
Abstract
Impaired proteostasis is associated with normal aging and is accelerated in neurodegeneration. This impairment may lead to the accumulation of protein, which can be toxic to cells and tissue. In a subset of frontotemporal lobar degeneration with tau pathology (FTLD-tau) cases, pathogenic mutations in the microtubule-associated protein tau (MAPT) gene are sufficient to cause tau accumulation and neurodegeneration. However, the pathogenic events triggered by the expression of the mutant tau protein remain poorly understood. Here, we show that molecular networks associated with lysosomal biogenesis and autophagic function are disrupted in brains from FTLD-tau patients carrying a MAPT p.R406W mutation. We then used human induced pluripotent stem cell (iPSC)-derived neurons and 3D cerebral organoids from patients carrying the MAPT p.R406W mutation and CRISPR/Cas9, corrected controls to evaluate proteostasis. MAPT p.R406W was sufficient to induce morphological and functional deficits in the lysosomal pathway in iPSC-neurons. These phenotypes were reversed upon correction of the mutant allele with CRISPR/Cas9. Treatment with mTOR inhibitors led to tau degradation specifically in MAPT p.R406W neurons. Together, our findings suggest that MAPT p.R406W is sufficient to cause impaired lysosomal function, which may contribute to disease pathogenesis and serve as a cellular phenotype for drug screening.
Similar content being viewed by others

Introduction
Frontotemporal lobar degeneration with tau pathology (FTLD-tau) is characterized clinically by behavioral abnormalities along with memory loss and neuropathologically by the accumulation of intracellular tau protein. A subset of FTLD-tau cases occur by familial mechanisms in which mutations in the MAPT gene, which encodes the tau protein, are dominantly inherited.
Several mechanisms have been proposed to explain how the more than 50 reported MAPT mutations cause disease: abnormal MAPT splicing, altered microtubule binding kinetics, impaired degradation, or tau accumulation and aggregation, among others [1]. All MAPT mutations share an increased propensity for the tau protein to accumulate [2]. One such mutation, MAPT p.R406W, presents with Alzheimer disease-like progressive memory decline and exhibits a protracted clinical course that may last decades [3,4,5]. How MAPT p.R406W mutations lead to tau aggregation and precipitate their characteristic neurodegenerative changes is not clear.
Despite an incomplete picture of how the MAPT p.R406W mutation leads to disease, increasing evidence implicates impairment of the neuronal endolysosomal pathway in FTLD-tau [6,7,8,9,10,11,12,13]. However, how neurons expressing mutant tau degrade tau and whether mutant tau is sufficient to drive disrupted protein homeostasis (e.g., proteostasis) remains poorly understood.
Patient-derived induced pluripotent stem cells (iPSC) have emerged as a powerful tool to study the molecular mechanisms underlying neurodegenerative diseases [14,15,16,17]. iPSCs have the potential to more faithfully reflect the endogenous expression and splicing of genes (e.g., MAPT) compared to models that rely on overexpression, giving us a path towards understanding the factors that contribute to disease [18, 19]. To date, iPSC carrying MAPT mutations p.N279K, p.P301L, p.V337M and IVS10 + 16 have been described, and display phenotypes such as tau accumulation, tau hyperphosphorylation, tau insolubility, and vulnerability to specific cellular stressors [20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. In iPSC-neurons from MAPT p.R406W carriers, mutant neurons exhibit altered pre-synaptic function, tau mislocalization, altered tau phosphorylation and fragmentation [19, 35, 36].
Here, we show that iPSC-neurons from MAPT p.R406W carriers exhibit morphological and functional deficits in the lysosomal pathway that were reversed upon CRISPR/Cas9-mediated correction of the mutant allele. Additionally, we observed an increased co-localization of total and phosphorylated tau with lysosomal vesicles. Thus, our findings suggest that MAPT p.R406W is sufficient to cause altered lysosomal function, which may contribute to disease pathogenesis.
Materials and Methods
Patient consent
The Washington University School of Medicine Institutional Review Board reviewed the protocol of the Knight Alzheimer Disease Research Center (ADRC) Neuropathology Core, from which clinically and neuropathologically well-characterized brain tissues were obtained. As tissue was obtained postmortem it was exempt from IRB approval. Research participants provided autopsy consent limited to removal of the brain. All data were analyzed anonymously.
Skin biopsies were performed following written informed consent from the donor. The informed consent was approved by the Washington University School of Medicine Institutional Review Board and Ethics Committee (IRB 201104178 and 201306108).
Transcriptomics in human brain tissue
To evaluate the impact of MAPT p.R406W on lysosomal genes, we queried summary statistics from our previously generated bulk RNAseq data of the insular cortex of MAPT p.R406W (n = 2) and neuropathology-free controls (n = 2) [19]. Briefly, DNA libraries of individual samples were constructed using the TruSeq Stranded Total RNA Sample Prep with Ribo-Zero Gold kit (Illumina) and then sequenced by the HiSeq 4000 (Illumina). FASTQ files were aligned to human GRCh37 primary assembly. After alignment, Salmon (v. 0.7.2) was used to quantify expression levels of individual genes included in the GENCODE reference genome (GRCh37.75). Differential gene expression was performed using the R (v.3.4.2) package DESeq2 (v.1.18.1) as previously described [19]. From the summary statistics, we extracted genes that are regulated by Transcription Factor EB (TFEB), defined as: [1] containing a Coordinated Lysosomal Expression and Regulation (CLEAR) sequence or [2] being altered by overexpression of TFEB in vitro [37]. To determine whether genes that are regulated by TFEB and altered in MAPT p.R406W brains were enriched in specific functional pathways, gene enrichment analysis was performed using ToppGene [38].
iPSC generation and genome engineering
Dermal fibroblasts from MAPT p.R406W carriers (F11362 and F11421) were transduced with non-integrating Sendai virus carrying OCT3/4, SOX2, KLF4, and cMYC (Life Technologies) as previously described [19]. iPSCs that were heterozygous for MAPT p.R406W were edited to WT (F11362.1Δ1B06 and F11421.12Δ2A07) using CRISPR/Cas9 as previously reported [19, 39]. Mutation status was confirmed by Sanger sequencing (Supplemental Fig. 1). Cell lines were maintained in mTeSR medium (StemCell Technologies) on Matrigel. Cell lines were confirmed to be free of mycoplasma.
iPSC differentiation
MAPT p.R406W iPSC and isogenic controls were differentiated into neural progenitor cells (NPCs) as previously described [19]. Briefly, iPSCs were dissociated with Accutase (Life Technologies). iPSCs were then plated at 65,000 cells per well in Neural Induction Media (NIM; Stem Cell Technologies) in a 96-well v-bottom plate to form neural aggregates. After 5 days, neural aggregates were plated on Poly-L-Ornithine (PLO) and laminin-coated plates to form neural rosettes. After 5 to 7 days, neural rosettes were isolated by enzymatic selection and cultured as NPCs. NPCs were cultured on PLO and laminin-coated plates and terminal differentiation was initiated with the addition of cortical maturation medium (Neurobasal-A (Life Technologies) supplemented with B27 (Gibco), BDNF (Peprotech), GDNF (Peprotech), cAMP (Sigma) and L-glutamate (Sigma)). Neural cultures were maintained for six weeks, a time at which cells are Tuj1-positive, express robust levels of tau, and exhibit robust action potentials [18, 40].
