
Abstract
Sex-determining region Y-box 2 (SOX2), a well-known stemness biomarker, is highly expressed in a variety of cancers, including human highly invasive bladder cancer (BC). However, the role of SOX2 may vary in different kinds of malignancy. In the present study, we discovered that ChlA-F, a novel conformation derivative of isolate Cheliensisin A (Chel A), remarkably inhibits the invasive ability of human invasive BC cells through downregulation of SOX2 protein expression. We found that ChlA-F treatment dramatically decreases SOX2 protein expression in human high-grade invasive BC cells. Ectopic expression of SOX2 reversed ChlA-F inhibition of cell invasion ability in human bladder cancer cells, suggesting that SOX2 is a major target of ChlA-F during its inhibition of human BC invasion. Mechanistic studies revealed that ChlA-F downregulates SOX2 at both the protein degradation and protein translation levels. Further studies revealed that ChlA-F treatment induces HuR protein expression and that the increased HuR interacts with USP8 mRNA, resulting in elevation of USP8 mRNA stability and protein expression. Elevated USP8 subsequently acts as an E3 ligase to promote SOX2 ubiquitination and protein degradation. We also found that ChlA-F treatment substantially increases c-Jun phosphorylation at Ser63 and Ser73, initiating miR-200c transcription. The increased miR-200c directly binds to the 3′-UTR of SOX2 mRNA to suppress SOX2 protein translation. These results present novel mechanistic insight into understanding SOX2 inhibition upon ChlA-F treatment and provide important information for further exploration of ChlA-F as a new therapeutic compound for the treatment of highly invasive/metastatic human BC patients.
Similar content being viewed by others


Introduction
Worldwide, bladder cancer (BC) is the ninth most common cancer, with more than 79,030 novel cases and 16870 deaths in the USA in the year 2017 [1]. Initially, 70% of all cases are diagnosed as non-muscle-invasive bladder cancer (NMIBC), with 20–30% of tumors progressing into more aggressive and potentially lethal forms, and the remaining 30% as muscle-invasive bladder cancer (MIBC) [2]. MIBC is a life-threatening disease, which is responsible for almost 100% of death from this disease. Therefore, it is of extreme importance to identify new molecular targets that specifically regulate the pathological process of BC invasion for improving management of invasive BC patients.
Cheliensisin A (Chel A), a styryl-lactone isolated from Goniothalamus cheliensis Hu, has been shown to possess potent antitumor activities both in vitro and in vivo [3, 4]. The tendency to hydrolyze and poor water solubility limits the possibility of Chel A becoming a potential drug candidate. To address these issues, ChlA-F, a novel C8 fluoride conformation-derivative of Chel A, is synthesized and has been identified as being appropriate for a druggable candidate with preferable anticancer potency [5]. Our recent studies reveal that ChlA-F possesses the significant potency on anchorage-independent growth inhibition through induction of autophagy in a Sestrin-2 (SESN2)-dependent fashion in human high-grade invasive bladder cancer (BC) cells [5]. The current study aimed to evaluate the effect of ChlA-F on human BC cell invasion and elucidate the molecular mechanism(s) underlying this action.
Cancer cells are considered to be capable of limitless proliferation and self-renewal, similar to stem cells. Moreover, a small number of cancer cells express stem cell markers and possess the stem cell-like ability to maintain tumor growth and metastasis [6]. Stemness-associated genes such as SOX2, Nanog, Oct-4, and CD44 are involved in the maintenance of embryonic stem cell stemness, and more recent studies have demonstrated their expression in certain kinds of human tumors, where they regulate tumorigenesis and metastasis [7]. SOX2 has been described as an essential embryonic stem cell marker and necessary factor for induced cellular reprogramming [8, 9]. Recently, research about SOX2 has switched from focusing on embryogenesis and development to focusing on its function in disease, especially in cancer. These studies have strongly suggested that SOX2 promotes invasion, migration, and metastasis in squamous cell carcinoma, melanoma, colorectal cancer, glioma, gastric cancer, ovarian cancer, and hepatocellular carcinoma [7, 10,11,12,13,14]. Although the involvement of SOX2 in BC stem-like properties have been reported in several studies [8, 9, 15], the inhibition of SOX2 in the mediation of ChlA-F attenuating human BC cell invasion has not been examined to the best of our knowledge. Given our most recent studies show that stem-like property is associated with human BC cell invasion [16], we explored the potential effect of ChlA-F, a new compound with high activity on the inhibition of human high invasive BC cells, on SOX2 expression and cell invasion in human high invasive BC cells.
Materials and methods
Plasmids and reagents
The c-Jun dominant negative mutant (TAM67) was described in our previous studies [17, 18]. The GFP-HUR expression vector was a generous gift from Dr. Imed-Eddine Gallouzi (McGill University Health Center, McGill University, Montreal, Canada). shRNA constructs against human USP8 (RMM4431-2004108) and HuR were purchased from Open Biosystems (Pittsburg, PA, USA). The SOX2 overexpression construct pSin-EF2-SOX2, the miR-200c overexpression construct, the miR-145 inhibitor and miR-200c inhibitor were purchased from Addgene (Cambridge, MA, USA). The human miR-200c promoter (−1771 to −9) was cloned into the pGL3-basic luciferase reporter. The human SOX2 mRNA 3'-UTR luciferease reporter and SOX2 3′-UTR mutant luciferase reporter (miR-200c binding site mutated) were cloned into the pMIR luciferase reporter. Plasmids were prepared using the Plasmid Preparation/Extraction Maxi kit from QIAGEN (Valencia, CA, USA). Antibodies specific against p-c-Jun S63 (2361S), p-c-Jun Ser73 (3270S), total-JUN (9165S), c-Fos (2250S), c-Jun(D) (5000S), Elk-1 (9182S), E2F1 (3742S), GAPDH (5174S), GFP (2956S), and SOX2 (23064S) were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies specific against Sp1 (sc-H225) and HuR (sc-5261) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody against HNRPD/AUF1 (ARP40238_T100) was purchased from Aviva (San Diego, CA, USA). Antibodies against β-Actin (A5441) and Nucleolin (N2662) were obtained from Sigma-Aldrich Corporation (St. Louis, MO, USA). Bafilomycin A1 (BAF) (sc-201550) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Actinomycin D (Act D) (50-76-0) was purchased from Fisher Scientific (Pittsburgh, PA, USA). The chemicals cycloheximide (CHX) and MG132 were purchased from Calbiochem (San Diego, CA, USA). The inhibitor SP600125 was bought from Calbiochem (San Diego, CA, USA).
General information for chemical procedures
ChlA-F with purity >99% was prepared according to a developed protocol (see Patent ZL201310034985.5 and PCT/CN2014/071751). Briefly, a solution of cheliensisine A (548 mg, 2 mM) in DCM (5 mL) at −78 °C under N2 was added drop-wise to a solution of BF3·OEt2 (202 μL) in DCM. The mixture was stirred at this temperature for 0.5 h until no starting material was detected. The mixture was then quenched by a saturated aqueous NaHCO3 solution. The aqueous phase was extracted with dichloromethane (30 mL × 3). Combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated. The crude product was purified by flash chromatography on silica gel to produce ChlA-F as white foam (53%). Mass spectra and high-resolution mass spectra were performed on a VG Auto Spec-3000 or a Finnigan MAT 90 instrument. 1H-nuclear magnetic resonance (NMR) and 13C-NMR experiments were performed on Bruker AM-400 DRX-500 and DRX-600 NMR spectrometers at ambient temperature.
