
Abstract
Obesity induces accumulation of adipose tissue macrophages (ATMs) and ATM-driven inflammatory responses that promote the development of glucose and lipid metabolism disorders. ClC-3 chloride channel/antiporter, encoded by the Clcn3, is critical for some basic cellular functions. Our previous work has shown significant alleviation of type 2 diabetes in Clcn3 knockout (Clcn3−/−) mice. In the present study we investigated the role of Clcn3 in high-fat diet (HFD)-induced obesity and ATM inflammation. To establish the mouse obesity model, both Clcn3−/− mice and wild-type mice were fed a HFD for 4 or 16 weeks. The metabolic parameters were assessed and the abdominal total adipose tissue was scanned using computed tomography. Their epididymal fat pad tissue and adipose tissue stromal vascular fraction (SVF) cells were isolated for analyses. We found that the HFD-fed Clcn3−/− mice displayed a significant decrease in obesity-induced body weight gain and abdominal visceral fat accumulation as well as an improvement of glucose and lipid metabolism as compared with HFD-fed wild-type mice. Furthermore, the Clcn3 deficiency significantly attenuated HFD-induced ATM accumulation, HFD-increased F4/80+ CD11c+ CD206− SVF cells as well as HFD-activated TLR-4/NF-κB signaling in epididymal fat tissue. In cultured human THP-1 macrophages, adenovirus-mediated transfer of Clcn3 specific shRNA inhibited, whereas adenovirus-mediated cDNA overexpression of Clcn3 enhanced lipopolysaccharide-induced activation of NF-κB and TLR-4. These results demonstrate a novel role for Clcn3 in HFD-induced obesity and ATM inflammation.
Similar content being viewed by others


Introduction
Obesity is mainly characterized by adipocyte hypertrophy (increase in fat cell size) and the accumulation of adipose tissue macrophages (ATMs) in humans and rodents, which induce chronic, low-grade inflammation, and insulin resistance (IR) [1]. In obese adipose tissue, ATMs undergo polarization from the M2 (anti-inflammatory) to M1 (pro-inflammatory) phenotype, which is considered a biomarker of macrophage-driven adipose tissue inflammation in IR, with elevated F4/80+ and CD11c+ M1-like macrophages [2]. Hypertrophied adipocytes and immune cells, such as M1-like macrophages, shift the secretion of cytokines and adipokines toward the inflammatory profile (e.g., MCP-1 and IL-6) [3], leading to the impairment of insulin sensitivity through activation of pro-inflammatory signaling, such as activation of TLR-4 and NF-κB [4]. Furthermore, the depletion of CD11c+ M1-like macrophages causes a marked reduction in pro-inflammatory macrophages in obese adipose tissue, which protects against obesity-associated IR [5]. However, the molecular details that underlie ATM-driven inflammatory responses within obese adipose tissue remain uncertain.
The ClC-3 chloride channel/antiporter, encoded by Clcn3, is critical for several basic cellular functions, such as cell volume regulation, proliferation, apoptosis, differentiation, and β cell insulin secretion [6,7,8,9]. Accumulating evidence from animal models and human patients of inflammatory bowel disease also suggests that ClC-3 is important for inflammatory processes [10]. Knockdown or genetic deletion of Clcn3 attenuates the NF-κB activity and inflammatory responses induced by different cytokines, such as TNF-α and IL-1β, in the vascular system and lung [11], thus highlighting the role of Clcn3 deficiency in restricting inflammatory responses, particularly under diseased conditions.
Our previous study has shown that the loss of Clcn3 significantly decreases the plasma levels of glucose, triglyceride (TG), and free fatty acid (FFA) and protects against IR compared with wild-type mice in a type 2 diabetes mouse model induced by a high sucrose/fat diet following low-dose streptozotocin [12]. However, it is not clear whether and how Clcn3 is involved in the essential functions of ATMs during obesity. In the present study, we established a high fat diet (HFD)-induced obesity model in Clcn3−/− (KO) mice and investigated the effects of Clcn3 on HFD-induced obesity and obesity-associated ATM inflammation. Collectively, our results demonstrated that Clcn3 is associated with HFD-induced obesity and macrophage-mediated inflammatory responses in visceral fat tissue during obesity.
Materials and methods
Animals
All experimental procedures were approved by the Sun Yat-sen University Animal Care and Use Committee and were in accordance with the “Guide for the Care and Use of Laboratory Animals” issued by the Ministry of Science and Technology of China. Clcn3−/− (KO) and littermate control (WT) mice were kindly provided by Dr. Dean Burkin from the Nevada Transgenic Center (University of Nevada School of Medicine, Nevada, USA). Heterozygous 129/SvJ-C57BL/6 offspring were used to establish breeding colonies as previously described [13].
Mice (8 weeks old) were fed a high-fat diet (HFD, 60% calories from fat, D12492; Research Diets Inc., USA) or a normal chow diet (NC, 10% calories from fat, D12450; Research Diets Inc., USA) for 4 or 16 weeks. At the end of the diet intervention, blood samples, livers, and epididymal fat pads were collected and stored.
Measurements of metabolic parameters
The following experiments were performed as previously described [12]. The serum total cholesterol (TC), triglyceride (TG), free fatty acid (FFA), and C-reactive protein (CRP) levels were detected using an automated biochemical analyzer (Hitachi 7180, Japan). The fasting blood glucose levels were measured using a glucometer (Roche Diagnostics, USA). The serum level of insulin was measured with a mouse insulin ELISA kit (Nanjing Jiancheng Bioengineering Institute, China). For the intraperitoneal glucose tolerance test (PGTT), mice fasted overnight were injected with D-glucose (2 g/kg body weight; Sigma-Aldrich, USA). For the insulin tolerance test (ITT), mice were fasted for 5 h with free access to water and injected with insulin (0.75 units/kg body weight; Novolin R, Novo Nordisk, Copenhagen, Denmark). Blood was collected from the tail at specific time intervals, and glucose was measured using a glucometer (Roche Diagnostics, USA).