Organoid generation
Human iPSCs were differentiated into 3D cerebral organoids as previously described [33, 41]. iPSCs were cultured as single cells in 10 µM of Y-27632 supplemented E8 media and transferred into individual wells of an AggreWell™800 plate (Stem Cell Technologies). The cells were incubated at 37 °C and 5% CO2 for 24 h. Organoids were then washed and transferred to an ultra-low attachment 10 cm plates in E6 medium supplemented with 2.5 μM Dorsomorphin (DM) (Tocris), 10 μM SB431542 (Tocris,) and 2.5 µM XAV-939 (Tocris). Media was replaced daily. On day 6 in suspension, media was replaced with neural medium (NM) containing Neurobasal-A (Life Technologies) supplemented with B27 supplement without vitamin A (Life Technologies), GlutaMax (Life Technologies), Anti-A (Life Technologies), 20 ng/ml FGF2 (R&D Systems) and 20 ng/ml EGF (Peprotech). Organoids were then cultured for an additional 19 days with daily medium changes in the first 10 days, and every other day for the final 9 days. To promote differentiation of the neural progenitors into neurons, FGF2 and EGF were replaced with 20 ng/ml BDNF (Peprotech) and 20 ng/ml NT3 (Peprotech) beginning at day 25. Beyond day 43, medium changes were performed every four days using only NM lacking growth factors.
Antibodies
The following antibodies were used in this study: Tau5 (total tau; generously provided by Dr. Lester Binder), DAPI (Sigma), Alexa488 anti-rabbit (Life Technologies, Carlsbad, CA, USA), Alexa568 anti-mouse (Life Technologies, Carlsbad, CA, USA), beta III Tubulin (Promega), LAMP1 (Abcam, ab24170), AT180 (ptau-Thr231; Thermo Fisher Scientific, MN1040), EEA1 (Abcam, ab2900), Cathepsin D (generously provided by Dr. Stuart Kornfeld, Washington University in St Louis), Rab7 (Cell Signaling Technology, 9367S), TFEB (Abcam, ab270604), pTFEB (pSer122, Cell Signaling Technology, 86843S) and Ubiquitin (Cell Signaling Technology, 3933S).
Immunoblotting
Cell lysates were extracted in lysis buffer (50 mM Tris pH 7.6, 1 mM EDTA, 150 mM NaCl, 1% TritonX-100, phosphatase and protease inhibitors (Millipore-Sigma)) and incubated on ice for 5 min. Lysates were then centrifuged at 14,000 × g for 10 min at 4 °C, and the resulting supernatant was saved for analysis. Total protein levels were assayed by bicinchoninic acid assay (BCA) assay (Thermo-Fisher). Standard sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed in 4–12% Criterion Tris-HCl gels (Bio-Rad) with 10 µg of total protein loaded in each well. Samples were boiled in Laemmli sample buffer prior to electrophoresis. Gels were transferred to polyvinylidene fluoride (PVDF) membranes, which were probed with the antibodies listed above. Antibodies were visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo) or Lumigen ECL Ultra (TMA-6) according to manufacturer’s instructions. Immunoblots were exposed on a Syngene G:Box iChemi XT Gel Documentation System and imaged using GeneSnap software according to manufacturer’s instructions. Band intensity was analyzed using GeneTools, and band intensity was expressed relative to the normalized control within each blot.
Immunostaining
To evaluate the impact of MAPT p.R406W on the endolysosomal pathway, culture media was aspirated, and cells were washed and fixed with 4% paraformaldehyde (Sigma, St Louis, MO, USA). Cells were washed and permeabilized with permeabilization buffer (0.1% Triton X-100 in PBS). Cells were then blocked in 3% bovine serum albumin (BSA; Sigma, St Louis, MO, USA) and incubated with primary and secondary antibodies diluted in 1% BSA. Immunostained cells were then imaged (Nikon Eclipse 80i fluorescent microscope) and acquired by using Metamorph Molecular Devices software. Confocal images were acquired by using Zeiss LSM 880 II Airyscan FAST Confocal Microscope.
Image quantification
Fiji, an ImageJ-based image analysis software [42], was used to quantify the distribution and size of LAMP1-positive vesicles. The extent of distribution of LAMP1-positive vesicles in each cell was determined by measuring the average distance between the nuclear membrane and LAMP1-stained vesicles present in the neurite in the axon hillock as described by Ouyang and colleagues [43]. To do this, the ‘straight line’ tool was selected to draw a straight line from the center of the nuclear membrane to the LAMP1 staining seen in the neurite in the axon hillock. The ‘measure’ tool was used to define the distance in micron. The size of the individual LAMP1-positive vesicles in the cell body was measured by using the plugin “find edges” in Fiji software to mark the edges. The diameter of the individual LAMP1-positive vesicles was then measured. In addition to measuring size, “find edges” allowed for the quantification of total LAMP1-positive vesicles.
Co-localization analysis was performed using Metamorph (Molecular Devices) software. The LAMP1-stained area was selected in the cell body of the neuron. The percent area of the red that overlapped with the green puncta area was calculated using Metamorph colocalization function. Pearson’s correlation coefficient (r) was used to quantify the association between LAMP1 and tau or tau phosphorylated at Thr231 (ptau). Values range from zero to one, where zero represents no pixels are co-localized and one represents co-localization of all pixels.
Lysosomal activity assay
A fluorometric assay was performed to test the impact of MAPT p.R406W on activity of lysosomal enzymes. Cell lysates were homogenized in buffer containing 150 mM NaCl, 10 mM Tris 7.5, 1 mM DTT, and 0.2% Triton X-100. The substrate of β-glucuronidase was added to the cell lysate separately. β-glucuronidase cleavage was measured at 448 nm emission and 365 nm excitation in a fluorescence spectrophotometer using a standard curve ranging from 0.02 to 5 mM of 4-methylumbelliferone [44]. Protein quantification from three technical replicates and three biological replicates were averaged and expressed as mean ± standard error of the mean (SEM). Statistical difference was measured using an unpaired Student’s t-test.
To evaluate lysosomal acidity, six-week-old neurons were incubated for 5 min with 5 nM of LysoTracker® Red DND-99 diluted in the cortical maturation medium. Live cell images were acquired immediately [45]. Fluorescence intensity of LysoTracker Red staining in the soma was quantified by Fiji. Briefly, the fluorescence intensity of LysoTracker® in the soma of each cell was measured and then corrected for background fluorescence resulting in the Corrected Total Cell Fluorescence (CTCFs) values. CTCFs values were calculated for each cell and graph was plotted by normalizing CTCF relative to the isogenic, wild-type average value.