To perform in vitro experiments, ChlA-F was dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich Corporation, 67-68-5) to make an 8 mM stock concentration and further diluted in DMEM/Ham’s F12 medium with a final DMSO concentration of 0.1% (v/v) for cell culture experiments. The same amount of DMSO was used as a vehicle control in all experiments.
Cell culture and transfections
Human BC cell lines T24T and U5637 were described in our previous studies [17, 19]. T24T cells were cultured at 37 °C in a 5% CO2 incubator in a 1:1 mixture of DMEM/Ham’s F12 medium supplemented with 5% FBS, 2 mmol/L l-glutamine, and 25 mg/mL gentamycin. U5637 cells were cultured in RPMI 1640 supplemented with 10% FBS, 2 mmol/L l-glutamine, and 25 mg/mL of gentamycin. Cell lines were authenticated every 6–12 months by verifying viability, recovery, growth, morphology and chemical response as well as testing STR loci and sex using the PowerPlex® 16 HS System provided by Genetica DNA Laboratories (Burlington, NC 27215 USA), as described in our previous studies [17, 20]. Transfections were carried out with specific plasmid constructs using PolyJet™ DNA in vitro Transfection Reagent (SignaGen Laboratories, Gaithersburg, MD) according to the manufacturer’s instructions. Stable transfection selection of SOX2, shUSP8, shHuR, TAM67, miR-145 inhibitor, miR-200c inhibitor, and miR-200c in T24T cells was performed with puromycin (0.2–0.3 μg/mL) or G418 (500–1000 μg/mL), depending on the antibiotic resistance plasmid transfected, and surviving stable transfectants were pooled as a stable mass culture, as described in our previous studies [17, 20].
Western blot
T24T and U5637 cells and their transfectants were extracted with cell lysis buffer (10 mmol/L Tris-HCl, pH 7.4, 1% SDS, and 1 mmol/L Na3VO4), and protein concentrations were determined using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). Cell extracts were subjected to SDS-PAGE and then transferred to polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA, USA). The protein band specifically bound to the primary antibody was detected by Typhoon FLA 7000 (GE Healthcare, Chicago, IL, USA) using an alkaline phosphatase-linked secondary antibody and an enhanced chemifluorescence. The Western blotting system was described in our previous studies [21].
RT-PCR and quantitative real-time PCR
Total RNA was extracted with TRIzol reagent (Invitrogen) after ChlA-F treatment according to the manufacturer’s instructions, and cDNAs were synthesized with the Thermo-Script RT-PCR system (Invitrogen). mRNA amounts were measured by quantitative real-time PCR. The primers for human SOX2 were 5′-CAT GCA CCG CTA CGA CGT GAG-3′ (Forward) and 5′-TGG GAG GAA GAG GTA ACC ACA GG-3′ (Reverse). The primers for human USP4 were 5′-GCC TTT ACC TGA TGA GTT TG-3′ (Forward) and 5′-TGC TCT TTG CCT TCT TCC TG-3′ (Reverse). The primers for human USP8 were 5′-GGT TCT GGA CCA GCT CTT AC-3′ (Forward) and 5′-CTG CCA CTT CAC CTT TAT GC-3′ (Reverse). The primers for human USP22 were 5′-GAC TGC CTG CGA CGA TTC A-3′ (Forward) and 5′-TGG CTG AGT GTT CAA ATC G-3′ (Reverse). PCR products were separated on 2% agarose gels, stained with ethidium bromide, and imaged under UV light with the Alpha Innotech SP Image system (Alpha Innotech Corporation, San Leandron, CA, USA).
Total microRNA was extracted using the miRNeasy Mini Kit (Qiagen, Valencia, CA, USA). Analysis of miR-429, miR-200b, miR-21, miR-200c, miR-182, and miR-129 expression was conducted using the miScript PCR Starter Kit (Qiagen, Valencia, CA, USA) and miScript PCR kit II RT Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol, with U6 used as the endogenous normalizer. Initial activation was performed at 95 °C for 15 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 55 °C for 30 s, and extension at 70 °C for 30 s. Cycle threshold (CT) values were determined, and the relative expression of microRNAs was calculated by using the values of 2−△△CT, as described in our publications [22, 23].
Luciferase reporter assay
Cells were transfected with the indicated luciferase reporter in combination with the pRL-TK vector (Promega, Fitchburg, WI, USA) as an internal control and subjected to ChlA-F (4 or 8 μM) for the indicated time. Luciferase activity was determined with a microplate luminometer, as described in previous studies [22,23,24]. All experiments were performed in triplicate, with results expressed as the mean ± standard error.
Cell invasion assay
The invasion assay was performed using the BD BioCoat™ Tumor Invasion System (BD Falcon, NY, USA) according to the manufacturer’s instruction. Briefly, cells (3 × 104) were simultaneously seeded onto chamber inserts coated with Matrigel™ matrix (invasion) or uncoated (migration) in triplicate in 400 μL of a serum-free, 1:1 mixture of DMEM/Ham’s F12 medium. Inserts were placed into wells containing 1 mL of medium supplemented with 10% FBS. Cells were incubated for 24 h, and then cells on both the inside and outside of the chamber were fixed with 3.7% formalin for 5 min, washed twice, treated with 100% methanol for 20 min, washed twice again, and then stained with Giemsa (1:20 in PBS) for 30 min in the dark. The number of migrated cells attached to the other side of the insert was counted under a light OLYMPUS CKX41 microscope in three random fields at 200× magnification. The number of migrated and invasive cells per image was determined using Image J software. Data are presented as the percentage of invasion through the BD Matrigel™ matrix and membrane relative to the migration of cells through the uncoated membrane. Data shown are representative of three independent experiments.
RNA-IP assay
Transient transfectants of 293T (pEGFP-C1, GFP-HuR) cells were cultured in 10-cm dishes until 80–90% confluent. Culture medium was replaced with DMEM containing 0.1% FBS for 12 h, and cells were then lysed in Polysome Lysis Buffer (10 mM HEPES pH 7; 100 mM KCl; 5 mM MgCl2; 25 mM EDTA; 0.5% IGEPAL; 2 mM DTT; 50 units/mL RNase OUT; 50 units/mL SUPERase-In; 0.2 mg/mL heparin; and complete proteinase inhibitor) on ice. The lysate was incubated with normal IgG/Protein A/G plus-agarose or anti-GFP agarose (Santa Cruz Biotechnology, Inc.) at 4 °C for 12 h. Agarose beads were collected by centrifugation, washed three times with NET2 buffer, extracted with Trizol, and subjected to RT- PCR.
[35S]-methionine pulse assays
T24T cells were seeded into 10-cm dishes and cultured for 24 h. Cells were exposed to ChlA-F (8 μM) for the indicated time and then incubated with methionine–cysteine-free DMEM (Gibco-BRL, Grand Island, NY) containing 2% FBS (Gibco-BRL, Grand Island, NY) for 30 min. Next, we pretreated the cells with MG132 (10 μM) for 30 min and then incubated with 2% FBS methionine–cysteine-free DMEM containing [35S]-labeled methionine/cysteine (250 μCi/dish, Trans 35S-label; Perkin Elmer, Boston, MA) for the indicated time periods. Cells were extracted with lysis buffer (Cell Signaling, Beverly, MA) containing complete protein inhibitor mixture (Roche, Branchburg, NJ) on ice for 10 min. Five hundred milligram of total lysate was incubated with the anti-SOX2 monoclonal antibody (Cell Signaling Technology) for 2 h at 4 °C. Next, Protein A/G plus-agarose beads (Santa Cruz Biotechnology, Inc.) that were pre-cleared by 20 mg/mL BSA for 2 h were added to the mixture and incubated with agitation for an additional 2 h at 4 °C. The immunoprecipitated samples were washed with cell lysis buffer five times, incubated at 100 °C for 5 min, and then subjected to SDS-PAGE analysis. [35S]-labeled SOX2 protein was detected with the PhosphorImager Typhoon FLA 7000 (GE, Pittsburgh, PA).