Micro-computed tomography (Micro-CT)
Lower abdominal computed tomography (CT, Inveon Micro-PET/CT, Siemens, Germany) scans were performed after 4 or 16 weeks of the NC or HFD. Mice were anesthetized with pentobarbital (50 mg/kg) prior to the Micro-CT scanning. The lower abdomen was defined as the level from the fourth lumbar vertebra (L4) to the coccygeal vertebra. The total lower abdomen adipose tissue volume and body volume were calculated using the regions of interest (ROI) by the Inveon Research Workplace software. Adipose tissue was defined by thresholding from −600 to −350 hounsfield unit (HU) on CT scans with manually segmentation. The ratio of the total adipose tissue volume/body volume was calculated [14].
Immunohistochemistry staining
The epididymal white adipose tissue (eWAT) and the liver tissue were fixed in 4% paraformaldehyde (Sigma-Aldrich, USA). The liver sections were stained with Hematoxylin and Eosin (H&E) as previously described [15]. The macrophage accumulation in eWAT sections was determined by immunohistochemical staining using an antibody against F4/80 (Spring Bioscience, China). The integral optical density (IOD) of the F4/80 cells (IOD sum/area) in each section was measured using Image-Pro Plus 6.0 (Media Cybernetics Corporation, Rockville, MD, USA). The average IOD of at least three section values was obtained as the mean IOD of a sample.
Flow cytometry analysis
To block mouse Fc receptors, the cell suspension was incubated with FcBlock in Staining Buffer (5 μg/mL at a cell density of 1 × 106/mL, BD Biosciences, USA) for 15 min on ice. The pellets were subsequently labeled with fluorophore-conjugated antibodies, including APC-eFluor® 780–conjugated F4/80 (eBioscience, USA), PE-conjugated CD11c (eBioscience, USA), and Alexa Fluor® 647-conjugated CD206 (Biolegend, China). The background fluorescence was quantified using an APC IgG к isotype control (eBioscience, USA), PE IgG isotype control (Invitrogen, USA), and Alexa Fluor® 647 IgG к isotype control (BioLegend, USA). Cells were measured on a Beckman Gallios flow cytometer and analyzed using Kaluza software (Beckman Coulter, USA).
Adipose stromal vascular fraction isolation
Epididymal WAT was excised and minced in PBS with 0.5% BSA. It was subsequently incubated in digestion buffer (DMEM, 12.5 mM HEPES [pH 7.4], 2% BSA, and 10 mg collagenase type I [Sigma-Aldrich, USA]) for 40 min at 37 °C in a shaking bath at 150 r/min. The digested tissue was filtered through a 250 μm nylon sieve and centrifuged at room temperature at 500 × g for 5 min. The pellet was washed with RBC lysis solution (Beyotime, China) and centrifuged at 500 × g for 5 min to remove free erythrocytes. The cell suspension was washed with PBS and spun at 300 × g for 5 min to obtain stromal vascular fraction (SVF) cells. The SVF provides a rich source of ATMs that exhibit a typical macrophage phenotype and are positive for F4/80, CD11c and CD206.
Cell culture
Human THP-1 monocytes (American Type Culture Collection, USA) were cultured in RPMI-1640 medium that contained 10% (v/v) fetal bovine serum (FBS) and 1% (w/v) penicillin/streptomycin. For monocyte–macrophage differentiation, cells were seeded at a density of 2 × 105 cells/mL and exposed to 100 nM phorbol-12-myristate-13-acetate (PMA) (Sigma-Aldrich, USA) for 72 h.
Adenoviral infection experiments
The adenovirus expressing a small hairpin RNA sequence (Ad-shClcn3) targeting the human Clcn3 and recombinant adenoviral vector overexpressing guinea pig Clcn3 cDNA (Ad- Clcn3) was successfully constructed by Shanghai Sunbio Biomedical Technology (Shanghai, China). The sequence of the shRNA against the human Clcn3 (Gene ID: NM_001243374.1) was 5′-CCACGACTGGTTTATCTTT-3′, which has an identity of 100% with Homo sapiens Clcn3 mRNA. The control shRNA (Neg) shows no significant homology to known human genes. The sequence of the negative control was 5′-TTCTCCGAACGTGTCACGT-3′. The guinea pig cDNA of Clcn3 was generously gifted by Dr. Dayue Duan (University of Nevada, Reno, USA) for constructing Ad-Clcn3. Adenoviral infection was carried out as previously described [16]. The adenoviral vector (Vector) without expressing Clcn3 was employed as the negative control. Cells at 50% confluence were infected with Ad-shClcn3 or Ad-Clcn3 for 6 h and then washed and incubated in fresh medium for an additional 30 h prior to the addition of LPS (100 ng/mL) for 12 h.
Quantitative real-time RT-PCR
The quantitative real-time PCR experiment was conducted as previously described [12]. Total RNA was extracted from eWAT or SVF cells using Trizol reagent (Invitrogen, USA). RNA was reverse-transcribed using the Transcriptor cDNA Synthesis Kit (Roche, USA). Quantitative RT-PCR was performed using the FS Universal SYBR Green Master (Roche, USA) according to the manufacturer’s protocol. The fold change in the mRNA expression of each gene was determined by the 2−ΔΔCt method using GAPDH mRNA as an internal control. The primer sequences are provided in Supplementary Table 1.
