according to the formula 0.5236 × r12 × r2 (r1 < r2) [33].
The detailed description for FACS Analysis and Colony Formation Assay has been added in Supplementary data.
Collection of clinical mammary tissues
Human BCa tissues and corresponding adjacent tissues were obtained from the First Hospital of China Medical University. All samples were obtained with patients’ informed contents.
Immunohistochemical experiments
Formalin-fixed paraffin-embedded sections of breast tissue specimens were prepared from clinical mastectomy specimens in the First Hospital of China Medical University. Tissue sections were dewaxed, rehydrated, removed endogenous peroxidase, boiled for antigen retrieval, followed by incubation with anti-USP22 antibody and streptavidin-perosidase-conjugated second antibodies (Fuzhou Maixin Biotech. Co., Ltd). The signals were visualized with diaminobenzidine and the nuclei were counterstained with hematoxylin as previously described [44]. Images were taken with an Olympus microscope. The collection of BCa samples was approved by the Human Research Ethics Committee of the first affiliated hospital to China Medical University.
Statistics
All statistical analysis was performed using the SPSS statistical software program. For the real-time PCR and luciferase assay, two-sided Student’s t test was used to determinate the significant difference. For IHC, Mann–Whitney U test was used to determinate the significant difference between breast hyperplasia and malignant BCa tissues in different clinical stages. Overall survival curves were plotted according to the Kaplan–Meier method with the log-rank test applied for comparison. Respective P values as a measure of significance are indicated.
References
Manavathi B, Dey O, Gajulapalli VNR, Bhatia RS, Bugide S, Kumar R. Derailed estrogen signaling and breast cancer: an authentic couple. Endocr Rev. 2013;34:1–32.
Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002;2:101–12.
Yang J, AlTahan A, Jones DT, Buffa FM, Bridges E, Interiano RB, et al. Estrogen receptor-alpha directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc Natl Acad Sci USA. 2015;112:15172–7.
Stender JD, Nwachukwu JC, Kastrati I, Kim Y, Strid T, Yakir M, et al. Structural and molecular mechanisms of cytokine-mediated endocrine resistance in human breast cancer cells. Mol cell. 2017;65:1122–35.e1125.
Bhatt S, Stender JD, Joshi S, Wu G, Katzenellenbogen BS. OCT-4: a novel estrogen receptor-alpha collaborator that promotes tamoxifen resistance in breast cancer cells. Oncogene. 2016;35:5722–34.
De Marchi T, Foekens JA, Umar A, Martens JWM. Endocrine therapy resistance in estrogen receptor (ER)-positive breast cancer. Drug Discov Today. 2016;21:1181–8.
Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. Cracking the estrogen receptor’s posttranslational code in breast tumors. Endocr Rev. 2011;32:597–622.
Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113:207–19.
Zhou W, Slingerland JM. Links between oestrogen receptor activation and proteolysis: relevance to hormone-regulated cancer therapy. Nat Rev Cancer. 2014;14:26–38.
Jin K, Park S, Teo WW, Korangath P, Cho SS, Yoshida T, et al. HOXB7 Is an ERalpha cofactor in the activation of HER2 and Multiple ER target genes leading to endocrine resistance. Cancer Discov. 2015;5:944–59.
Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27–33.
Wade MA, Jones D, Wilson L, Stockley J, Coffey K, Robson CN, et al. The histone demethylase enzyme KDM3A is a key estrogen receptor regulator in breast cancer. Nucleic Acids Res. 2015;43:196–207.
Gao WW, Xiao RQ, Zhang WJ, Hu YR, Peng BL, Li WJ, et al. JMJD6 licenses ERalpha-dependent enhancer and coding gene activation by modulating the recruitment of the CARM1/MED12 co-activator complex. Mol Cell. 2018;70:340–57.e348.
Zhao Y, Lang G, Ito S, Bonnet J, Metzger E, Sawatsubashi S, et al. A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol Cell. 2008;29:92–101.