Statistical analysis
The Student’s T-test was used to determine significant differences, and p < 0.05 was considered as a significant difference between compared groups.
Results
ChlA-F blocks BC cell invasion through downregulation of SOX2 expression
Given our most recent studies show that stem-like property is associated with human BC cell invasion [16], we explored the potential effect of ChlA-F on SOX2 expression and cancer cell invasion in human high invasive BC cells. To this end, T24T and U5637 cells were treated with the indicated dose of ChlA-F for 24 h, which resulted in downregulation of SOX2 (Fig. 1a). To further evaluate the contribution of SOX2 downregulation to ChlA-F inhibition of BC, T24T, and U5637 cells were transfected with a SOX2 overexpression construct (Fig. 1b). As shown in Fig. 1c, ChlA-F treatment dramatically inhibited both exogenous and endogenous SOX2 protein expression both in T24T and U5637 cells.

ChlA-F inhibits invasion through downregulation of SOX2 protein expression in bladder cancer cells. a T24T and U5637 cells were treated with ChlA-F at the indicated concentrations for 24 h. SOX2 protein expression was determined by Western blot. b, c T24T and U5637 cells stably transfected with SOX2 plasmid were seeded into 6-well plates and treated with or without 8 µM ChlA-F for 24 h. d, e The invasion abilities of T24T (Vector) and T24T (SOX2) cells treated with or without 8 µM ChlA-F were evaluated by using a BD BioCoat TM Matrigel TM Invasion Chamber. The scale bar was 200 μM. f, g The invasion abilities of U5637 (Vector) and U5637 (SOX2) cells treated with or without 8 µM ChlA-F were evaluated by using a BD BioCoat TM Matrigel TM Invasion Chamber. The scale bar was 200 μM. The asterisk (*) indicates a significant difference between cells treated with or without 8 µM ChlA-F. h The whole cell lysate of T24T(KOSOX2) cells and T24T (Vector) cells that were treated with or without ChlA-F at the indicated concentrations for 24 h were subjected to Western blot to determine SOX2 protein expression. i, j The invasion abilities of the indicated cells were subjected to transwell invasion assay by using a BD BioCoat TM Matrigel TM Invasion Chamber. The scale bar was 200 μM. The asterisk (*) indicates a significant decrease from T24T(Vector) cells treated with 8 µM ChlA-F. The symbol (#) indicates a significant decrease from T24T(Vector) cells treated with vehicle control
Cell migration is an essential cellular process refers that cells move from one location to another location [25], while cell invasion defines the ability of cells to become motile and to navigate through the extracellular matrix within a tissue or to infiltrate neighboring tissues [26]. Cancer cells that become invasive disseminate to secondary sites and form metastases [26]. To test whether ChlA-F was able to inhibit either migration and/or invasion in human BC cells, the BD BioCoat™ Tumor Invasion System was employed to evaluate the effects of ChlA-F on both migration and invasion. Consistent with the inhibition of SOX2 protein expression, ChlA-F profoundly inhibited cell invasion without showing the observable effect on cell migration in both T24T(Vector) and T24T cells overexpressing SOX2, T24T(SOX2), cells (Fig. 1d, e). This finding was also observed in U5637(Vector) and U5637 cells overexpressing SOX2, U5637(SOX2), cells (Fig. 1f, g). Moreover, we also knocked out the SOX2 gene using CRISPR/Cas9 systems in T24T cells to mimic the biologic effects of ChlA-F treatment. As shown in Fig. 1h, the stable SOX2 knockout transfectant T24T(KOSOX2) was identified for comparing the invasion abilities of the stable transfectants. The knockout of SOX2 remarkably decreased the T24T cell invasion abilities, but not migration, in comparison to its vector transfectants (Fig. 1i, j), indicating that knockout of SOX2 could mimic the inhibitory effect of ChlA-F on BC cell invasion. These results strongly reveal that ChlA-F specific inhibits invasion without affecting migration accompanied by attenuation of both endogenous and exogenous SOX2 expression in both human BC T24T and U5637 cells.
ChlA-F downregulates SOX2 expression at the level of protein degradation by increasing USP8 protein expression
To investigate the underlying mechanisms of SOX2 regulation, we examined the effect of ChlA-F on SOX2 mRNA expression. As shown in Fig. 2a, ChlA-F treatment did not show a noticeable effect on SOX2 mRNA expression both in T24T and U5637 cells. This result was inconsistent with the result obtained for SOX2 protein and suggested that ChlA-F inhibition of SOX2 protein expression occurs at the level of protein degradation or translation. Given that CHX is a commonly used protein synthesis inhibitor, it was employed to block new protein synthesis, which allowed us to determine the degradation of synthesized proteins in the cells. To determine if the regulation of SOX2 by ChlA-F occurs at the protein degradation level, T24T cells were treated with CHX alone or in combination with ChlA-F at indicated time periods. We observed that SOX2 protein degradation rates were markedly increased when cells were co-incubated with ChlA-F plus CHX in comparison to cells incubated with CHX alone (Fig. 2b). In eukaryotic cells, the ubiquitin-proteasome pathway and lysosomal proteolysis mediate protein degradation. We first examined if autophagy could induce SOX2 protein degradation, and the results showed no apparent differences in SOX2 protein expression between cells co-treated with ChlA-F and BAF and cells treated with ChlA-F alone (Fig. S1), suggesting that ChlA-F-mediated SOX2 protein degradation might occur through the ubiquitin-proteasome pathway. To test this possibility, MG132, a proteasome inhibitor, was employed. The inhibition of proteasome-dependent axis by MG132 reversed ChlA-F inhibition of SOX2 protein, revealing that the proteasome-dependent pathway is participating in ChlA-F-inhibited SOX2 protein degradation (Fig. 2c). Given that UBE2S has been reported to regulate SOX2 protein ubiquitination [27], we first evaluated the potential effect of ChlA-F on UBE2S expression and the result was negative as shown in Fig. S2. Ubiquitin-like domain of ubiquitin Specific Peptidase 4 (USP4) could bind to the catalytic domain of ubiquitination enzyme, by which USP4 can inhibit ubiquitination enzyme activity [28], whereas USP8 can remove conjugated ubiquitin from proteins and therefore plays an important regulatory role at the level of protein turnover by preventing degradation [29]. USP22 has also been reported to regulate SOX2 expression [30]. To determine which is the upstream regulator mediating SOX2 protein degradation by ChlA-F, USP4, USP8, USP22 mRNA level were first evaluated following ChlA-F treatment. As shown in Fig. 2d, USP8 mRNA was upregulated in T24T cells following ChlA-F treatment, whereas USP4 and USP22 mRNA showed only minimal alterations. We also detected USP8 protein expression by Western Blot. Consistent with mRNA results, USP8 protein expression was remarkably upregulated after treatment with ChlA-F (Fig. 2e). To test the relationship of USP8 induction with SOX2 downregulation following ChlA-F treatment, we used shRNAs to knockdown USP8 expression in human BC T24T cell lines and evaluated the effect on SOX2 protein expression. We found that knockdown of USP8 increased the basal levels of SOX2 protein and reversed ChlA-F-induced downregulation of SOX2 (Fig. 2f). Accordingly, SOX2 protein degradation was inhibited in T24T (shUSP8) cells compared with T24T (Nonsense) cells following ChlA-F treatment (Fig. 2g). Besides, to determine the role of USP8 induction in ChlA-F inhibition of BC cell invasion, we evaluated cell invasion in T24T (Nonsense) and T24T (shUSP8) stable transfectants. Knockdown of USP8 attenuated ChlA-F inhibition of BC cell invasion but did not affect BC cell migration (Fig. 2h, i). Taken together, our results demonstrated that ChlA-F induces USP8 expression, promoting SOX2 protein degradation and leading to inhibition of human BC cell invasion.