Western blotting
eWAT or THP-1 macrophages were harvested using RIPA lysis buffer (Beyotime, China), and immunoblots were performed as previously described [12]. Briefly, the samples were separated on 8% SDS polyacrylamide gels and then transferred to the nitrocellulose membrane. The membrane was incubated with the corresponding primary antibodies overnight, followed by appropriate horseradish peroxidase-conjugated secondary antibodies for 1.5 h. The bands were detected by an enhanced chemiluminescence (ECL) detection system (Beyotime, China). The images of the chemiluminescence were captured with an imaging system (Bio-Rad, Hercules, CA, USA). The optical densities were normalized to those of β-actin, and the fold difference for each target protein was calculated as the ratio of the target protein expression/β-actin expression (ImageJ 1.42q software, National Institutes of Health, USA). The primary antibodies are shown as follows: ClC-3 (1:200, Alomone Labs, Israel), TLR-4 (1:500, Santa Cruz Biotechnology, USA), NF-κB p65 (1:1000, Cell Signaling Technology, USA), p-NF-κB p65 (1:1000, Cell Signaling Technology, USA), IRS-1 (1:500, Santa Cruz Biotechnology, USA), p-IRS-1 (1:500, Santa Cruz Biotechnology, USA), and β-actin (1:2000, Cell Signaling Technology, USA).
Statistical analysis
All data are expressed as the mean ± SEM. Unpaired Student’s t-test was used to compare 2 groups. A one-way analysis of variance (ANOVA), followed by a Bonferroni multiple comparison post hoc test with a 95% CI, was used for 3 or more groups. The statistical significance was evaluated via analysis of variance using Prism 4 software (GraphPad Software, San Diego, CA, USA). P < 0.05 was considered statistically significant.
Results
Loss of Clcn3 protects against diet-induced obesity
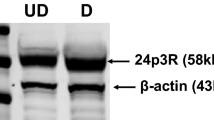
There was no protein expression of ClC-3 in the WAT, liver and skeletal muscle in KO mice, as detected by Western blotting (Fig. S1a). The HFD-induced obesity model was established in Clcn3+/+ wild-type (WT) and Clcn3−/− knockout (KO) mice. As shown in Fig. 1a, b, the Clcn3 mRNA and protein levels were significantly increased in eWAT from obese mice after the 4-week-HFD compared with those of the age-matched NC-fed groups (P < 0.05). Moreover, more significant increases in the mRNA and protein levels of Clcn3 were shown in eWAT from WT-HFD mice after the 16-week-HFD compared with those of the age-matched NC-fed groups (P < 0.01). There was no difference in body weight between WT and KO mice fed the NC; moreover, there was no difference in the total calorie intake among all groups during the feeding period (Fig. 1c, d). These results might exclude the influence from global Clcn3 knockout on eating behavior that may be due to the degeneration of hippocampal neurons where adipocyte derived leptin has binding sites [17]. Body weight in WT mice progressively increased from 4 weeks of administration of the HFD onwards, which was significantly decreased by Clcn3 deficiency from 6 weeks of administration of the HFD onwards (Fig. 1d). Measurement of the eWAT weight and quantification of the abdominal fat depot demonstrated that the eWAT weight (normalized to body weight) markedly increased from 4 weeks of the HFD onwards (P < 0.05 vs age-matched WT-NC groups), which was significantly decreased by Clcn3 deficiency at the end of the 16-week-HFD (Fig. 1e, P < 0.01 vs age-matched WT-HFD groups). Similar inhibitory effects of Clcn3 deficiency on the HFD-induced increase in the absolute and relative abdominal visceral adipose tissue (VAT) volumes were measured and quantified by Micro-CT (Fig. 1f). The HFD induced an increase in the absolute and relative abdominal VAT volume from 4 weeks of the HFD onwards (P < 0.01 vs age-matched WT-NC groups), which was significantly decreased in KO-HFD mice after the 16-week-HFD (Fig. 1f, P < 0.01 vs age-matched WT-HFD groups).

Clcn3−/− (KO) mice are protected against diet-induced obesity. Eight-week-old KO mice and their Clcn3+/+ (WT) littermates were fed the NC or HFD. Western blotting (a) and qRT-PCR (b) analyses showed a significant increase in protein and mRNA expression of Clcn3 in eWAT of HFD-fed WT mice (WT-HFD) for 4 or 16 weeks compared with those of age-matched NC-fed WT mice (WT-NC). *P < 0.05, **P < 0.01 vs age-matched WT-NC group, n = 6 mice per group. Weekly food intake (c) and body weight (d) were measured every 2 weeks. *P < 0.05 vs age-matched WT-NC group, #P < 0.05 vs age-matched KO-HFD group, n = 10 mice per group. e The weights of eWAT from KO and WT mice were measured and normalized to body weight. *P < 0.05, **P < 0.01, n = 10 mice per group. f A lower abdomen computed tomography scan was performed, and representative image slices (L6) are shown. The adipose tissue volume and the total adipose tissue volume/total body volume ratio were calculated. **P < 0.01, n = 6 mice per group
A HFD can induce ectopic fat storage and activate macrophage-driven inflammatory responses in the liver, resulting in the occurrence of obesity-related hepatic steatosis and insulin resistance. We also set out to characterize hepatic changes in WT and KO mice. Interestingly, no substantial differences in the liver weight normalized to the body weight or hepatic steatosis were observed between the WT-HFD and KO-HFD groups fed a HFD for 4 or 16 weeks, as visualized by H&E staining (Fig. S2). These data suggested that Clcn3 deficiency may not affect HFD-induced fatty liver.