Atanassov BS, Evrard YA, Multani AS, Zhang Z, Tora L, Devys D, et al. Gcn5 and SAGA regulate shelterin protein turnover and telomere maintenance. Mol Cell. 2009;35:352–64.
Lin Z, Yang H, Kong Q, Li J, Lee SM, Gao B, et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol Cell. 2012;46:484–94.
Atanassov BS, Dent SY. USP22 regulates cell proliferation by deubiquitinating the transcriptional regulator FBP1. EMBO Rep. 2011;12:924–30.
Gennaro VJ, Stanek TJ, Peck AR, Sun Y, Wang F, Qie S, et al. Control of CCND1 ubiquitylation by the catalytic SAGA subunit USP22 is essential for cell cycle progression through G1 in cancer cells. Proc Natl Acad Sci USA. 2018;115:E9298–307.
Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Investig. 2005;115:1503–21.
Glinsky GV. Genomic models of metastatic cancer: functional analysis of death-from-cancer signature genes reveals aneuploid, anoikis-resistant, metastasis-enabling phenotype with altered cell cycle control and activated Polycomb Group (PcG) protein chromatin silencing pathway. Cell Cycle. 2006;5:1208–16.
Liu Y, Yang Y, Xu H, Dong X. Implication of USP22 in the regulation of BMI-1, c-Myc, p16INK4a, p14ARF, and cyclin D2 expression in primary colorectal carcinomas. Diagnostic Mol Pathol. 2010;19:194–200.
Lv L, Xiao XY, Gu ZH, Zeng FQ, Huang LQ, Jiang GS. Silencing USP22 by asymmetric structure of interfering RNA inhibits proliferation and induces cell cycle arrest in bladder cancer cells. Mol Cell Biochem. 2011;346:11–21.
Yang DD, Cui BB, Sun LY, Zheng HQ, Huang Q, Tong JX, et al. The co-expression of USP22 and BMI-1 may promote cancer progression and predict therapy failure in gastric carcinoma. Cell Biochem Biophys. 2011;61:703–10.
Piao S, Liu Y, Hu J, Guo F, Ma J, Sun Y, et al. USP22 is useful as a novel molecular marker for predicting disease progression and patient prognosis of oral squamous cell carcinoma. PLoS ONE. 2012;7:e42540.
Schrecengost RS, Dean JL, Goodwin JF, Schiewer MJ, Urban MW, Stanek TJ, et al. USP22 regulates oncogenic signaling pathways to drive lethal cancer progression. Cancer Res. 2014;74:272–86.
Ning Z, Wang A, Liang J, Xie Y, Liu J, Feng L, et al. USP22 promotes the G1/S phase transition by upregulating FoxM1 expression via beta-catenin nuclear localization and is associated with poor prognosis in stage II pancreatic ductal adenocarcinoma. Int J Oncol. 2014;45:1594–608.
Xiao H, Tian Y, Yang Y, Hu F, Xie X, Mei J, et al. USP22 acts as an oncogene by regulating the stability of cyclooxygenase-2 in non-small cell lung cancer. Biochem biophys Res Commun. 2015;460:703–8.
Tang B, Liang X, Tang F, Zhang J, Zeng S, Jin S, et al. Expression of USP22 and Survivin is an indicator of malignant behavior in hepatocellular carcinoma. Int J Oncol. 2015;47:2208–16.
Zhang Y, Yao L, Zhang X, Ji H, Wang L, Sun S, et al. Elevated expression of USP22 in correlation with poor prognosis in patients with invasive breast cancer. J Cancer Res Clin Oncol. 2011;137:1245–53.
Tang B, Tang F, Li B, Yuan S, Xu Q, Tomlinson S, et al. High USP22 expression indicates poor prognosis in hepatocellular carcinoma. Oncotarget. 2015;6:12654–67.
Wang H, Li YP, Chen JH, Yuan SF, Wang L, Zhang JL, et al. Prognostic significance of USP22 as an oncogene in papillary thyroid carcinoma. Tumour Biol. 2013;34:1635–9.