To evaluate proteolytic activity in lysosomal compartments, six-week-old neurons were incubated for 4 h with 10 µg/mL of DQ™ Red BSA diluted in the cortical maturation medium. Cells were washed once with HBSS solution. Live cell images were acquired immediately. Fluorescence intensity of DQ™ Red BSA staining in the soma was quantified by Fiji as described above. CTCFs values were calculated for each cell and graph was plotted by normalizing CTCF relative to the wild-type average value.
Torin treatment of iPSC-neurons
After 6 weeks in culture, iPSC-neurons maintained in a 48-well plate were washed with DMEM/F12 and treated with 250 nM Torin or DMSO control. After 5 h of treatment, cell culture media was collected, centrifuged at 1000 rpm for 10 min at 4 °C to remove cell debris, and flash frozen. Cells were collected in PBS and pelleted.
Immunoprecipitation and Mass Spectrometry (IP/MS) to quantify tau fragments
IP/MS for tau fragments was performed as previously described [40]. Briefly, CNBr-activated Sepharose beads (GE Healthcare 17-0430-01) were crosslinked to Tau1 (mouse monoclonal, provided by Drs. Nicholas Kanaan) and HJ8.5 antibodies (mouse monoclonal, provided by Dr. David Holtzman). Samples are spiked with 5 ng 15N labeled recombinant 2N4R tau (gift from Dr. Guy Lippens), and tau concentration is calculated using this internal standard.
Soluble tau was immunoprecipitated in detergent (1% NP-40), chaotropic reagent (5 mM guanidine), and protease inhibitors (Roche Complete Protease Inhibitor Cocktail). 30 µL of 50% slurry of the tau antibody beads were rotated with the solution for 90 min at room temperature. The beads were washed one time in 0.5 M guanidine and two times in 25 mM triethyl ammonium bicarbonate buffer (TEABC, Fluka 17902). The bound tau was digested on-beads with 400 ng MS grade trypsin (Promega, V5111) or AspN (Promega, V162A) for 16 h at 37 °C. Digests were loaded onto TopTip C18 (Glygen, TT2C18.96), desalted, and eluted per manufacturer’s instructions.
A 5 µL aliquot of the peptide resuspension was subjected to nano-Acquity LC and MS analysis. The nano-Acquity LC (Waters Corporation, Milford, MA) was fitted with HSS T3 75 µm x 100 mm, 1.8 µm column and a flow rate of 0.5 µL/min of a gradient of solution A and B was used to separate the peptides. Solution A was composed of 0.1% formic acid in MS-grade water and solution B was composed of 0.1% formic acid in acetonitrile. Peptides were eluted from the column with a gradient of 2% to 20% of solution B in 8 min, then 20% to 40% solution B for another 3 min before ramping up to 85% solution B in another 3 min to clean the column. The Orbitrap Fusion/Fusion Lumos was equipped with a Nanospray Flex electrospray ion source (Thermo Scientific, San Jose, CA). Peptide ions sprayed from a 10 µm SilicaTip emitter (New Objective, Woburn, Ma) into the ion–source were targeted and isolated in the quadrupole and were then fragmented by HCD and ion fragments were detected in the Orbitrap (resolution of 60,000, mass range 150–1200 m/z). Monitoring of hydrophilic peptides (SSRcalc < 9, all without leucine) for peptide profiling was performed on a HSS T3 300 µm x 100 mm, 1.8 µm column at a flow rate of 4ul/min with an elution occurring with a 2% to 12% solution B gradient and a spray operating on a 30 µm SilicaTip emitter.

RNAseq was performed in brains from MAPT p.R406W and neuropathology-free controls and reported in Jiang et al. [19]. A Principal component analysis reveals transcriptome-wide differences between MAPT p.R406W (n = 2) and neuropathology-free control brains (n = 16). B We tested whether genes that contain a CLEAR sequence or that are differentially expressed in the presence of TFEB in vitro were differentially expressed in human brains from MAPT p.R406W carriers compared with normal controls. Volcano plot showing log2 fold change between MAPT p.R406W and control brains and the –log10 p-value for each gene. Red nodes: genes with p < 0.05.
Results
Lysosomal gene expression is altered in brains from MAPT p.R406W patients
Prior studies have demonstrated that lysosomal proteins, such as TFEB, LAMP1 and Cathepsin D, are elevated in brains from FTLD-tau patients [46, 47]. TFEB is a transcription factor that regulates lysosomal biogenesis by coordinating expression of lysosomal genes [48]. To determine whether TFEB gene expression is altered in FTLD-tau caused by MAPT p.R406W, we performed differential gene expression analysis in bulk transcriptomic data obtained from a brain region affected early in disease pathogenesis, the insular cortex, from MAPT p.R406W carriers (n = 2) and unrelated controls (n = 2) (Fig. 1) [19]. TFEB expression was significantly elevated in MAPT p.R406W carriers compared with unrelated controls (Table 1). TFEB regulates expression of genes containing a CLEAR-box sequence (5′-GTCACGTGAC-3) as well as some genes that lack this recognition sequence [37]. We analyzed genes with a CLEAR-box sequence (Supplemental Table 1) and genes that change as a function of TFEB overexpression (Supplemental Table 2). We found that approximately one-third of all genes regulated by TFEB are differentially expressed in brains from MAPT p.R406W carriers (Fig. 1, Table 1). These differentially expressed genes are enriched in pathways associated with hydrolase activity (GO:0016798; p = 1.15E−18) and ATPase activity (GO:0046961; p = 2.81E–06). TFEB has also been implicated in autophagosome formation [49]. Expression of ATG5, ATG12, and ATG16L1 genes, which form a complex that is required in the early stages of autophagosome formation, were significantly lower in MAPT p.R406W carriers compared with unrelated controls (Table 1). ATG2A and ATG2B, which recruit other ATG proteins to the p62-positive autophagosome formation site, were also differentially expressed (Table 1) [50]. Together, these findings point to a molecular imbalance in TFEB-mediated and autophagosome pathways in MAPT p.R406W brains.
MAPT p.R406W is sufficient to alter lysosomal protein levels in human neurons
To determine whether MAPT p.R406W is sufficient to disrupt protein clearance machinery, we leveraged human iPSC isolated from MAPT p.R406W carriers [18, 19] (Supplemental Fig. 1). We also used CRISPR/Cas9 genome-edited, isogenic controls to evaluate whether cellular phenotypes were driven specifically by the mutant allele [18, 19] (Supplemental Fig. 1). Two independent MAPT p.R406W donors and their isogenic, corrected controls (MAPT WT) were then differentiated into cortical neurons using a growth factor-based approach (Fig. 2A; see Methods). At 6 weeks in culture, iPSC-neurons are enriched for Tuj1-positive cells (Fig. 2B), produce spontaneous action potentials, form functional synapses and display a profile of tau isoform expression that is similar to tau found in the central nervous system [19, 40]. After 6 weeks in cortical maturation medium, neurons exhibited significantly elevated total (Tau5) and ptau (pThr231; AT180; Supplemental Fig. 2). All data points were collected from multiple biological replicates across three independent differentiations from two independent MAPT p.R406W donor lines.