Clcn3 deficiency ameliorates HFD-induced glucose and lipid metabolism disorders
We subsequently investigated whether Clcn3 deficiency affects HFD-induced glucose and lipid metabolism disorders. Consistent with our previous findings [12], KO mice were largely protected from the HFD-induced elevation of fasting insulin (FI) and fasting blood glucose (FBG) levels compared with the age-matched WT littermates after the 16-week-HFD (Fig. 2a, b). When ITT and PGTT were performed, KO-HFD mice had a smaller area under the curve (AUC) than their corresponding WT-HFD littermates, indicating that Clcn3 deficiency could improve glucose metabolism disorders and impair insulin sensitivity during HFD-induced obesity (Fig. 2c, d). Serine 307 phosphorylation of mouse insulin receptor substrate-1 (IRS-1) is a common mechanism of insulin signaling that contributes to IR [18]. As shown in Fig. 2e, the Western blotting data indicated that Clcn3 deficiency markedly increased the downregulation of phospho-IRS-1 (Ser307) in eWAT induced by the HFD, suggesting that Clcn3 may play a crucial role in regulating impaired insulin signaling during obesity. In addition, the loss of Clcn3 markedly decreased the HFD-induced elevation in the serum TG and FFA levels; however, there was no significant change in the HFD-induced total TC level (Table 1). Taken together, these findings indicated that Clcn3 deficiency improved insulin sensitivity, glucose, and lipid metabolism in diet-induced obese mice.

KO mice are protected against HFD-induced glucose and lipid metabolism disorders. The levels of fasting insulin (a) and fasting blood glucose (b) were measured in serum samples. **P < 0.01 vs WT-NC group. ##P < 0.01 vs KO-HFD group, n = 10 mice per group. ITT (c), PGTT (d) and area under the curve (AUC) analyses of KO and WT mice fed the NC or HFD for 4 or 16 weeks are shown. *P < 0.05, **P < 0.01 vs WT-NC group, #P < 0.05, ##P < 0.01 vs KO-HFD group, n = 10 mice per group. e The levels of p-IRS-1 (serine 307) and IRS-1 in eWAT of KO and WT mice were measured by Western blotting, and β-actin was used as the loading control. **P < 0.01 vs WT-NC group. ##P < 0.01 vs KO-HFD group, n = 6 mice per group
Clcn3 deficiency reduces HFD-induced ATM accumulation and F4/80+CD11c+CD206− ATMs
HFD-fed obese mice exhibit the accumulation of adipose tissue macrophages (ATMs) and an increase in M1-like ATMs in eWAT, which are biomarkers of adipose tissue inflammation during the development of IR [19]. We performed the immunofluorescent staining of ClC-3 and F4/80, a typical macrophage marker in eWAT isolated from WT-HFD mice after the 16-week-HFD, indicating a cell type-specific location of Clcn3 in ATMs (Fig. S3). Immunohistochemical staining showed that the HFD induced an increase of the total numbers of F4/80+ macrophages in eWAT from WT-HFD mice compared with the age-matched WT-NC at the end of the 16-week-HFD, which could be markedly reduced by Clcn3 deficiency (Fig. 3a). There was no significant difference in the total number of F4/80+ eWAT macrophages among WT and KO mice fed the NC or HFD for 4 weeks (Fig. 3a). Flow cytometry analysis of eWAT and SVF further identified that Clcn3 deficiency markedly reduced the HFD-induced increase in F4/80 positive cells within visceral adipose tissue during obesity (Fig. 3b). These results indicated that the loss of Clcn3 decreases obesity-induced ATM accumulation, and this effect may be related to the time duration of the HFD.

KO mice exhibit a decreased number of ATMs in eWAT from obese mice after 16-week-HFD. a Histological analysis of F4/80 staining in eWAT of KO and WT mice was used to determine ATM accumulation (scale bars represent 100 μm). The mean IODs of macrophages stained with an anti-F4/80 antibody were obtained. b Flow cytometry was used to quantify the effect of Clcn3 deficiency on F4/80+ macrophage accumulation in SVF cells isolated from eWAT of KO and WT mice fed the NC or HFD for 16 weeks. **P < 0.01, n = 6–8 mice per group
We further investigated whether Clcn3 deficiency affects pro-inflammatory and anti-inflammatory ATM subpopulations following HFD consumption. The flow cytometry analysis demonstrates that the cell numbers of typical M1 ATMs (F4/80+CD11c+CD206− cells) in the SVF from eWAT were significantly reduced in KO-HFD mice compared with their corresponding WT-HFD group (Fig. 4a). The number of typical M2 ATMs (F4/80+CD11c−CD206+ cells) was not significantly different between the KO-HFD and WT-HFD groups (Fig. 4a). qRT-PCR analysis for various macrophage markers yielded similar results, showing that Clcn3 deficiency significantly decreased the enhanced transcript levels of F4/80 and CD11c induced by the 16-week-HFD, compared with the corresponding WT-HFD mice (P < 0.01, Fig. 4b, c). There is no significant difference in the F4/80 and CD11c mRNA expression between the WT-HFD and KO-HFD groups at the end of the 4-week-HFD. Of note, the Clcn3 deficiency induced a significant increase in the CD206 mRNA expression after the 16-week-HFD (P < 0.05, Fig. 4d). These results indicate that ATMs from Clcn3−/− mice are biased toward an anti-inflammatory phenotype and suggest that Clcn3 may be involved in obesity-induced ATM polarization.