Kongsema M, Zona S, Karunarathna U, Cabrera E, Man EP, Yao S, et al. RNF168 cooperates with RNF8 to mediate FOXM1 ubiquitination and degradation in breast cancer epirubicin treatment. Oncogenesis. 2016;5:e252.
Xu K, Shimelis H, Linn DE, Jiang R, Yang X, Sun F, et al. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer Cell. 2009;15:270–82.
Duong V, Boulle N, Daujat S, Chauvet J, Bonnet S, Neel H, et al. Differential regulation of estrogen receptor alpha turnover and transactivation by Mdm2 and stress-inducing agents. Cancer Res. 2007;67:5513–21.
D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med. 2011;17:1636–40.
Fan S, Ma YX, Wang C, Yuan R-Q, Meng Q, Wang J-A, et al. p300 Modulates the BRCA1 inhibition of estrogen receptor activity. Cancer Res. 2002;62:141–51.
Fan M, Park A, Nephew KP. CHIP (carboxyl terminus of Hsc70-interacting protein) promotes basal and geldanamycin-induced degradation of estrogen receptor-alpha. Mol Endocrinol. 2005;19:2901–14.
Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–44.
Weathington NM, Mallampalli RK. Emerging therapies targeting the ubiquitin proteasome system in cancer. J Clin Investig. 2014;124:6–12.
Xia X, Liao Y, Guo Z, Li Y, Jiang L, Zhang F, et al. Targeting proteasome-associated deubiquitinases as a novel strategy for the treatment of estrogen receptor-positive breast cancer. Oncogenesis. 2018;7:75.
Chauhan D, Tian Z, Nicholson B, Kumar KGS, Zhou B, Carrasco R, et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22:345–58.
Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010;70:9265–76.
Zou R, Zhong X, Wang C, Sun H, Wang S, Lin L, et al. MDC1 enhances estrogen receptor-mediated transactivation and contributes to breast cancer suppression. Int J Biol Sci. 2015;11:992–1005.
Wang C, Sun H, Zou R, Zhou T, Wang S, Sun S, et al. MDC1 functionally identified as an androgen receptor co-activator participates in suppression of prostate cancer. Nucleic Acids Res. 2015;43:4893–908.
Acknowledgements
We appreciate Dr Yanshu Li, Dr Yunlong Huo, and Dr Tao Wen for helpful technique support. We thank Dr Shigeaki Kato (Soma Central Hospital, Fukushima, Japan) for providing ERα, its truncated mutants, and pERE-tk-luc plasmids, as well as important comments for draft. We thank Dr Yujie Sun (Nanjing Medical University, Nanjing, China) for MCF-7, T47D, BT474, and MDA-MB-231 cells, which were purchased from American Type Culture Collection.
Funding
This study was supported by the National Natural Science Foundation of China (31871286 for YZ, 81872015 for CW, 31701102 for SW, 81702800 for RZ, and 81502438 for TZ); 973 Program Grant from the Ministry of Science and Technology of China (2013CB945201); Ministry of Education fund innovation team (IRT 13101); Foundation for Special Professor of Liaoning Province for YZ (the fifth batch).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Y. Shi
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, S., Zhong, X., Wang, C. et al. USP22 positively modulates ERα action via its deubiquitinase activity in breast cancer. Cell Death Differ 27, 3131–3145 (2020). https://doi.org/10.1038/s41418-020-0568-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-0568-2
This article is cited by
-
USP22 supports the aggressive behavior of basal-like breast cancer by stimulating cellular respiration
Cell Communication and Signaling (2024)
-
USP22 regulates APL differentiation via PML-RARα stabilization and IFN repression
Cell Death Discovery (2024)
-
USP32 deubiquitinase: cellular functions, regulatory mechanisms, and potential as a cancer therapy target
Cell Death Discovery (2023)
-
USP22 upregulates ZEB1-mediated VEGFA transcription in hepatocellular carcinoma
Cell Death & Disease (2023)
-
MYSM1 acts as a novel co-activator of ERα to confer antiestrogen resistance in breast cancer
EMBO Molecular Medicine (2023)