ChlA-F attenuates SOX2 protein expression through accumulating USP8 expression. a T24T and U5637 cells were treated with 4 or 8 µM ChlA-F for 24 h. The cells were then used to extract total RNA using Trizol reagent. Real-time PCR was carried out to detect SOX2 mRNA expression. b Cycloheximide (CHX) (50 µg/mL) with or without ChlA-F (8 µM) was added to the culture medium and incubated for the indicated time. SOX2 protein expression was determined by Western blot. β-Actin was used as an internal control. c T24T cells were treated with or without 8 µM ChlA-F and with or without 10 µM MG132 for 24 h. SOX2 protein levels were assessed by Western blot. d The mRNA expression levels of USP4, USP8, and USP22 were determined by real-time PCR after T24T cells treated with 4 µM or 8 µM ChlA-F for 12 h. e T24T cells were treated with ChlA-F (µM) at the indicated doses for 24 h, and USP8 protein levels were determined by Western blot. f T24T (shUSP8) and T24T (Nonsense) cells were exposed to 8 µM ChlA-F for 24 h. USP8 and SOX2 protein expression were determined by Western blot. g T24T (shUSP8) and T24T (Nonsense) cells were treated with 8 µM ChlA-F, then analyzed for SOX2 protein degradation rates in the presence of cycloheximide (CHX) (50 µg/mL) over different time periods. The middle panel, same with top panel, was separated two parts derived from two cell transfectants to make the data easily comparable between two transfectants. h The invasion abilities of T24T (Nonsense) and T24T (shUSP8) cells treated with or without 8 µM ChlA-F were evaluated by using a BD BioCoatTM MatrigelTM Invasion Chamber. Migration ability was determined using the empty insert membrane without the matrigel, while invasion ability was evaluated by using the same system with Matrigel applied. Scale bar was 200 μM. i The invasion rate was normalized to the insert control according to the manufacturer’s instructions. Results were presented as T24T (Nonsense) cells relative to T24T (shUSP8) cells, which were exposed to 8 µM of ChlA-F for 24 h. The asterisk (*) indicates a significant difference between the cells
ChlA-F increases HuR protein expression to enhance USP8 mRNA stability
The upregulation of USP8 expression by ChlA-F treatment might occur at the transcriptional, post-transcriptional, or translational level. To further investigate the potential mechanisms underlying USP8 accumulation, Actinomycin D (Act D) was used to block new mRNA synthesis and thus facilitates to evaluate the already synthesized RNA degradation or RNA stability [31, 32]. To further determine if ChlA-F regulates USP8 expression at the mRNA stability level, T24T cells were treated with Act D (20 μg/mL) with or without ChlA-F for the indicated different time periods, and real-time PCR was used to evaluate USP8 abundance, normalized to that of the relatively stable GAPDH mRNA. The results indicated that treatment of T24T cells with ChlA-F in the presence of Act D led to an increase in the USP8 mRNA stability (half-life≈24 h) in comparison to cells treated with Act D alone (half-life≈10 h) (Fig. 3a), indicating that ChlA-F positively promotes USP8 mRNA stability.

ChlA-F enhances USP8 mRNA stabilization by promoting HuR expression. a T24T cells were treated with Act D (20 μg/mL) for the indicated time periods with or without ChlA-F (8 μM). Total RNA was isolated and subjected to real-time PCR analysis for USP8 mRNA expression. b T24T and U5637 cells were treated with ChlA-F(µM) at the indicated doses, and cell extracts were subjected to Western blot to determine Nucleolin, AUF1, and HuR protein levels. β-Actin was used as an internal control. c T24T (shHuR) and T24T (Nonsense) cells were treated with or without 8 µM ChlA-F for 24 h. Cell extracts were subjected to Western blot to determine HuR, USP8 and SOX2 protein levels. β-Actin was used as an internal control. d USP8 mRNA expression was determined by real-time PCR in T24T (shHuR) and T24T (Nonsense) cells treated with or without 8 µM ChlA-F for 24 h. The symbol “#” indicates a significant difference between Vector transfectant and shHuR stable transfectant following ChlA-F treatment (p < 0.05). e 293T cells were seeded into 10-cm dishes. After cell density reached 70–80%, the cells were transient transfected with either GFP-vector or GFP-HuR constructs as indicated. 48 h after transfection, the transfectants were extracted for an RNA-IP with anti-GFP antibody to assay HuR binding to USP8 mRNA, as described in “Materials and methods”. XIAP [56] and β-Actin were used as positive control and negative control respectively for HuR binding
Several RNA-binding proteins, such as Nucleolin, AUF1, and HuR, have been reported to bind to their target mRNAs and regulate their stability [33,34,35]. To test the potential contribution of these RNA-binding proteins to ChlA-F upregulation of USP8 mRNA stability, the effects of ChlA-F on the protein expression of Nucleolin, AUF1, and HuR were evaluated in both T24T and U5637 cells. As shown in Fig. 3b, ChlA-F treatment led to the accumulation of HuR protein in a dose-dependent manner, whereas it did not show an observable effect on the expression of Nucleolin or AUF1. To further determine the role of HuR in the regulation of USP8 mRNA stability, an shRNA specifically targeting human HuR was used to knockdown HuR expression in T24T cells (Fig. 3c). HuR knockdown abolished the upregulatory effects of ChlA-F on the expression of USP8 mRNA and protein and reversed ChlA-F-induced downregulation of SOX2 expression in T24T cells (Fig. 3c, d). Moreover, RNA-IP assays showed that HuR could specifically bind to USP8 mRNA (Fig. 3e). Thus, our results suggest that HuR directly binds to USP8 mRNA and increases its stability.
The induction of miR-200c, not miR-145, is crucial for ChlA-F inhibition of SOX2 protein translation and BC cell invasion
To determine if effects on translation explain the decreased SOX2 protein levels in T24T cells treated with ChlA-F, a [35S]-methionine short-term pulse-labeling assay was performed to examine the SOX2 protein translational process. As shown in Fig. 4a, the incorporation of [35S]-methionine/cysteine into newly synthesized SOX2 protein was reduced in T24T cells following ChlA-F treatment, indicating that synthesis of SOX2 protein was impaired by ChlA-F. Several studies have suggested that miRNAs repress gene expression by binding to complementary sequences in the 3′ untranslated region (3′-UTR) of their target mRNAs to inhibit the initiation step of translation [36, 37]. To elucidate the mechanisms involved in ChlA-F inhibition of SOX2 protein translation, a SOX2 3′-UTR luciferase reporter was transiently co-transfected with pRL-TK into T24T and U5637 cells. The transfectants were used to test the potential effect of ChlA-F on SOX2 mRNA 3′-UTR activity. We found that ChlA-F treatment leads to a marked reduction of SOX2 mRNA 3′-UTR activity in a dose-dependent manner in both T24T and U5637 cells. (Fig. 4b). Based on these results, we used the TargetScan and miRcode databases to screen for potential miRNA binding sites in the 3′-UTR regions of SOX2 mRNA. As shown in Fig. 4c, this analysis indicated that miR-200b, miR-200c, miR-429, miR-145, miR-21, miR-182, and miR-129 have potential binding sites in the 3′-UTR regions of SOX2 mRNA. To identify which of these miRNAs was responsible for regulation of SOX2 translation, we evaluated their expression via real-time PCR in T24T cells treated with ChlA-F. We found that ChlA-F treatment specifically induces the expression of miR-200c and miR-145 (Fig. 4d), indicating that they might be involved in downregulation of SOX2 translation followed ChlA-F treatment.