KO mice exhibit reduced macrophage accumulation and a lower percentage of F4/80+CD11c+CD206− SVF cells from eWAT of obese mice. a Flow cytometry was used to quantify effects of Clcn3 deficiency on F4/80+CD11c+CD206− and F4/80+CD11c−CD206+ macrophage subpopulations in SVF cells isolated from eWAT of KO and WT mice fed the NC or HFD for 16 weeks. **P < 0.01, n = 6 mice per group. The mRNA levels of F4/80 (b), CD11c (c), and CD206 (d) were analyzed by qRT-PCR in SVF of eWAT from KO and WT mice fed the NC or HFD for 4 or 16 weeks. *P < 0.05, **P < 0.01, n = 6/group
Clcn3 deficiency decreases systemic and adipose tissue inflammation induced by HFD
To determine whether Clcn3 deficiency results in different inflammatory profiles during obesity, we also measured the levels and expression of cytokines and adipokines in the serum or eWAT from KO and WT mice (Fig. 5). The transcript levels of the examined systemic cytokines and adipose tissue cytokines (or adipokines) were not significantly different between the WT-NC and KO-NC groups, indicating that Clcn3 deficiency had no influence on the basal production of these cytokines and chemokines in the absence of a dietary challenge (Fig. 5). The serum of HFD-fed WT mice showed an increase in C-reactive protein (CRP), indicating the induction and progression of low-grade adipose tissue inflammation. These changes were largely prevented in Clcn3−/− mice after the 16-week-HFD (Fig. 5a). qRT-PCR analysis showed that Clcn3 deficiency significantly decreased the HFD-induced increase in mRNA expression of MCP-1, TLR-4, and IL-6 in eWAT (Fig. 5b–d). Interestingly, Clcn3 deficiency significantly attenuated the HFD-induced increase in MCP-1 and TLR-4 as early as 4 weeks after HFD feeding, while it had no significant effects on the HFD-induced change in TNF-α and adiponectin (Fig. 5e, f). Tamoxifen at 10 µM has commonly been used to inhibit ClC-3-dependent Cl− currents and volume-sensitive Cl− movement in many cell types, including RAW macrophages [20]. The qRT-PCR data demonstrated that tamoxifen causes a significant decrease in Clcn3 and inhibits the LPS-induced increase in MCP-1, TLR-4, and IL-6 in F4/80+ SVF cells isolated from mouse eWAT (Fig. S4). These results support that the Clcn3-dependent Cl− currents may be involved in regulating ATM inflammation. Collectively, these findings indicated that the production of pro-inflammatory mediators induced by a HFD is lower locally and systemically in KO mice than in the corresponding WT mice, suggesting a decrease in the levels of adipose inflammation in KO mice during obesity.

KO mice demonstrate reduced levels of inflammatory mediators. a The serum CRP level was measured in KO mice and their WT littermates fed a NC or a HFD for 4 or 16 weeks. The mRNA levels of MCP-1 (b), TLR-4 (c), IL-6 (d), TNF-α (e), and adiponectin (f) in eWAT were measured in KO mice and their WT littermates fed the NC or HFD. *P < 0.05, **P < 0.01, n = 6/group. g Protein expression of TLR-4 and activation of NF-κB (as indicated by the ratio of phospho-NF-κB p65 to total NF-κB p65) were measured in eWAT from KO mice and their corresponding WT littermates. **P < 0.01, n = 6–8 mice per group
Clcn3 deficiency reduces HFD-induced activation of TLR-4 and NF-κB in eWAT
The TLR4/NF-κB signaling pathway is enhanced in HFD-induced elevation of the LPS level and adipose tissue inflammation that links obesity to IR [4]. The transcript level of TLR-4 was alleviated in eWAT from KO mice fed a HFD (Fig. 5c). HFD feeding increased the phospho-p65 and TLR-4 protein levels in eWAT from WT mice, which were reduced in KO mice (Fig. 5g). The activation of TLR-4 and NF-κB was further investigated in macrophages derived from the THP-1 cell line, which was infected with Ad-Clcn3 shRNA or Ad-Clcn3, respectively, and then treated with LPS. LPS potently increased the NF-κB activation and TLR-4 protein expression, which were largely abolished by Clcn3 knockdown and further potentiated by Clcn3 overexpression in THP-1 macrophages (Fig. 6a, b). Collectively, these results indicated that Clcn3 deficiency may ameliorate adipose tissue inflammation via suppressing the activation of TLR-4 and NF-κB.

Clcn3 regulates LPS-induced activation of TLR-4/ NF-κB in THP-1 macrophages. The effects of adenoviral Clcn3-shRNA knockdown (a) and Clcn3 overexpression (b) on protein expression of TLR-4 and phospho-NF-κB p65/total NF-κB p65 were detected by Western blotting. The respective control shRNA (Neg) and the adenoviral vector (Vector) without Clcn3 expression were employed. *P < 0.05, **P < 0.01, n = 6 independent experiments
Discussion
In the present study, we have provided the first and strong evidence that downregulation of Clcn3 was a crucial protective factor against diet-induced obesity because Clcn3 knockout attenuated HFD-induced body weight gain, abdominal adipose volume expansion, and glucose and lipid disorders. Notably, the 4-week-HFD was shown to increase the mRNA and protein expressions of Clcn3 in adipose tissue, as well as several pro-inflammatory cytokines (such as MCP-1 and TLR-4) characteristic of adipose tissue inflammation, which could be significantly reduced in KO mice. These findings suggest that Clcn3 may be an early contributor to the development of obesity and obesity-related adipose tissue inflammation.
Macrophage-related chronic inflammation has been implicated as a major pathogenic mechanism of adipose tissue dysfunction and consequent IR. Clcn3 deficiency has recently been reported to prevent atherosclerotic lesion development via suppressing the uptake of ox-LDL in macrophages [21], directly linking Clcn3 to atherosclerotic lesion formation through the regulation of macrophage functions. Increased ATMs contribute to systemic IR by creating a pro-inflammatory environment. A HFD induces an increase in F4/80+ macrophage accumulation from both the recruitment of macrophages from the blood stream and the proliferation of itself, and it promotes the pro-inflammatory M1 macrophage polarization within abdominal visceral fat and the secretion of pro-inflammatory adipokines. Accumulating evidence in obese mouse models has documented that the inhibition of M1 macrophage polarization is an effective strategy for attenuating adipose tissue inflammation and IR [5, 22]. Consistent with these reports, our present study provides direct evidence showing that Clcn3 may play an important role in obesity-associated chronic ATM inflammation because the loss of Clcn3 significantly reduced the HFD-induced increase in the cell numbers of ATMs and F4/80+CD11c+CD206− ATMs during obesity.