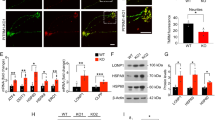
A iPSC from a MAPT mutation carrier and CRISPR/Cas9-corrected control (wild-type (WT)) were differentiated into cortical neurons and cultured for 6 weeks prior to analysis. B Representative immunostaining for Tuj1 (green) and DAPI (blue) illustrates that at 6 weeks in culture, cells are enriched for neurons. Scale bar, 10 microns. C Immunoblots of cell lysates (10 μg total protein) were probed with LAMP1, Cathepsin D and ubiquitin antibodies. D Quantification of protein analyte levels in the MAPT p.R406W neurons and isogenic controls. Graph represents mean ± SEM. Significance was determined using an unpaired, t-test. *p < 0.05; **p < 0.01. ****p < 0.0001.
To determine whether the MAPT p.R406W mutation induces downstream protein degradation pathways, we measured TFEB, phospho-TFEB, Rab7, Lysosomal Associated Membrane Protein 1 (LAMP1), Cathepsin D and ubiquitinated proteins in cell lysates from MAPT p.R406W neurons and isogenic, control neurons. We observed a modest increase in proteins involved in lysosomal biogenesis (total TFEB, phospho-TFEB, and Rab7 [51]) in MAPT p.R406W neurons, however, the difference compared to isogenic, control neurons did not reach statistical significance (Supplemental Fig. 2). We found that LAMP1 and the active form of Cathepsin D were significantly elevated in the MAPT p.R406W neurons compared to isogenic, control neurons (Fig. 2C, D). LAMP1 is a glycoprotein that resides on lysosomal membranes and plays a role in lysosomal integrity, pH, and catabolism [52]. Cathepsin D is a lysosomal aspartyl protease [53]. Given the observation that lysosomal protein machinery was elevated in MAPT p.R406W neurons, we sought to determine whether there was evidence of broader cellular and proteostatic stress by measuring ubiquitinated proteins. High molecular weight ubiquitinated proteins were significantly elevated in MAPT p.R406W neurons compared to isogenic, control neurons (Fig. 2C, D). Thus, MAPT p.R406W is sufficient to produce altered expression of lysosomal and proteostatic stress in human neurons.
MAPT p.R406W alters lysosomal morphology in human neurons
Alterations in lysosomal protein levels in iPSC-neurons suggests that the MAPT p.R406W mutation impacts lysosomal function. To begin to investigate the impact of MAPT p.R406W on lysosomal function, we evaluated LAMP1-positive vesicles in Tuj1-positive MAPT p.R406W neurons and isogenic controls. We observed a distinct pattern of LAMP1 staining between the MAPT p.R406W and the isogenic, control neurons (Fig. 3A). To quantify these differences, we measured three parameters (summarized in Fig. 3B): vesicle distribution (Fig. 3C), vesicle size (Fig. 3D) and vesicle number (Fig. 3E). Analyses were performed in Tuj1-positive cells (Supplemental Fig. 3).

Human iPSC-neurons neurons from a MAPT p.R406W mutation carrier and the isogenic, CRISPR/Cas9-corrected control (wild-type (WT)) were differentiated into cortical neurons and cultured for 6 weeks prior to analysis. A Representative immunostaining for LAMP1 (red) and DAPI (blue) illustrates altered LAMP1-positive vesicles in the MAPT p.R406W neurons compared with isogenic controls. Scale bar, 10 microns. White arrows indicate LAMP1-positive vesicles in the neurites of MAPT p.R406W neurons. B Diagram of quantification of LAMP1-positive vesicles represented in C–E. All quantification was performed in Tuj1-positive cells. C Distance of LAMP1-positive vesicles from the nuclear membrane is significantly greater in MAPT p.R406W neurons (n = 27 cells) compared to isogenic controls (n = 60 cells). D The size of LAMP1-positive vesicles in the soma is significantly larger in MAPT p.R406W neurons (n = 10 cells) compared with the isogenic controls (n = 12 cells). E The number of LAMP1-positive vesicles within the soma is significantly reduced in MAPT p.R406W neurons (n = 11 cells) compared to isogenic controls (n = 11 cells). Graphs represent mean ± SEM. Significance was determined using an unpaired, t-test. *p < 0.05; **p < 0.01; ****p < 0.0001.
Active lysosomes are clustered around the perinuclear region, while immature lysosomes are localized near the cell periphery [54, 55]. LAMP1-positive vesicle distribution reflects the distance of vesicles between the nuclear membrane and the cell periphery (neurite). We found that the distribution of LAMP1-positive vesicles in Tuj1-positive neurons was significantly greater in MAPT p.R406W neurons compared with isogenic, control neurons (Fig. 3C). LAMP1-positive vesicles in the MAPT p.R406W neurons were also larger in size (Fig. 3D) and fewer in number (Fig. 3E) compared to isogenic, control neurons. We replicated this finding in an independent donor carrying the MAPT p.R406W mutation and its corresponding isogenic control (Supplemental Fig. 4). To determine whether the MAPT p.R406W mutation produce a generalized defect in vesicle trafficking, we analyzed the distribution and size of early endosomes by monitoring Early Endosome Antigen 1 (EEA1)-positive vesicles in these neurons. We found no difference between the MAPT p.R406W and isogenic, control neurons (Supplemental Fig. 5). Thus, MAPT p.R406W neurons exhibit defects in lysosomal morphology.
MAPT p.R406W is sufficient to alter lysosomal function in human neurons
To determine whether the observed morphological changes in LAMP1-positive vesicles in MAPT p.R406W neurons translates to altered lysosomal function, we evaluated vesicle acidity using LysoTracker. Immature and dysfunctional lysosomal vesicles are less acidic [56]. LysoTracker staining was diffuse in MAPT p.R406W neurons, while distinct, large puncta were observed in the soma in isogenic, control neurons (Fig. 4A). Upon quantification of the LysoTracker fluorescence in the soma of individual neurons (Fig. 4B), we observed a significant decrease in LysoTracker intensity in the soma of MAPT p.R406W neurons compared to the isogenic, control neurons (Fig. 4C). We replicated this finding in an independent donor line with its isogenic control (Supplemental Fig. 6A, B). Thus, MAPT p.R406W neurons have fewer LAMP1-positive vesicles (Fig. 3E), and these vesicles are less acidic than control neurons.

Human iPSC-neurons neurons from a MAPT p.R406W mutation carrier and isogenic, CRISPR/Cas9-corrected control (wild-type (WT)) were differentiated into cortical neurons and cultured for 6 weeks prior to analysis. A MAPT p.R406W neurons exhibit reduced LysoTracker staining. Live cells were incubated with Lysotracker and were imaged as described in Methods. Representative images of LysoTracker-stained neurons are shown in gray scale for clarity. Scale bar, 10 microns. Lower panel represents magnification of the cells in the black box. B Schematic of the quantification of LysoTracker staining in MAPT p.R406W neurons (n = 78 cells) and isogenic controls (n = 46 cells). C Quantification of the intensity of LysoTracker staining in individual soma. Graphs represent mean ± SEM. Significance was determined using an unpaired, t-test. ***p < 0.001.