ChlA-F inhibits SOX2 protein translation by increasing miR-200c levels. a After treatment with or without ChlA-F (8 µM), T24T cells were treated with MG132 (10 µM) for 30 min. Newly synthesized SOX2 protein was monitored by a pulse assay using 35S-labeled methionine/cysteine. b A SOX2 3′-UTR-driven luciferase reporter and a TK reporter were transiently transfected into T24T cells treated with or without ChlA-F (4 or 8 µM). The luciferase activity of each transfectant was evaluated, and the bars show mean ± SD from three independent experiments. The asterisk (*) indicates a significant decrease compared with control transfectants (p < 0.05). c Potential microRNA binding sites in SOX2 mRNA 3′-UTR were predicted using the TargetScan, Pictar, and miRcode databases. d Quantitative real-time PCR was used to measure miRNA expression. The bars show mean ± SD from three independent experiments. The asterisk (*) indicates a significant increase in comparison with T24T control cells (p < 0.05). e Quantitative real-time PCR was employed to evaluate miR-200c expression in U5637 cells. The bars show mean ± SD from three independent experiments. The asterisk (*) indicates a significant increase in comparison with U5637 control cells (p < 0.05). f Schematic of the miR-200c binding sites and its mutant of the pMIR-report-SOX2 3′-UTR luciferase reporter. g Attenuation of ChlA-F inhibition of SOX2 mRNA 3′-UTR activity in the miR-200c binding site mutant of pMIR-report-SOX2 3′-UTR transfectants in comparison to ChlA-F inhibition of SOX2 mRNA 3′-UTR reporter activity in wild-type reporter transfectants. The asterisk (*) indicates a significant decrease in comparison with the vehicle control group. The symbol “&” indicates a significant difference between WT SOX2 3′UTR transfectant and Mutant SOX2 3′UTR transfectant upon ChlA-F treatment (p < 0.05)
To test if miR-200c and/or miR-145 inhibit SOX2 translation, T24T cells were first stably transfected with miR-145 inhibitor. Unexpectedly, inhibition of miR-145 expression by its inhibitor slightly decreased SOX2 level, suggesting that miR-145 inhibitor was not able to increase SOX2 protein expression. This result, together with the data showing that miR-145 overexpression showed no observable effect on SOX2 protein expression in T24T cells (Fig. S5), promoted us to exclude the possible involvement of miR-145 in the inhibition of SOX2 translation. Since miR-200c was induced by ChlA-F in both T24T and U5637 cells (Fig. 4d, e), the point mutation at miR-200c binding site in wild-type SOX2 mRNA 3′-UTR luciferase reporter was constructed as displayed in Fig. 4f. Both WT and mutant of SOX2 mRNA 3′-UTR luciferase reporter were then transfected into T24T cells, respectively. As shown in Fig. 4g, ChlA-F treatment profoundly inhibited WT SOX2 mRNA 3′-UTR luciferase reporter activity, whereas mutation of miR-200c binding site impaired the reporter activity in response to ChlA-F treatment, indicating that miR-200c binding site is required for ChlA-F inhibition of SOX2 mRNA 3′-UTR activity. Therefore, miR-200c and its inhibitor were stably transfected into T24T cells, respectively, and the levels of miR-200c were evaluated by real-time PCR, as shown in Fig. 5a, c. As expected, ectopic overexpression of miR-200c dramatically decreased SOX2 protein expression (Fig. 5b) and the introduction of miR-200c inhibitor reversed ChlA-F downregulation of SOX2 protein expression (Fig. 5d). Consistently, the miR-200c inhibitor attenuated ChlA-F inhibition of cellular invasion without affecting their migration in T24T cells (Fig. 5e–g). Taken together, our results revealed that ChlA-F treatment induces miR-200c expression, resulting in reduced SOX2 protein expression and mediating inhibition of the invasive ability of human BC cells.

miR-200c plays an vital role in the downregulation of SOX2 protein by ChlA-F. a, b Overexpression of miR-200c in T24T cells was evaluated by real-time PCR, and SOX2 protein expression was determined by Western blot. c miR-200c inhibitor lentivirus was used to infect T24T cells, and knockdown efficiency was determined by real-time PCR. Results are presented as the mean ± SD of triplicates. The asterisk (*) indicates a significant decrease as compared to Vector control (p < 0.05). d T24T (Vector) and T24T (miR200c inhibitor) cells were treated with or without 8 µM ChlA-F. SOX2 protein expression was determined by Western blot. β-Actin was used as a protein loading control. e The invasion abilities of T24T (Vector) and T24T (miR200c inhibitor) cells treated with or without 8 µM ChlA-F were evaluated by using a BD BioCoat TM Matrigel TM Invasion Chamber. Migration ability was determined by using the empty insert membrane without Matrigel. Invasion ability was assessed by using the same system with Matrigel applied. The scale bar was 200 μM. f, g The invasion and migration rates were normalized with the insert control according to the manufacturer’s instructions. The asterisk (*) indicates a significant difference between cells treated with or without 8 µM ChlA-F
JNK pathway activation is responsible for ChlA-F-mediated miR-200c transcription
miRNA biogenesis is regulated at multiple levels, including transcription, maturation by Drosha and Dicer, modification by RNA editing, RNA methylation, uridylation, adenylation, Argonaute-loading, and RNA decay [38]. Therefore, we transfected a wild-type, miR-200c-promoter-driven luciferase reporter into T24T cells to evaluate the effect of ChlA-F on miR-200c transcription. As expected, ChlA-F treatment led to a distinct increase in miR-200c promoter transcription in a dose-dependent manner (Fig. 6a). To identify the transcription factor(s) involved in ChlA-F-induced miR-200c transcription, bioinformatics analysis of the miR-200c promoter was performed. We found several potential transcription factors binding sites, including E2F-1, AP-1, Elk-1, SOX2, and SP1, in the miR-200c promoter region, as shown in Fig. 6b. To identify the specific transcription factor(s) involved in the modulation of miR-200c transcription, the effect of ChlA-F on related transcription factor protein abundance/activation was determined. We found that ChlA-F treatment specifically induced c-Jun phosphorylation at Ser63 and Ser73, whereas it did not lead to any observable increases in c-Fos, E2F-1, SP1, or Elk-1, suggesting that c-Jun activation is involved in the transcriptional activity of the miR-200c promoter following ChlA-F treatment (Fig. 6c). SOX2 also has binding sites in the miR-200c promoter region, so we tested if SOX2 could regulate miR-200c transcription. As shown in Fig. 6d, knockout of SOX2 decreased miR-200c levels, which is inconsistent with the fact that ChlA-F downregulated SOX2 but increased miR-200c expression. Thus, a stable transfectant of the dominant-negative mutant JUN (TAM67) was used to address the role of c-Jun in ChlA-F-induced miR-200c transcription and expression. TAM67 is a truncation mutant of c-Jun that lacks the major transactivation domain of c-Jun (amino acids 1–122) but retains the DNA-binding and leucine zipper domains, and therefore a TAM67 heterodimer binds DNA but does not activate transcription of c-Jun [39]. Expectedly, blockage of JUN activation by ectopic expression of TAM67 abolished the upregulatory effects of ChlA-F on miR-200c promoter activity (Fig. 6e) and expression (Fig. 6f), as well as abolishing the inhibitory effects of ChlA-F on SOX2 protein expression (Fig. 6g). Mitogen-activated protein kinases (MAPKs) are widely expressed in multicellular organisms, with critical roles in multiple biological processes, such as cell proliferation, death, differentiation, migration, and invasion. Multiple extracellular stimuli can activate MAPKs and lead to a broad intracellular response through the MAPK signaling pathway [40]. Therefore, we analyzed the activation of kinases upstream of c-JUN upon ChlA-F treatment. As shown in Fig. 6h, activation of JNK, MEK, and ERK was observed in accordance with c-Jun activation following ChlA-F treatment in both T24T and U5637 cells. Since JNK is a well-known an upstream kinase responsible for c-Jun phosphorylation and activation [41], we employed JNK inhibitor SP600125 to evaluate its effect on miR-200c transcription and SOX2 expression. The results indicated that treatment of cells with JNK inhibitor SP600125 markedly attenuated the phosphorylated level of JNKs and c-Jun induced by ChlA-F (Fig. 6i), and such inhibition of c-Jun activation reversed ChlA-F inhibition of SOX2 expression (Fig. 6i). Consistently, the inhibition of JNK and c-Jun by SP600125 also abolished miR-200c promoter activity (Fig. 6j). Collectively, these results suggest that JNK pathway activation is responsible for ChlA-F-mediated miR-200c transcription.