Obesity produces a state of chronic and low grade inflammation accompanied by elevated levels of pro-inflammatory cytokines. In this study, we observed that the transcript levels of several key inflammatory cytokines (e.g., MCP-1 and IL-6) in eWAT were increased following HFD feeding in combination with a high serum CRP, thus confirming that adipose tissue serves as a primary source of inflammatory responses during obesity. The presented results demonstrated that upregulation of TNF-α or downregulation of adiponectin induced by a HFD were not affected by Clcn3 deficiency. The result in Fig. S2 demonstrates that global Clcn3 deficiency did not affect HFD-induced injury to the mouse liver, suggesting that the influence of Clcn3 deficiency on obesity may be restricted to adipose tissue. Thus, it is likely that the effect of Clcn3 on the local transcript levels of different types of inflammatory cytokines is also limited. In addition, Clcn3 deficiency tended to increase the adiponectin mRNA expression after the 16-week-HFD; however, there is no significant difference between the WT-HFD and KO-HFD groups. It has been found that there is an association between adipose tissue hypertrophy, a lower adiponectin level and aging [23]. Therefore, further investigation is required to examine the time-dependent influences of Clcn3 on age-related adipokine levels during obesity or type 2 diabetes. In addition, the expression of IL-6 was used to define adipose tissue inflammation. However, recent studies have demonstrated that a significant proportion of obese patients did not express typical inflammatory cytokines, such as IL-6 and TNF-α [24]. Moreover, oncostatin M (OSM), a member of the IL-6 family, has been found to change the phenotypes of adipose tissue macrophages toward an anti-inflammatory M2 type. OSM may thus serve as a novel target for obesity and obesity-induced metabolic syndrome [25, 26]. Due to the increased complexity of obesity-related inflammatory cytokines indicated in adipose tissue, further investigation is required to evaluate the effects of Clcn3 deficiency on the inflammatory cytokine profile during obesity.
TLR-4 has been demonstrated to be critical in adipose tissue inflammation, in which it directly targets NF-κB and, in turn, induces the release of pro-inflammatory adipokines [4, 27]. Of note, Clcn3 deficiency significantly suppressed the HFD-induced increase in the TLR-4 mRNA and protein levels in eWAT at the early stage of obesity (after 4 weeks of the HFD), suggesting that Clcn3 may be a novel mechanism that underlies obesity-induced adipose tissue inflammation through TLR-4 signaling.
LPS is an important endogenous inflammatory mediator that underlies adipose tissue dysfunction during diet-induced obesity [28]. A HFD induces an increase in the level of LPS that could cause abdominal ATM-driven inflammation, thus contributing to the impaired glucose metabolism during obesity [29]. Consistent with these findings, we observed a significant increase in the serum CRP level accompanied by altered lipid metabolism in obese mice following feeding a HFD, which could be abolished by Clcn3 deficiency. LPS has been reported to promote the secretion of pro-inflammatory cytokines through the activation of TLR-4 and its downstream NF-κB signaling in adipose tissue inflammation [30]. Recently, the knockdown of Clcn3 has been demonstrated to inhibit LPS-induced inflammation in RAW 264.7 cells by blocking the activation of TLR-4/NF-κB signaling [31]. In agreement with these findings, we further demonstrated that Clcn3 deficiency remarkably suppressed the activation of TLR-4/NF-κB in eWAT from obese mice following a HFD. Collectively, Clcn3 may mediate local adipose tissue inflammatory responses and systemic inflammation in obese mice via the activation of TLR-4/NF-κB signaling pathways.
Previous studies in Clcn3−/− mice have demonstrated that there is a decrease in systemic insulin levels or β cell insulin exocytosis, although the exact molecular mechanism remains debated [7, 17, 32, 33]. The data from our lab are in agreement with these previous reports and support that the Clcn3 gene may be involved in the regulation of insulin secretion [12]. With the exception of the rest insulin levels, our results indicated there were no abnormalities in the fasting glucose level, FFA and TG levels, glucose tolerance (PGTT), and insulin sensitivity (ITT and HOMA-IR) in global Clcn3−/− mice. These findings are consistent with a previous report that hyperglycemia or rebound hypoglycemia were not present following a glucose load in Clcn3−/− mice [34]. Apart from the pancreas, global Clcn3−/− mice show a degeneration of hippocampal neurons and an increase in the systemic leptin level [17], which raise another concern regarding the neuronal influences on the eating behaviors of global Clcn3−/− mice. Our results demonstrated that there is no significant difference in the body weight between WT and KO mice fed the NC; moreover, there was no significant difference in the total calorie intake among all groups during the feeding period. These findings suggested that the improvement in HFD-induced glucose and lipid disorders in KO mice may not be due to the Clcn3 deficiency on pancreatic insulin secretion and eating behaviors, and there must be another mechanism involved in this process. Furthermore, our present data showed that Clcn3 deficiency significantly increased the down-regulation of the serine 307 phosphorylation of IRS-1 induced by the HFD in eWAT, indicating that Clcn3 deficiency may recover the impaired insulin signaling. Similar beneficial effects of Clcn3 deficiency on disrupted glucose and lipid metabolism have been found in a high sucrose/fat, low-dose STZ induced T2DM mouse model [12]. Together, these findings raised the interesting possibility that Clcn3 might play a more general role in glucose and lipid metabolism disorders during the development of T2DM. Because the global Clcn3 knockout produces complex compensatory changes, further investigation in Clcn3 conditional knockout mouse models will be helpful to elucidate the role of Clcn3 in obesity and obesity-associated adipose tissue inflammation and metabolic changes.