Next, we evaluated the impact of MAPT p.R406W on cargo uptake and proteolytic degradation (Fig. 5A). DQ-BSA is a fluorogenic stain that is internalized and fluoresces upon degradation by proteolytic activity in the lysosome (Fig. 5B) [57]. MAPT p.R406W neurons exhibited dense and punctate staining while isogenic, control neurons produced more diffuse staining (Fig. 5A). Upon quantification, we found that there is a significant increase in the DQ-BSA fluorescence intensity in the soma of MAPT p.R406W neurons compared to isogenic controls (Fig. 5C). We next measured the activity of a lysosomal hydrolase enzyme, β-glucuronidase, which degrades glycosaminoglycans like heparan sulfate. We observed a significant increase in β-glucuronidase activity in MAPT p.R406W neurons compared with isogenic controls (Figs. 5D, E). We replicated this finding in an independent donor line (Supplemental Fig. 6C, D). Together, these results suggest that MAPT p.R406W leads to changes in acidity and proteolysis in degradative vesicles in human neurons.

Human iPSC-neurons neurons from a MAPT p.R406W mutation carrier and isogenic, CRISPR/Cas9-corrected control (wild-type (WT)) were differentiated into cortical neurons and cultured for 6 weeks prior to analysis. A MAPT p.R406W neurons exhibit increased DQ-BSA fluorescence. Live cells were incubated with DQ-BSA and were imaged as described in Methods. Representative images of DQ-BSA-stained neurons are shown in gray scale for clarity. Scale bar, 10 microns. Lower panel represents magnification of the cells in the black box. B Diagram of DQ-BSA mechanism. C Quantification of the intensity of DQ-BSA staining in soma from MAPT p.R406W neurons (n = 79 cells) and isogenic controls (n = 69 cells). D Diagram of secondary elevation of lysosomal enzymes. E Enzyme activity of β-Glucuronidase measured in cell lysates from MAPT p.R406W neurons and isogenic controls. Enzymatic activity was normalized to the total protein. Graphs represent mean ± SEM. Significance was determined using an unpaired, t-test. *p < 0.05; ****p < 0.0001.
Total tau and phosphorylated tau accumulate in lysosomes in MAPT p.R406W neurons
Tau phosphorylated at Thr231 (ptau) is enriched in tauopathy brains, particularly those carrying the MAPT p.R406W mutation [58,59,60]. We observed a significant increase in total tau and ptau by immunoblot in iPSC-neurons from MAPT p.R406W carriers (Supplemental Fig. 2). Specific tau post-translational modifications (e.g., phosphorylation and acetylation) have been reported to differentially clog and accumulate in lysosomes [10, 61]. Hence, we next sought to determine whether total tau and ptau accumulate in these dysfunctional lysosomes. Neurons were stained with LAMP1 and total tau (Tau5) or ptau (pThr231; AT180) and imaged by confocal microscopy. Tau-positive cells were then quantified for the extent of co-localization with LAMP1. We observed an increase in the co-localization of LAMP1 with total tau (Fig. 6A, upper panel) and LAMP1 with ptau (Fig. 6A, lower panel) in MAPT p.R406W neurons. Upon quantification, we detected a significant increase in the co-localization of tau (Fig. 6B) and ptau (Fig. 6C) with LAMP1-positive vesicles compared with the isogenic controls.

iPSC from a MAPT p.R406W mutation carrier and CRISPR/Cas9-corrected control (wild-type (WT)) were differentiated into cortical neurons and cultured for 6 weeks prior to analysis. A Representative confocal images showing colocalization of LAMP1 (red) and total tau (Tau5; green) or ptau (AT180; green) in the MAPT p.R406W neurons compared with isogenic controls. Scale bar, 10 microns. B Colocalization of LAMP1 and total tau in tau-positive soma (WT, n = 146 cells; p.R406W, n = 95 cells) C Colocalization of LAMP1 and ptau in ptau-positive soma (WT, n = 73 cells; p.R406W, n = 39 cells). Graphs represent mean ± SEM. Significance was determined using an unpaired, t-test. **p < 0.01; ****p < 0.0001.
MAPT p.R406W cerebral organoids phenocopy lysosomal alterations
Based on the alteration in lysosomal structure and function in the MAPT p.R406W neurons, we hypothesized that the mutant neurons are undergoing cellular and proteostatic stress. To determine whether we could replicate our findings from the 2D, planar culture in 3D, we generated cerebral organoids from a MAPT p.R406W carrier and isogenic control. Culturing neurons in 3D facilitates self-organization, layering similar to what is observed in the developing human cortex, and formation of complex synaptic connections [33, 41, 62]. Following previously published protocols [41], iPSCs were patterned into neural aggregates and then cultured in suspension for 2 months prior to analysis (Fig. 7A). Similar to the 2D cultures, we observed a significant increase in β-glucuronidase activity in the MAPT p.R406W organoids compared to the isogenic controls (Fig. 7B). We also found that LAMP1 and Cathepsin D were significantly increased in the MAPT p.R406W organoids compared to the isogenic controls (Fig. 7C, D).

A Schematic of organoid generation. iPSC from a MAPT mutation carrier and CRISPR/Cas9-corrected control (wild-type (WT)) were differentiated into cortical organoids and cultured for 2 months. B Enzyme activity of β-Glucuronidase measured in organoid lysates (10 μg protein) from MAPT p.R406W and corrected control organoids. Enzymatic activity was normalized to total protein. Graphs represent mean ± SEM. C Immunoblots of cell lysates from organoids were probed with LAMP1 and Cathepsin D (active form shown) antibodies. D Quantification of protein analyte levels in the MAPT p.R406W organoids and isogenic controls. Graphs represent mean ± SD. Significance was determined using an unpaired, t-test. *p < 0.05.
mTOR inhibition reduces tau in MAPT p.R406W neurons
Tau is degraded by lysosomal pathways [10, 63, 64], and here, we show that MAPT p.R406W neurons and 3D cerebral organoids exhibit disrupted lysosomal function. Thus, we asked whether pharmacologically targeting protein degradation pathways would enhance tau degradation. mTOR is the catalytic subunit of mTORC1 and mTORC2, which plays a role in lysosomal biogenesis, activity, and positioning. MAPT p.R406W and isogenic, control neurons were treated with a dual mTORC1 and mTORC2 inhibitor, torin-1, or DMSO control, and cell lysates were analyzed by IP/MS. Torin-1 treatment led to a significant reduction in intracellular tau in MAPT p.R406W neurons without altering tau levels in control neurons (Fig. 8).

iPSC-derived neurons from MAPT p.R406W and isogenic, controls were cultured for 6 weeks prior to treatment with torin-1 or DMSO, control for 5 h. Tau was measured in cells lysates using IP/MS. Graphs represent mean ± SEM. Significance was determined using an unpaired, t-test. *p < 0.05.
