
Abstract
DNA methylation (DNAm), the addition of a methyl group to a cytosine in DNA, plays an important role in the regulation of gene expression. Single-nucleotide polymorphisms (SNPs) associated with schizophrenia (SZ) by genome-wide association studies (GWAS) often influence local DNAm levels. Thus, DNAm alterations, acting through effects on gene expression, represent one potential mechanism by which SZ-associated SNPs confer risk. In this study, we investigated genome-wide DNAm in postmortem superior temporal gyrus from 44 subjects with SZ and 44 non-psychiatric comparison subjects using Illumina Infinium MethylationEPIC BeadChip microarrays, and extracted cell-type-specific methylation signals by applying tensor composition analysis. We identified SZ-associated differential methylation at 242 sites, and 44 regions containing two or more sites (FDR cutoff of q = 0.1) and determined a subset of these were cell-type specific. We found mitotic arrest deficient 1-like 1 (MAD1L1), a gene within an established GWAS risk locus, harbored robust SZ-associated differential methylation. We investigated the potential role of MAD1L1 DNAm in conferring SZ risk by assessing for colocalization among quantitative trait loci for methylation and gene transcripts (mQTLs and tQTLs) in brain tissue and GWAS signal at the locus using multiple-trait-colocalization analysis. We found that mQTLs and tQTLs colocalized with the GWAS signal (posterior probability >0.8). Our findings suggest that alterations in MAD1L1 methylation and transcription may mediate risk for SZ at the MAD1L1-containing locus. Future studies to identify how SZ-associated differential methylation affects MAD1L1 biological function are indicated.
Similar content being viewed by others

Introduction
Schizophrenia (SZ) is a severe neuropsychiatric disorder with complex etiology. Heritability estimates for SZ from twin studies are consistently ~80% [1], thus suggesting a substantial genetic contribution to its etiology. Genome-wide association studies (GWAS) have identified many single-nucleotide polymorphisms (SNPs) associated with SZ, although each SNP has only a small effect on risk for the disorder [2]. A recent large-scale GWAS meta-analysis identified SNPs at 270 distinct genetic risk loci [3]. Heritability estimates from GWAS fall short of those predicted by twin studies, thus suggesting that other forms of genetic variation contribute to risk for SZ. Indeed, recent studies have found a high burden of both rare SNPs and rare copy number variants in individuals diagnosed with SZ [4, 5].
SZ-associated SNPs often alter local DNA methylation (DNAm) [6,7,8]. DNAm, the addition of a methyl group to a cytosine in DNA, stably affects gene expression via interaction with transcription factor binding [9]. DNAm is associated with both increased and decreased gene expression as well as other forms of gene regulation, including splicing and alternative promoter usage [9,10,11]. Changes in DNAm, acting through effects on gene expression, represent one potential mechanism by which SZ-associated SNPs can confer risk.
The superior temporal gyrus (STG) is a region of the brain critical for auditory processing. In individuals with SZ, altered STG function is associated with auditory verbal hallucinations and impaired auditory sensory processing. Impaired auditory processing further contributes to phonologic dyslexia and difficulty recognizing and expressing spoken emotional tone (prosody) in SZ [12].
In this study, we investigated genome-wide SZ-associated differential methylation in the STG. To this end, we used Illumina Infinium MethylationEPIC BeadChip microarrays (EPIC arrays) to measure DNAm at ~850,000 sites across the genome in the STG from 44 subjects with SZ and 44 non-psychiatric comparison (NPC) subjects. We applied tensor composition analysis (TCA) to extract cell-type-specific DNAm signals from brain tissue-level data. These analyses identified several genes that harbored cell-type-specific differences in DNAm between SZ and NPC subjects including mitotic arrest deficient 1-like 1 (MAD1L1), a gene within one of the 270 SZ risk loci identified in the largest GWAS study to date and one of 130 genes thought highly likely to explain the association between GWAS loci and SZ [3]. Our subsequent analyses focused on MAD1L1. To gain insight into the potential role of MAD1L1 DNAm in conferring SZ risk, we identified methylation and transcript quantitative trait loci (mQTLs and tQTLs) for MAD1L1 in postmortem cerebral cortex using publicly available data [7, 13] and performed multiple-trait-colocalization (MOLOC) analysis to assess for statistical colocalization [14], or shared genetic traits, using the methylation and transcription phenotypes and the GWAS signal at the MAD1L1-containing locus.
Materials and methods
Postmortem brains
Tissue was obtained from postmortem brains recovered and processed as described previously [15, 16]. Briefly, brains were retrieved during routine autopsies at the Allegheny County Medical Examiner’s Office, Pittsburgh, PA, USA, following informed consent from next-of-kin. An independent committee of experienced clinicians made consensus Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition diagnoses, or determined the absence thereof, based on clinical records and collateral history obtained via structured interviews with surviving relatives [17]. The right hemisphere was blocked coronally and the resultant slabs snap frozen and stored at −80 °C. Slabs containing the STG were identified and the STG was removed as a single block from each of the slabs in which it was present. Samples containing all six cortical layers of STG (planum temporale), but excluding the adjacent white matter, were harvested. All procedures were approved by the University of Pittsburgh Committee for the Oversight of Research and Clinical Training Involving Decedents and the Institutional Review Board for Biomedical Research.