Volume-sensitive Cl− channels/currents have been reported to be crucial for adipocyte hypertrophy, β cell excitability and inflammatory responses driven by immune cells and may thus be associated with adipose dysfunction [35, 36]. Clcn3 has been reported to encode the Cl− channel or regulate the volume-regulated Cl− movement or Cl− current in many cell types. The presented results from Clcn3−/− mice support the relation between an abnormal Cl− signal and adipose tissue dysfunction during obesity and T2DM. The inhibitory effect of tamoxifen on the LPS-induced increase in the transcript levels of inflammatory cytokines (e.g., MCP-1, TLR-4, and IL-6) in F4/80+ SVF cells isolated from mouse eWAT suggested a novel role of an abnormal Cl− signal in ATM inflammation. However, SWELL 1 (leucine-rich repeats containing 8A, LRRC8A) has recently been reported to be an essential component of volume-regulated ion channels (VRACs) in adipocytes and β cells, and may serve as an endogenous regulator of insulin sensitivity and glucose homeostasis [35, 37,38,39]. In addition, bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells [40]. Therefore, the molecular identity for volume-sensitive Cl− channels remains controversial. The discrepancy regarding the Cl− channel molecular component in different cell types and distinct cell components within adipose tissue (e.g., adipocytes, preadipocytes, immune cells, and endothelial cells) suggests that the molecular components of volume-sensitive Cl− channels in adipose tissue should be complicated. Caution should be employed in investigating the relation between Clcn3 and Cl− signaling, particularly under pathological conditions.
There may be limitations to the classification scheme of macrophages using CD11c because CD11c has also recently been found on adipose tissue dendritic cells (ATDCs) [41]. It has been reported that ATDCs may play a role in obesity-associated adipose inflammation [42, 43]. Therefore, our presented results could not exclude the influence of Clcn3 deficiency on ATDCs. Further investigation with specific macrophage markers, such as CD64, will be helpful to differentiate ATMs from ATDCs.
In conclusion, the present study identified that Clcn3 is a novel mechanism to regulate HFD-induced obesity and adipose tissue inflammatory responses. Clcn3 may thus be involved in the development of diet-induced obesity and associated lipid and glucose metabolism disorders. These results also suggest that modulation of Clcn3 may provide an appealing therapeutic target for obesity and type 2 diabetes.
References
Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–101.
Odegaard JI, Chawla A. Mechanisms of macrophage activation in obesity-induced insulin resistance. Nat Clin Pract Endocrinol Metab. 2008;4:619–26.
Shimobayashi M, Albert V, Woelnerhanssen B, Frei IC, Weissenberger D, Meyer-Gerspach AC, et al. Insulin resistance causes inflammation in adipose tissue. J Clin Invest. 2018;128:1538–50.
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–25.
Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8:301–9.
Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature. 1997;390:417–21.
Deriy LV, Gomez EA, Jacobson DA, Wang X, Hopson JA, Liu XY, et al. The granular chloride channel ClC-3 is permissive for insulin secretion. Cell Metab. 2009;10:316–23.
Guan YY, Wang GL, Zhou JG. The ClC-3 Cl- channel in cell volume regulation, proliferation and apoptosis in vascular smooth muscle cells. Trends Pharmacol Sci. 2006;27:290–6.
Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Annu Rev Physiol. 2010;72:95–121.
Huang LY, He Q, Liang SJ, Su YX, Xiong LX, Wu QQ, et al. ClC-3 chloride channel/antiporter defect contributes to inflammatory bowel disease in humans and mice. Gut. 2014;63:1587–95.
Yang H, Huang LY, Zeng DY, Huang EW, Liang SJ, Tang YB, et al. Decrease of intracellular chloride concentration promotes endothelial cell inflammation by activating nuclear factor-kappaB pathway. Hypertension. 2012;60:1287–93.
Huang YY, Huang XQ, Zhao LY, Sun FY, Chen WL, Du JY, et al. ClC-3 deficiency protects preadipocytes against apoptosis induced by palmitate in vitro and in type 2 diabetes mice. Apoptosis. 2014;19:1559–70.
Zheng LY, Li L, Ma MM, Liu Y, Wang GL, Tang YB, et al. Deficiency of volume-regulated ClC-3 chloride channel attenuates cerebrovascular remodelling in DOCA-salt hypertension. Cardiovasc Res. 2013;100:134–42.
Luu YK, Lublinsky S, Ozcivici E, Capilla E, Pessin JE, Rubin CT, et al. In vivo quantification of subcutaneous and visceral adiposity by micro-computed tomography in a small animal model. Med Eng Phys. 2009;31:34–41.
Luciani A, Dechoux S, Deveaux V, Poirier-Quinot M, Luciani N, Levy M, et al. Adipose tissue macrophages: MR tracking to monitor obesity-associated inflammation. Radiology. 2012;263:786–93.
Liang GZ, Cheng LM, Chen XF, Li YJ, Li XL, Guan YY, et al. ClC-3 promotes angiotensin II-induced reactive oxygen species production in endothelial cells by facilitating Nox2 NADPH oxidase complex formation. Acta Pharmacol Sin. 2018;39:1725–34.
Maritzen T, Keating DJ, Neagoe I, Zdebik AA, Jentsch TJ. Role of the vesicular chloride transporter ClC-3 in neuroendocrine tissue. J Neurosci. 2008;28:10587–98.
Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277:1531–7.
McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48.
Burow P, Klapperstuck M, Markwardt F. Activation of ATP secretion via volume-regulated anion channels by sphingosine-1-phosphate in RAW macrophages. Pflugers Arch. 2015;467:1215–26.
Tao J, Liu CZ, Yang J, Xie ZZ, Ma MM, Li XY, et al. ClC-3 deficiency prevents atherosclerotic lesion development in ApoE−/− mice. J Mol Cell Cardiol. 2015;87:237–47.
Zhuge F, Ni Y, Nagashimada M, Nagata N, Xu L, Mukaida N, et al. DPP-4 inhibition by linagliptin attenuates obesity-related inflammation and insulin resistance by regulating M1/M2 macrophage polarization. Diabetes. 2016;65:2966–79.