Discussion
The MAPT p.R406W mutation causes autosomal dominant FTLD-tau. However, the pathogenic events triggered by the expression of MAPT p.R406W are not fully understood. Current cellular and animal models used to study FTLD-tau and other tauopathies have several limitations. Most cellular and transgenic animal models rely on overexpression of a mutant transgene comprising a single tau isoform, which may produce effects that are a function of protein overexpression and possible off-target effects rather than a disease-relevant phenotype. Thus, our understanding of how tau is metabolized in the human brain has been obtained from experimental paradigms that do not fully capture physiological conditions relevant to human tauopathies. Here, we show that lysosomal biogenesis, autophagosome machinery, and downstream protein degradation pathways are disrupted in FTLD-tau brains expressing the MAPT p.R406W mutation and that the MAPT p.R406W is sufficient to induce these changes in human neurons and cerebral organoids, leading to altered lysosomal function. Finally, we show that treatment with mTORC1 and mTORC2 inhibitors reduce tau levels specifically in MAPT p.R406W neurons.
FTLD-tau caused by MAPT p.R406W results in an overall defect in lysosomal machinery. TFEB, which is the master regulator of lysosomal function, was found to be elevated in the FTLD-tau brain tissue homogenates [46]. This increase could reflect changes in neurons and glia. Similarly, we found that TFEB transcript levels were elevated in the MAPT p.R406W brain tissue. TFEB promotes expression of lysosomal genes and autophagosome formation [37, 49, 65], and many of these genes are also differentially expressed in MAPT p.R406W brains. Modulation of lysosomal pathways in mouse models of tauopathy modifies tau clearance and pathology [11,12,13]. It has been shown that overexpression of TFEB in mouse models of tauopathy (PS19 and rTg4510) activates the lysosomal pathways, leading to reduced tau aggregation and lipofuscin granules and restored synaptic function [12, 13]. TFEB has also been shown to play an essential role in mediating lysosomal-mediated exocytosis and spreading of tau [66]. Additionally, reduced expression of ATG genes suggest that autophagy is compromised in MAPT p.R406W brain tissue. ATG5-ATG12-ATG16L1 complex is required at the upstream stages of autophagosomes formation. ATG2a and ATG2b are involved in recruiting additional ATG proteins at the p62‐positive autophagosome‐formation site [50, 67, 68]. Thus, multiple stages of autophagosome formation may be disrupted in FTLD-tau brains.
Tau is degraded through several pathways, including the ubiquitin/proteasome pathway and the lysosome pathway [8, 9]. High molecular weight ubiquitinated proteins were elevated in the MAPT p.R406W neurons. This phenotype has been reported in other stem cell models of FTLD-tau and suggests that the cells are undergoing cellular and proteostatic stress [24]. Interestingly, mutant tau protein is unable to efficiently activate the lysosome pathway [10]. Our current findings show that MAPT p.R406W is sufficient to lead to enhanced co-localization of total and phosphorylated forms of tau with LAMP1-positive degradative vesicles. This could be due the inefficient activation of the lysosome pathway, reduced degradative capacity within the vesicles, or a feedback loop in which both occur. Consistent with prior reports [36], we do not observe bona-fide aggregated material in MAPT p.R406W neurons. With aging, the observed build-up of tau in dysfunctional lysosomes could lead to neurofibrillary tangles.
Efficient bulk cargo degradation and recycling is critical for the homeostasis of the polarized neurons and occurs in the cell body [54]. Endosomes and immature lysosomes travel along the neurites and fuse with the active, LAMP1-positive, lysosomes in the soma [69]. LAMP1-vesicles were distributed away from the nuclear membrane towards the neurites in MAPT p.R406W neurons, and this defect was reversed upon correction of the mutant allele. Interestingly, similar phenotypes have been reported in cell lines from Parkinson’s disease patients [70]. LAMP1-vesicle distribution could be due to defects in vesicle trafficking; however, the absence of changes in early endosome distribution as well as the altered lysosomal size suggests there are specific functional defects in the lysosome.
Lysosomes represent a heterogeneous population of vesicles that differ in size, function, and maturity. In addition to a reduction in acidity, we observed an increase in lysosomal enzymes and proteolytic activity in mutant neurons, which is consistent with a phenotype described in lysosomal storage disorders. Secondary elevation of lysosomal enzyme activity, when evaluated in cell lysates under the correct buffer conditions, is a compensatory mechanism found in cells from patients and cell models of lysosomal storage diseases [71, 72]. Loss-of-function mutations in lysosomal genes lead to the accumulation of undegraded material that changes the pH and environment in the lysosomal affecting its degradative capacity [72,73,74]. MAPT p.R406W neurons exhibit an increase in active Cathepsin D along with increased DQ-BSA staining, pointing to more proteolytic activity in the cell. However, the observed elevated proteolytic activity occurs in an environment with enlarged LAMP1-positive vesicles, reduced acidity, and an increase in β-glucuronidase. Together, these data suggest that a number of proteolytic enzymes are building up in vesicles that lack degradative capacity. Phenotypes consistent with those observed in lysosomal storage diseases are increasingly reported in neurodegenerative disease [70, 75,76,77,78,79,80,81].
In conclusion, we used iPSC-derived neurons and cerebral organoids to demonstrate that lysosomal dysfunction in FTLD-tau brains may be triggered by the MAPT mutation. Targeting these defects by increasing the lysosomal-mediated degradation will likely be effective in treating multiple tauopathies [77].
Data availability
The datasets analyzed during the current study are available in the Synpase repository: https://www.synapse.org/#!Synapse:syn12181323.
References
van Swieten J, Spillantini MG. Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol. 2007;17:63–73.
Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta. 2005;1739:240–50.
Reed LA, Grabowski TJ, Schmidt ML, Morris JC, Goate A, Solodkin A, et al. Autosomal dominant dementia with widespread neurofibrillary tangles. Ann Neurol. 1997;42:564–72.
van Swieten JC, Stevens M, Rosso SM, Rizzu P, Joosse M, de Koning I, et al. Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol. 1999;46:617–26.
Lindquist SG, Holm IE, Schwartz M, Law I, Stokholm J, Batbayli M, et al. Alzheimer disease-like clinical phenotype in a family with FTDP-17 caused by a MAPT R406W mutation. Eur J Neurol. 2008;15:377–85.
Morimoto RI, Cuervo AM. Proteostasis and the aging proteome in health and disease. J Gerontol A Biol Sci Med Sci 2014;69:S33–8.
Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–97.
Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol. 2013;105:49–59.
Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17:22–35.
Caballero B, Wang Y, Diaz A, Tasset I, Juste YR, Stiller B, et al. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell. 2018;17:e12692.
Zhu M, Zhang S, Tian X, Wu C. Mask mitigates MAPT- and FUS-induced degeneration by enhancing autophagy through lysosomal acidification. Autophagy 2017;13:1924–38.
Wang H, Wang R, Carrera I, Xu S, Lakshmana MK. TFEB Overexpression in the P301S Model of Tauopathy mitigates increased PHF1 levels and Lipofuscin Puncta and rescues memory deficits. eNeuro. 2016;3. https://doi.org/10.1523/ENEURO.0042-16.2016.
Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y, et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med. 2014;6:1142–60.
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012;482:216–20.
Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008;321:1218–21.
Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267–80.