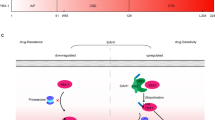
ChlA-F increases miR-200c expression by promoting the expression of c-Jun. a The miR-200c promoter-driven luciferase reporter and TK reporter were transiently transfected into T24T cells treated with various concentrations of ChlA-F (µM) for 12 h. The luciferase activity of each transfectant was evaluated, and the bars show mean ± SD from three independent experiments (p < 0.05). b Schematic representation of transcription factor binding sites in the human miR-200c promoter region from −1771 to −9. c T24T cells were treated with ChlA-F at the indicated doses for 24 h, and the cell extracts were subjected to Western blot to determine transcription factor expression. β-Actin was used as an internal control. d miR-200c expression was determined by real-time PCR in T24T SOX2 knockout cells and T24T(vector) cells. e, f Stable transfectants of TAM67 and its scramble vector were treated with 8 µM ChlA-F. miR-200c promoter activity was evaluated by a luciferase assay (e). miR-200c expression was determined by real-time PCR (f). The symbol “#” indicates a significant difference in comparison with scramble Vector transfectant upon ChlA-F treatment (p < 0.05). g Cells stably transfected with Vector or TAM67 cells were treated with or without 8 µM ChlA-F, and cell extracts were subjected to Western blot to determine the expression of c-Jun and SOX2. h Activation of JNK, MEK, and ERK in the MAPK signaling pathway was determined in accordance with c-Jun activation following ChlA-F treatment in T24T and U5637 cells. i T24T cells were treated with or without 8 µM ChlA-F and with or without SP600125 (25 μM) for 24 h. The cell lysates were subjected to Western blot assay for determination protein levels using specific antibodies as indicated. j Co-transfectants of the miR-200c promoter and TK in T24T cells were treated with or without 8 µM ChlA-F and with or without SP600125 (25 μM) for 24 h. Then miR-200c promoter activity was evaluated by a luciferase assay. The symbol “#” indicates a significant difference between with or without SP600125 following ChlA-F treatment (p < 0.05). k Schematic summary of molecular mechanisms underlying the anticancer activity of ChlA-F on human bladder cancers
Discussion
ChlA-F was recently found to have an anticancer effect, although the mechanism remains unclear. Our studies recently demonstrated that treatment with ChlA-F significantly inhibits anchorage-independent growth of human BC cells by induction of autophagy in a Sestrin2-dependent fashion [5]. Furthermore, ChlA-F inhibits the invasive ability of human BC cells via attenuation of MMP2 expression. These studies indicated that ChlA-F possesses remarkable anticancer activity in cancer cells. In the present study, we showed that ChlA-F treatment dramatically decreases SOX2 expression at both the protein degradation and translation levels in human high-grade invasive BC cells, inhibiting human BC cell invasion. ChlA-F treatment induced expression of HuR protein, increasing HuR interaction with USP8 mRNA and resulting in elevation of USP8 mRNA stability and subsequent SOX2 protein degradation. ChlA-F treatment profoundly increased c-Jun phosphorylation at Ser63 and Ser73, promoting transcriptional induction of miR-200c by ChlA-F treatment and in turn suppressing SOX2 protein translation by directly targeting the SOX2 mRNA 3′-UTR. Collectively, our results present novel mechanistic insight into an essential role of SOX2 in human BC invasion as diagrammed in Fig. 6k and further suggest a potential role for SOX2 in BC prognosis and treatment.
SOX2, a self-renewal transcription factor crucial to maintaining pluripotency in embryonic stem cells, has been reported to play an important role in the development of numerous malignancies [14, 42,43,44,45,46]. SOX2 is involved in many oncogenic biological processes, including cancer cell proliferation, migration, and invasion. Although the role of SOX2 in human BC cell invasion remains unclear, a relationship between SOX2 expression and invasion has been documented in other malignancies. Recently, it has been reported that SOX2 targets fibronectin1 or phosphorylates Src kinase to promote cell migration and invasion in ovarian cancer [14, 42]. In prostate cancer cells, both acute and chronic exposure to hypoxia can result in elevated expression levels of SOX2, thereby promoting cell invasion and sphere formation [43]. In cervical squamous cell carcinoma, SOX2 is overexpressed and contributes to cervical cancer cell migration and invasion in vitro [45]. Furthermore, loss of miR-638 in vitro promotes cell invasion and a mesenchymal-like transition by directly targeting SOX2 in colorectal carcinoma cells [46]. Here, we found that ChlA-F treatment decreases both endogenous and exogenous SOX2 protein expression in human BC cells. Mechanistic studies showed that ChlA-F specifically triggers expression of the RNA-binding protein, stabilizing USP8 mRNA and promoting SOX2 protein degradation. Further, treatment with ChlA-F activated the MAPK/JNK/c-Jun cascade and increased miR-200c transcription, mediating SOX2 protein translation inhibition. The mechanism described above mediates ChlA-F downregulation of SOX2 protein expression, inhibiting BC cell invasion.