Miller KN, Burhans MS, Clark JP, Howell PR, Polewski MA, DeMuth TM, et al. Aging and caloric restriction impact adipose tissue, adiponectin, and circulating lipids. Aging Cell. 2017;16:497–507.
Rakotoarivelo V, Lacraz G, Mayhue M, Brown C, Rottembourg D, Fradette J, et al. Inflammatory cytokine profiles in visceral and subcutaneous adipose tissues of obese patients undergoing bariatric surgery reveal lack of correlation with obesity or diabetes. EBioMedicine. 2018;30:237–47.
Komori T, Morikawa Y. Oncostatin M in the development of metabolic syndrome and its potential as a novel therapeutic target. Anat Sci Int. 2018;93:169–76.
Komori T, Tanaka M, Senba E, Miyajima A, Morikawa Y. Deficiency of oncostatin M receptor beta (OSMRbeta) exacerbates high-fat diet-induced obesity and related metabolic disorders in mice. J Biol Chem. 2014;289:13821–37.
Suganami T, Tanimoto-Koyama K, Nishida J, Itoh M, Yuan X, Mizuarai S, et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol. 2007;27:84–91.
Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes. 2010;59:3049–57.
Hersoug LG, Moller P, Loft S. Role of microbiota-derived lipopolysaccharide in adipose tissue inflammation, adipocyte size and pyroptosis during obesity. Nutr Res Rev. 2018;31:1–11.
Rodriguez-Calvo R, Serrano L, Coll T, Moullan N, Sanchez RM, Merlos M, et al. Activation of peroxisome proliferator-activated receptor beta/delta inhibits lipopolysaccharide-induced cytokine production in adipocytes by lowering nuclear factor-kappaB activity via extracellular signal-related kinase 1/2. Diabetes. 2008;57:2149–57.
Xiang NL, Liu J, Liao YJ, Huang YW, Wu Z, Bai ZQ, et al. Abrogating ClC-3 Inhibits LPS-induced Inflammation via blocking the TLR4/NF-kappaB Pathway. Sci Rep. 2016;6:27583.
Li DQ, Jing X, Salehi A, Collins SC, Hoppa MB, Rosengren AH, et al. Suppression of sulfonylurea- and glucose-induced insulin secretion in vitro and in vivo in mice lacking the chloride transport protein ClC-3. Cell Metab. 2009;10:309–15.
Jentsch TJ, Maritzen T, Keating DJ, Zdebik AA, Thevenod F. ClC-3--a granular anion transporter involved in insulin secretion? Cell Metab. 2010;12:307–8.
Dickerson LW, Bonthius DJ, Schutte BC, Yang B, Barna TJ, Bailey MC, et al. Altered GABAergic function accompanies hippocampal degeneration in mice lacking ClC-3 voltage-gated chloride channels. Brain Res. 2002;958:227–50.
Zhang Y, Xie L, Gunasekar SK, Tong D, Mishra A, Gibson WJ, et al. SWELL1 is a regulator of adipocyte size, insulin signalling and glucose homeostasis. Nat Cell Biol. 2017;19:504–17.
Bao J, Perez CJ, Kim J, Zhang H, Murphy CJ, Hamidi T, et al. Deficient LRRC8A-dependent volume-regulated anion channel activity is associated with male infertility in mice. JCI Insight. 2018;3:e99767.
Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, et al. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science. 2014;344:634–8.
Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, et al. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell. 2014;157:447–58.
Kang C, Xie L, Gunasekar SK, Mishra A, Zhang Y, Pai S, et al. SWELL1 is a glucose sensor regulating beta-cell excitability and systemic glycaemia. Nat Commun. 2018;9:367.
Milenkovic A, Brandl C, Milenkovic VM, Jendryke T, Sirianant L, Wanitchakool P, et al. Bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells. Proc Natl Acad Sci USA. 2015;112:E2630–9.
Stefanovic-Racic M, Yang X, Turner MS, Mantell BS, Stolz DB, Sumpter TL, et al. Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes. 2012;61:2330–9.
Cho KW, Zamarron BF, Muir LA, Singer K, Porsche CE, DelProposto JB, et al. Adipose tissue dendritic cells are independent contributors to obesity-induced inflammation and insulin resistance. J Immunol. 2016;197:3650–61.
Wu H, Perrard XD, Wang Q, Perrard JL, Polsani VR, Jones PH, et al. CD11c expression in adipose tissue and blood and its role in diet-induced obesity. Arterioscler Thromb Vasc Biol. 2010;30:186–92.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No 81773722, 81370897, 81471425, and 81370680), the CHINA-CANADA Joint Health Research Program from NSFC-CIHR (No 81361128011), the Guangdong Provincial Department of Science and Technology (No 2016A050502023), the Science and Technology Program of Guangzhou City (No 201607010255 and 201803010092), the Fundamental Research Funds for the Central Universities (No 17ykzd02), and the 111 Project (No B13037) .
Author information
Authors and Affiliations
Contributions
GLW designed the study. XMZ, MMM, CCJ, GLW, XLH, and JYD wrote the manuscript. MMM, CCJ, XLH, LS, HZ, ZXR, and JL performed the experiments. CCJ, XLH, XJW, and XQH analyzed the data; XQH, HSS, and YYG revised the manuscript. GLW and XMZ are the guarantors of this work and, as such, had full access to all the data in this study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Ma, Mm., Jin, Cc., Huang, Xl. et al. Clcn3 deficiency ameliorates high-fat diet-induced obesity and adipose tissue macrophage inflammation in mice. Acta Pharmacol Sin 40, 1532–1543 (2019). https://doi.org/10.1038/s41401-019-0229-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0229-5
Keywords
This article is cited by
-
Clues and new evidences in arterial hypertension: unmasking the role of the chloride anion
Pflügers Archiv - European Journal of Physiology (2022)