Iovino M, Patani R, Watts C, Chandran S, Spillantini MG. Human stem cell-derived neurons: a system to study human tau function and dysfunction. PloS One. 2010;5:e13947.
Karch CM, Kao AW, Karydas A, Onanuga K, Martinez R, Argouarch A, et al. A comprehensive resource for induced pluripotent stem cells from patients with primary Tauopathies. Stem Cell Rep. 2019;13:939–55.
Jiang S, Wen N, Li Z, Dube U, Del Aguila J, Budde J, et al. Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl Psychiatry. 2018;8:265.
Wren MC, Zhao J, Liu CC, Murray ME, Atagi Y, Davis MD, et al. Frontotemporal dementia-associated N279K tau mutant disrupts subcellular vesicle trafficking and induces cellular stress in iPSC-derived neural stem cells. Mol Neurodegener. 2015;10:46.
Iovino M, Agathou S, Gonzalez-Rueda A, Del Castillo Velasco-Herrera M, Borroni B, Alberici A, et al. Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 2015;138:3345–59.
Sposito T, Preza E, Mahoney CJ, Seto-Salvia N, Ryan NS, Morris HR, et al. Developmental regulation of tau splicing is disrupted in stem cell-derived neurons from frontotemporal dementia patients with the 10 + 16 splice-site mutation in MAPT. Hum Mol Genet. 2015;24:5260–9.
Ehrlich M, Hallmann AL, Reinhardt P, Arauzo-Bravo MJ, Korr S, Ropke A, et al. Distinct neurodegenerative changes in an induced pluripotent stem cell model of frontotemporal dementia linked to mutant TAU protein. Stem Cell Rep. 2015;5:83–96.
Hallmann AL, Arauzo-Bravo MJ, Mavrommatis L, Ehrlich M, Ropke A, Brockhaus J, et al. Astrocyte pathology in a human neural stem cell model of frontotemporal dementia caused by mutant TAU protein. Sci Rep. 2017;7:42991.
Seo J, Kritskiy O, Watson LA, Barker SJ, Dey D, Raja WK, et al. Inhibition of p25/Cdk5 attenuates Tauopathy in mouse and iPSC models of frontotemporal dementia. J Neurosci. 2017;37:9917–24.
Sohn P, Tracy T, Huang C, Yan R, Camargo C, Mok S, et al. Pathogenic Tau impairs axon initial segment plasticity and excitability homeostasis. Neuron. 2019;104:458–70.e5.
Silva MC, Ferguson FM, Cai Q, Donovan KA, Nandi G, Patnaik D, et al. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife. 2019;8:e45457.
Biswas MHU, Almeida S, Lopez-Gonzalez R, Mao W, Zhang Z, Karydas A, et al. MMP-9 and MMP-2 contribute to neuronal cell death in iPSC Models of frontotemporal dementia with MAPT mutations. Stem Cell Rep. 2016;7:316–24.
Fong H, Wang C, Knoferle J, Walker D, Balestra ME, Tong LM, et al. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Rep. 2013;1:226–34.
Silva MC, Cheng C, Mair W, Almeida S, Fong H, Biswas MHU, et al. Human iPSC-derived neuronal model of Tau-A152T frontotemporal dementia reveals tau-mediated mechanisms of neuronal vulnerability. Stem Cell Rep. 2016;7:325–40.
Kim M, Kim HJ, Koh W, Li L, Heo H, Cho H, et al. Modeling of frontotemporal dementia using iPSC technology. Int J Mol Sci. 2020;21:5319.
Kopach O, Esteras N, Wray S, Abramov AY, Rusakov DA. Genetically engineered MAPT 10+16 mutation causes pathophysiological excitability of human iPSC-derived neurons related to 4R tau-induced dementia. Cell Death Dis. 2021;12:716.
Bowles KR, Silva MC, Whitney K, Bertucci T, Berlind JE, Lai JD, et al. ELAVL4, splicing, and glutamatergic dysfunction precede neuron loss in MAPT mutation cerebral organoids. Cell 2021;184:4547–63 .e17.
Esteras N, Rohrer JD, Hardy J, Wray S, Abramov AY. Mitochondrial hyperpolarization in iPSC-derived neurons from patients of FTDP-17 with 10+16 MAPT mutation leads to oxidative stress and neurodegeneration. Redox Biol. 2017;12:410–22.
Imamura K, Sahara N, Kanaan NM, Tsukita K, Kondo T, Kutoku Y, et al. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci Rep. 2016;6:34904.
Nakamura M, Shiozawa S, Tsuboi D, Amano M, Watanabe H, Maeda S, et al. Pathological progression induced by the frontotemporal Dementia-associated R406W Tau mutation in patient-derived iPSCs. Stem Cell Rep. 2019;13:684–99.
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science 2009;325:473–7.
Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–11.
Budde J, Martinez R, Hsu S, Wen N, Chen J, Coppola G, et al. Precision genome-editing with CRISPR/Cas9 in human induced pluripotent stem cells. 2017. https://doi.org/10.1101/187377.
Sato C, Barthelemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, et al. Tau kinetics in neurons and the human central nervous system. Neuron 2018;97:1284–98.e7.
Yoon SJ, Elahi LS, Pasca AM, Marton RM, Gordon A, Revah O, et al. Reliability of human cortical organoid generation. Nat Methods. 2019;16:75–8.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82.
Ouyang Q, Lizarraga SB, Schmidt M, Yang U, Gong J, Ellisor D, et al. Christianson syndrome protein NHE6 modulates TrkB endosomal signaling required for neuronal circuit development. Neuron 2013;80:97–112.
Griffey M, Bible E, Vogler C, Levy B, Gupta P, Cooper J, et al. Adeno-associated virus 2-mediated gene therapy decreases autofluorescent storage material and increases brain mass in a murine model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. 2004;16:360–9.
Chazotte B. Labeling lysosomes in live cells with LysoTracker. Cold Spring Harb Protoc. 2011;2011:pdb prot5571.
Martini-Stoica H, Cole AL, Swartzlander DB, Chen F, Wan YW, Bajaj L, et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J Exp Med. 2018;215:2355–77.
Piras A, Collin L, Gruninger F, Graff C, Ronnback A. Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration, and progressive supranuclear palsy. Acta Neuropathol Commun. 2016;4:22.
Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21:421–30.
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011;332:1429–33.
Velikkakath AK, Nishimura T, Oita E, Ishihara N, Mizushima N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell. 2012;23:896–909.
Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11:467–80.
Andrejewski N, Punnonen EL, Guhde G, Tanaka Y, Lullmann-Rauch R, Hartmann D, et al. Normal lysosomal morphology and function in LAMP-1-deficient mice. J Biol Chem. 1999;274:12692–701.
Steet R, Lee WS, Kornfeld S. Identification of the minimal lysosomal enzyme recognition domain in cathepsin D. J Biol Chem. 2005;280:33318–23.
Kulkarni VV, Maday S. Neuronal endosomes to lysosomes: A journey to the soma. J Cell Biol. 2018;217:2977–9.
Gowrishankar S, Ferguson SM. Lysosomes relax in the cellular suburbs. J Cell Biol. 2016;212:617–9.