The ubiquitin-specific protease (USP) family, with ~85 members, is the largest family of deubiquitination enzymes (DUBs) [47]. USP8 has been reported to accelerate degradation of epidermal growth factor receptor (EGFR) [48] upon EGF stimulation. Although USP8 has been previously reported to be implicated in the regulation of ubiquitin dynamics, its connection to the ChlA-F anticancer activity has never been explored. The results obtained from analysis of the normalized TCGA database revealed that USP8 expression was significantly downregulated in BCs (Fig. S6), while ChlA-F treatment remarkably increased USP8 protein expression, revealing that USP8 induction might be associated with anticancer activity of ChlA-F compound. This notion was greatly supported by our findings that knockdown of USP8 increased SOX2 protein stability and abolished the inhibition of cancer cell invasion by ChlA-F treatment. Given that USP8 is an endosome-associated deubiquitinating enzyme and regulates ubiquitination status and degradation of several plasma membrane proteins [29, 49], we anticipate that USP8 might indirectly downregulate SOX2 following ChlA-F treatment in human BC cells.
miRNAs were recently reported to regulate BC cell invasion and metastasis [50, 51]. miR-200c is considered to be an important regulator of suppression of tumorigenesis and cancer metastasis. For example, miR-200c induces epithelial differentiation and suppresses EMT by inhibiting translation of the EMT-activators ZEB1 and ZEB2 in intrahepatic cholangiocellular carcinoma, a type of primary liver cancer [52]. Moreover, miR-200c expression is lost and expression of its target genes ZEB1/ZEB2 is increased in highly invasive MDA-MB-231 breast cancer cells, facilitating cell invasion and distant metastasis [53]. Conversely, ZEB1 links activation of EMT and maintenance of stemness by suppressing stemness-inhibiting miRNAs, such as miR-200c, in pancreatic and colorectal cancer cells [54]. A similar negative feedback loop of miRNA target genes also appears in the regulation of miR-200c and its target gene SOX2 [55]. It has been shown that miR-200c negatively regulates SOX2 by binding the SOX2 3′UTR. In turn, SOX2 binds to specific transcription factor-binding sites of the miR-200c promoter and inhibits its transcription [55]. Here, we found that miR-200c directly binds to the SOX2 3′UTR and inhibits SOX2 protein translation following ChlA-F treatment. However, miR-200c was unexpectedly reduced in SOX2 knockout cells. This result is inconsistent with our finding that SOX2 was reduced and miR-200c was upregulated after ChlA-F treatment. Thus, our results suggest that SOX2, a downstream gene of miR-200c, is responsible for ChlA-F inhibition of BC cell invasion.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. Cancer J Clin. 2017;67:7–30.
Divrik RT, Sahin AF, Yildirim U, Altok M, Zorlu F. Impact of routine second transurethral resection on the long-term outcome of patients with newly diagnosed pT1 urothelial carcinoma with respect to recurrence, progression rate, and disease-specific survival: a prospective randomised clinical trial. Eur Urol. 2010;58:185–90.
Zhang J, Gao G, Chen L, Deng X, Li J, Yu Y, et al. Cheliensisin A inhibits EGF-induced cell transformation with stabilization of p53 protein via a hydrogen peroxide/Chk1-dependent axis. Cancer Prev Res. 2013;6:949–58.
Zhao D, Gong T, Fu Y, Nie Y, He LL, Liu J, et al. Lyophilized Cheliensisin A submicron emulsion for intravenous injection: characterization, in vitro and in vivo antitumor effect. Int J Pharm. 2008;357:139–47.
Hua X, Xu J, Deng X, Xu J, Li J, Zhu DQ, et al. New compound ChlA-F induces autophagy-dependent anti-cancer effect via upregulating Sestrin-2 in human bladder cancer. Cancer Lett. 2018;436:38–51.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511:246–50.
Zhu FY, Qian WQ, Zhang HJ, Liang Y, Wu MQ, Zhang YY, et al. SOX2 is a marker for stem-like tumor cells in bladder cancer. Stem Cell Rep. 2017;9:429–37.
Ooki A, Begum A, Marchionni L, VandenBussche CJ, Mao SF, Kates M, et al. Arsenic promotes the COX2/PGE2-SOX2 axis to increase the malignant stemness properties of urothelial cells. Int J Cancer. 2018;143:113–26.
Alonso MM, Diez-Valle R, Manterola L, Rubio A, Liu D, Cortes-Santiago N, et al. Genetic and epigenetic modifications of Sox2 contribute to the invasive phenotype of malignant gliomas. PLoS ONE. 2011;6:e26740.
Han X, Fang XF, Lou XY, Hua DS, Ding WC, Foltz G, et al. Silencing SOX2 induced mesenchymal-epithelial transition and its expression predicts liver and lymph node metastasis of CRC patients. PLoS ONE. 2012;7:e41335.
Girouard SD, Laga AC, Mihm MC, Scolyer RA, Thompson JF, Zhan Q, et al. SOX2 contributes to melanoma cell invasion. Lab Invest. 2012;92:362–70.
Sun C, Sun L, Li Y, Kang X, Zhang S, Liu Y. Sox2 expression predicts poor survival of hepatocellular carcinoma patients and it promotes liver cancer cell invasion by activating Slug. Med Oncol. 2013;30:503.
Lou X, Han X, Jin C, Tian W, Yu W, Ding D, et al. SOX2 targets fibronectin 1 to promote cell migration and invasion in ovarian cancer: new molecular leads for therapeutic intervention. OMICS. 2013;17:510–8.
Chen X, Xie RH, Gu P, Huang M, Han JL, Dong W, et al. Long noncoding RNA LBCS inhibits self-renewal and chemoresistance of bladder cancer stem cells through epigenetic silencing of SOX2. Clin Cancer Res. 2019;25:1389–403.
Zhu J, Huang G, Hua X, Li Y, Yan H, Che X, et al. CD44s is a crucial ATG7 downstream regulator for stem-like property, invasion, and lung metastasis of human bladder cancer (BC) cells. Oncogene. 2019;38:3301–15.
Liang Y, Zhu J, Huang H, Xiang D, Li Y, Zhang D, et al. SESN2/sestrin 2 induction-mediated autophagy and inhibitory effect of isorhapontigenin (ISO) on human bladder cancers. Autophagy. 2016;12:1229–39.
Wang Y, Xu J, Gao G, Li J, Huang H, Jin H, et al. Tumor-suppressor NFkappaB2p100 interacts with ERK2 and stabilizes PTEN mRNA via inhibition of miR-494. Oncogene. 2016;35:4080–90.
Zhang R, Che X, Zhang J, Li Y, Li J, Deng X, et al. Cheliensisin A (Chel A) induces apoptosis in human bladder cancer cells by promoting PHLPP2 protein degradation. Oncotarget. 2016;7:66689–99.
Zeng XR, Xu Z, Gu JY, Huang HS, Gao GX, Zhang XR, et al. Induction of miR-137 by isorhapontigenin (ISO) directly targets Sp1 protein translation and mediates its anticancer activity both in vitro and in vivo. Mol Cancer Ther. 2016;15:512–22.
Peng M, Wang J, Zhang D, Jin H, Li J, Wu XR, et al. PHLPP2 stabilization by p27 mediates its inhibition of bladder cancer invasion by promoting autophagic degradation of MMP2 protein. Oncogene. 2018;37:5735–48.
Xu J, Wang Y, Hua X, Xu J, Tian Z, Jin H, et al. Inhibition of PHLPP2/cyclin D1 protein translation contributes to the tumor suppressive effect of NFkappaB2 (p100). Oncotarget. 2016;7:34112–30.
Zeng X, Xu Z, Gu J, Huang H, Gao G, Zhang X, et al. Induction of miR-137 by Isorhapontigenin (ISO) Directly targets Sp1 protein translation and Mediates Its Anticancer Activity Both In Vitro and In Vivo. Mol Cancer Ther. 2016;15:512–22.