Colacurcio DJ, Nixon RA. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev. 2016;32:75–88.
Marwaha R, Sharma M. DQ-Red BSA trafficking assay in cultured cells to assess cargo delivery to lysosomes. Bio Protoc. 2017;7:e2571.
Schraen-Maschke S, Sergeant N, Dhaenens CM, Bombois S, Deramecourt V, Caillet-Boudin ML, et al. Tau as a biomarker of neurodegenerative diseases. Biomark Med. 2008;2:363–84.
Barthelemy NR, Li Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med. 2020;26:398–407.
Krishnamurthy PK, Johnson GV. Mutant (R406W) human tau is hyperphosphorylated and does not efficiently bind microtubules in a neuronal cortical cell model. J Biol Chem. 2004;279:7893–900.
Caballero B, Bourdenx M, Luengo E, Diaz A, Sohn PD, Chen X, et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat Commun. 2021;12:2238.
Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods. 2015;12:671–8.
Wang Y, Mandelkow E. Degradation of tau protein by autophagy and proteasomal pathways. Biochem Soc Trans. 2012;40:644–52.
Alquezar C, Schoch KM, Geier EG, Ramos EM, Scrivo A, Li KH, et al. TSC1 loss increases risk for tauopathy by inducing tau acetylation and preventing tau clearance via chaperone-mediated autophagy. Sci Adv. 2021;7:eabg3897.
Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–66.
Xu Y, Du S, Marsh JA, Horie K, Sato C, Ballabio A, et al. TFEB regulates lysosomal exocytosis of tau and its loss of function exacerbates tau pathology and spreading. Mol Psychiatry. 2020;26:5925–39.
Kishi-Itakura C, Koyama-Honda I, Itakura E, Mizushima N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J Cell Sci. 2014;127:4089–102.
Tang Z, Takahashi Y, Chen C, Liu Y, He H, Tsotakos N, et al. Atg2A/B deficiency switches cytoprotective autophagy to non-canonical caspase-8 activation and apoptosis. Cell Death Differ. 2017;24:2127–38.
Yap CC, Digilio L, McMahon LP, Garcia ADR, Winckler B. Degradation of dendritic cargos requires Rab7-dependent transport to somatic lysosomes. J Cell Biol. 2018;217:3141–59.
di Domenico A, Carola G, Calatayud C, Pons-Espinal M, Munoz JP, Richaud-Patin Y, et al. Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Rep. 2019;12:213–29.
Ohmi K, Kudo LC, Ryazantsev S, Zhao HZ, Karsten SL, Neufeld EF. Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc Natl Acad Sci USA. 2009;106:8332–7.
Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–34.
Benitez BA, Sands MS. Primary fibroblasts from CSPalpha mutation carriers recapitulate hallmarks of the adult-onset neuronal ceroid lipofuscinosis. Sci Rep. 2017;7:6332.
Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 2010;6:217–27.
Bahr BA. Lysosomal modulatory drugs for a broad strategy against protein accumulation disorders. Curr Alzheimer Res. 2009;6:438–45.
Zunke F, Mazzulli JR. Modeling neuronopathic storage diseases with patient-derived culture systems. Neurobiol Dis. 2019;127:147–62.
Hernandez I, Luna G, Rauch JN, Reis SA, Giroux M, Karch CM, et al. A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci Transl Med. 2019;11:eaat3005.
Zhou X, Paushter DH, Pagan MD, Kim D, Nunez Santos M, Lieberman RL, et al. Progranulin deficiency leads to reduced glucocerebrosidase activity. PLoS One. 2019;14:e0212382.
Valdez C, Wong YC, Schwake M, Bu G, Wszolek ZK, Krainc D. Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum Mol Genet. 2017;26:4861–72.
Jian J, Hettinghouse A, Liu CJ. Progranulin acts as a shared chaperone and regulates multiple lysosomal enzymes. Genes Dis. 2017;4:125–6.
Darios F, Stevanin G. Impairment of Lysosome function and autophagy in rare neurodegenerative diseases. J Mol Biol. 2020;432:2714–34.
Acknowledgements
We thank Carlos Cruchaga, Grant Galasso, Niko Nykanen, and Abhirami Iyer for thoughtful discussions of the manuscript and Dr. Stuart Kornfeld for providing the Cathepsin D antibody. We are grateful to the Neuracell core facility directed by Steven Lotz and Dr. Khadijah Onanuga of the Neural Stem Cell Institute for producing the iPSC-derived organoids. OH is an Archer Foundation Research Scientist. Confocal images were generated on a Zeiss LSM 880 Airyscan Confocal Microscope which was purchased with support from the Office of Research Infrastructure Programs (ORIP), a part of the NIH Office of the Director under grant OD021629. Diagrams were created with Biorender.com.
Funding
Funding provided by the National Institutes of Health (AG005681, AG066444, AG056293, NS110890, AG053303), Hope Center for Neurological Disorders (CMK), Rainwater Charitable Organization (CMK, ST), Chan Zuckerberg Initiative (CMK), Farrell Family Fund for Alzheimer’s Disease (CMK), McDonnell Science Grant for Neuroscience (CMK).
Author information
Authors and Affiliations
Contributions
SM designed experiments performed, analyzed immunostaining and immunoblotting, and wrote the manuscript. RM performed iPSC differentiation into neurons and revised the manuscript. MK performed colocalization analysis of LAMP1 with tau and ptau. OH analyzed data. BAB performed lysosomal enzyme assay, advised in data interpretation, and revised the manuscript. ST provided the organoids and revised the manuscript. KH and CS performed and analyzed IP/MS. CMK designed and supervised the study, performed the computational analyses, wrote the manuscript, and acquired funding. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
ST is president of StemCultures, scientific co-founder of Luxa Biotech, and has served on the scientific advisory boards of Sana Biotechnology and Blue Rock Therapeutics and as a consultant to Merck. KH is an Eisai-sponsored visiting researcher at Washington University and has received salary from Eisai. The remaining authors declare no competing interests.
Ethics approval and consent to participate
The Washington University School of Medicine Institutional Review Board reviewed the protocol of the Knight Alzheimer Disease Research Center (ADRC) Neuropathology Core, from which clinically and neuropathologically well-characterized brain tissues were obtained. As tissue was obtained postmortem it was exempt from IRB approval. Research participants provided autopsy consent limited to removal of the brain. All data were analyzed anonymously. Skin biopsies were performed following written informed consent from the donor. The informed consent was approved by the Washington University School of Medicine Institutional Review Board and Ethics Committee (IRB 201104178 and 201306108).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mahali, S., Martinez, R., King, M. et al. Defective proteostasis in induced pluripotent stem cell models of frontotemporal lobar degeneration. Transl Psychiatry 12, 508 (2022). https://doi.org/10.1038/s41398-022-02274-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-02274-5
This article is cited by
-
Long non-coding RNA SNHG8 drives stress granule formation in tauopathies
Molecular Psychiatry (2023)
-
Patients with sporadic FTLD exhibit similar increases in lysosomal proteins and storage material as patients with FTD due to GRN mutations
Acta Neuropathologica Communications (2023)