Jiang G, Wu AD, Huang C, Gu J, Zhang L, Huang H, et al. Isorhapontigenin (ISO) inhibits invasive bladder cancer formation in vivo and human bladder cancer invasion in vitro by targeting STAT1/FOXO1 Axis. Cancer Prev Res. 2016;9:567–80.
Trepat X, Chen Z, Jacobson K. Cell migration. Compr Physiol. 2012;2:2369–92.
Sibley DL, Charron A, Håkansson S, Mordue D. Invasion and intracellular survival by Toxoplasma. Madame Curie Bioscience Database [Internet]. Landes Bioscience 2013.
Wang J, Zhang Y, Hou J, Qian X, Zhang H, Zhang Z, et al. Ube2s regulates Sox2 stability and mouse ES cell maintenance. Cell Death Differ. 2016;23:393–404.
Zhou F, Xie F, Jin K, Zhang Z, Clerici M, Gao R, et al. USP4 inhibits SMAD4 monoubiquitination and promotes activin and BMP signaling. EMBO J. 2017;36:1623–39.
Yeates EF, Tesco G. The endosome-associated deubiquitinating enzyme USP8 regulates BACE1 enzyme ubiquitination and degradation. J Biol Chem. 2016;291:15753–66.
Sussman RT, Stanek TJ, Esteso P, Gearhart JD, Knudsen KE, Mcmahon SB. The epigenetic modifier ubiquitin-specific protease 22 (USP22) regulates embryonic stem cell differentiation via transcriptional repression of sex-determining region Y-box 2 (SOX2). J Biol Chem. 2013;288:24234–46.
Ayupe AC, Reis EM. Evaluating the stability of mRNAs and noncoding RNAs. Methods Mol Biol. 2017;1468:139–53.
Hao S, Baltimore D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol. 2009;10:281–8.
Abdelmohsen K, Gorospe M. RNA-binding protein nucleolin in disease. RNA Biol. 2012;9:799–808.
Gratacos FM, Brewer G. The role of AUF1 in regulated mRNA decay. Wiley Inter Rev RNA. 2010;1:457–73.
Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley Inter Rev RNA. 2010;1:214–29.
Hausser J, Zavolan M. Identification and consequences of miRNA-target interactions—beyond repression of gene expression. Nat Rev Genet. 2014;15:599–612.
Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321–33.
Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–24.
Thompson EJ, Gupta A, Stratton MS, Bowden GT. Mechanism of action of a dominant negative c-jun mutant in inhibiting activator protein-1 activation. Mol Carcinog. 2002;35:157–62.
Yang M, Huang CZ. Mitogen-activated protein kinase signaling pathway and invasion and metastasis of gastric cancer. World J Gastroenterol. 2015;21:11673–9.
Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–15.
Wang XJ, Ji XN, Chen JZ, Yan D, Zhang ZB, Wang QF, et al. SOX2 enhances the migration and invasion of ovarian cancer cells via Src kinase. PLoS ONE. 2014;9:e99594.
Bae KM, Dai Y, Vieweg J, Siemann DW. Hypoxia regulates SOX2 expression to promote prostate cancer cell invasion and sphere formation. Am J Cancer Res. 2016;6:1078–88.
Lee SH, Oh SY, Do SI, Lee HJ, Kang HJ, Rho YS, et al. SOX2 regulates self-renewal and tumorigenicity of stem-like cells of head and neck squamous cell carcinoma. Br J Cancer. 2014;111:2122–30.
Chang XH, Zhang J, Huang CL, Pang XA, Luo QS, Zhang HJ, et al. Sex-determining region Y-related high mobility group box (SOX)-2 is overexpressed in cervical squamous cell carcinoma and contributes cervical cancer cell migration and invasion in vitro. Tumor Biol. 2015;36:7725–33.
Ma KL, Pan XR, Fan PS, He YH, Gu J, Wang W, et al. Loss of miR-638 in vitro promotes cell invasion and a mesenchymal-like transition by influencing SOX2 expression in colorectal carcinoma cells. Mol Cancer 2014;13.
Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–97.
Mizuno E, Iura T, Mukai A, Yoshimori T, Kitamura N, Komada M. Regulation of epidermal growth factor receptor down-regulation by UBPY-mediated deubiquitination at endosomes. Mol Biol Cell. 2005;16:5163–74.
Alwan HA, van Leeuwen JE. UBPY-mediated epidermal growth factor receptor (EGFR) de-ubiquitination promotes EGFR degradation. J Biol Chem. 2007;282:1658–69.
Yang R, Liu M, Liang H, Guo S, Guo X, Yuan M, et al. miR-138-5p contributes to cell proliferation and invasion by targeting Survivin in bladder cancer cells. Mol Cancer. 2016;15:82.
Yu G, Yao WM, Xiao W, Li H, Xu H, Lang B. MicroRNA-34a functions as an anti-metastatic microRNA and suppresses angiogenesis in bladder cancer by directly targeting CD44. J Exp Clin Cancer Res. 2014;33:779.
Oishi N, Kumar MR, Roessler S, Ji J, Forgues M, Budhu A, et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology. 2012;56:1792–803.
Ahmad A, Aboukameel A, Kong D, Wang Z, Sethi S, Chen W, et al. Phosphoglucose isomerase/autocrine motility factor mediates epithelial-mesenchymal transition regulated by miR-200 in breast cancer cells. Cancer Res. 2011;71:3400–9.
Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–95.
Lu YX, Yuan L, Xue XL, Zhou M, Liu Y, Zhang C, et al. Regulation of colorectal carcinoma stemness, growth, and metastasis by an miR-200c-Sox2-negative feedback loop mechanism. Clin Cancer Res. 2014;20:2631–42.
Zhang X, Zou TT, Rao JN, Liu L, Xiao L, Wang PY, et al. Stabilization of XIAP mRNA through the RNA binding protein HuR regulated by cellular polyamines. Nucleic Acids Res. 2009;37:7623–37.
Acknowledgements
This work was partially supported by the grants from the Natural Science Foundation of China (NSFC91773391, NSFC81872587 and NSFC81702530); Wenzhou Science and Technology Bureau (Y20170028) and Key Discipline of Zhejiang Province in Medical Technology (First Class, Category A).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Del Sal
Rights and permissions
About this article

Cite this article
Hua, X., Huang, M., Deng, X. et al. The inhibitory effect of compound ChlA-F on human bladder cancer cell invasion can be attributed to its blockage of SOX2 protein. Cell Death Differ 27, 632–645 (2020). https://doi.org/10.1038/s41418-019-0377-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-019-0377-7
This article is cited by
-
Tumorigenesis of basal muscle invasive bladder cancer was mediated by PTEN protein degradation resulting from SNHG1 upregulation
Journal of Experimental & Clinical Cancer Research (2024)
-
Differential functions of RhoGDIβ in malignant transformation and progression of urothelial cell following N-butyl-N-(4-hydmoxybutyl) nitrosamine exposure
BMC Biology (2023)
-
Targeting the SOX2/CDP protein complex with a peptide suppresses the malignant progression of esophageal squamous cell carcinoma
Cell Death Discovery (2023)
-
Neuropilin-2 acts a critical determinant for epithelial-to-mesenchymal transition and aggressive behaviors of human head and neck cancer
Cellular Oncology (2023)
-
Molecular mechanism of lncRNA SNHG12 in immune escape of non-small cell lung cancer through the HuR/PD-L1/USP8 axis
Cellular & Molecular Biology Letters (2022)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}