Cohort membership
The cohort comprised 44 subjects with either SZ (N = 31) or schizoaffective disorder (N = 13), and 44 NPC subjects. Subjects diagnosed with SZ and schizoaffective disorder were grouped together for analysis, and referred to as SZ subjects, or the SZ group. In this study, as in our previous studies, we found that the diagnoses do not differ with respect to DNAm [18]. Each subject in the SZ group was matched with one NPC subject for sex, hemisphere, and as closely as possible for postmortem interval (PMI), age, and other characteristics (Table 1 and Supplementary Table 1).
DNA preparation and bisulfite conversion
DNA (~10 μg) was isolated from STG gray matter (~20 mg) using AllPrep DNA/RNA/Protein Mini Kit (Qiagen, Valencia, CA, USA) and bisulfite was converted using EZ-96 DNA Methylation Kit (Zymo Research, Irvine, CA, USA), both as per the manufacturer’s protocol.
DNA methylation arrays
DNAm is the addition of a methyl group to a cytosine in DNA. DNAm is observed within the context of cytosine-phosphate-guanine dinucleotides (CpGs), most commonly, but also within the context of cytosine-phosphate-H dinucleotides (CpHs, where H = cytosine, adenine, or thymine) [19, 20]. CpGs and CpHs are referred to as “DNAm sites” or “sites” in this manuscript. DNAm was measured at 866,091 sites using MethylationEPIC BeadChip Infinium array (EPIC array; Illumina, San Diego, CA, USA) as per the manufacturer’s protocol [21, 22]. A β-value, the proportion of a particular site that is methylated in a DNA sample, was determined for each site by taking the ratio of the methylated to unmethylated signal, using the formula: β value = intensity of the methylated signal/(intensity of the unmethylated signal + intensity of the methylated signal + 100). A 96-entry EPIC array was filled with samples from the 88 subjects, including replicate samples from eight subjects. Data are available for download from Gene Expression Omnibus (GEO; GSE144910).
Data processing and filtering
Data analyses were performed using the R software environment (www.r-project.org).
Color adjustment and background correction were performed using the bgAdjust2C method [23]. Normalization was performed using the preprocessQuantile function in the R package minfi [24]. The initial dataset comprised data from 1,051,815 probes corresponding to 866,091 DNAm sites for each subject. Multidimensional scaling (MDS) was used to visualize the degree of similarity among samples [25]. Prior to data filtering, samples were segregated strongly by sex in MDS space (Supplementary Fig. 1A). Data filtering involved removing all data points associated with a probe if the probe failed detection as indicated by a median detection p value >0.01 (probes corresponding to 12,350 DNAm sites), cross-reacted with multiple genomic regions (probes corresponding to 39,269 DNAm sites), contained an SNP within its binding site (probes corresponding to 27,395 DNAm sites), or interrogated a DNAm site on a sex chromosome (probes corresponding to 18,628 DNAm sites). Data from probes corresponding to 768,449 DNAm sites remained for downstream analysis (Supplementary Fig. 2). After data filtering, MDS using data from the 3000 most variable sites was performed and samples were no longer segregated by sex (Supplementary Fig. 1B), but segregation by race (Supplementary Fig. 1C) and age (Supplementary Fig. 1D, E) became evident. The replicate sample pairs from each of the eight subjects from which replicate samples were collected and assayed co-segregated in MDS space (Supplementary Fig. 1F), thus demonstrating the reproducibility of our approach. The β-values for each replicate pair were averaged for the downstream analyses.
Differential DNA methylation
Linear regression was used to identify differentially methylated sites (DMSs). DNAm, in the form of preprocessed and normalized β-values, was the dependent variable and diagnosis was the independent variable. Race, age, and PMI were included as covariates in the analysis. The MDS analysis described above supported the inclusion of race as a covariate. Most subjects in this cohort self-identified as either white or black; however, one subject self-identified as Asian Indian and, consistent with known genetic architecture [26], clustered with the subjects of European ancestry (Supplementary Fig. 1C) and was thus combined with the subjects that self-identified as white for analyses. The inclusion of age as a covariate is supported by the MDS analysis as well as existing literature that shows age has a robust effect on DNAm [27,28,29]. Though samples did not segregate by PMI in MDS space (data not shown), it was included as a covariate because the stability of many molecular measures has been found to be particularly sensitive to PMI [30, 31], and to maintain consistency with our previous study in which it was included as a covariate in our primary analyses [18].
Differential methylated regions (DMRs) were identified using the R package DMRcate [32]. DMRcate uses an approach based on tunable kernel smoothing of the differential methylation signal across the genome obtained in the site-based differential DNAm analysis described above. A Benjamini–Hochberg corrected false discovery rate (FDR) <0.1 for the smoothed signal was considered significant. Then regions with a maximum of 1000 base pairs containing at least two such significant sites were defined as DMRs.
Neuron and glia proportion estimates
The proportion of neurons and glia in each sample was estimated with CETS, an R package that uses β values from cell-type-specific sites to generate the estimation [33].
Neuron- and glia-specific differential DNA methylation
The CETS-estimated proportions of neurons and glia for each subject and TCA [34] were used to estimate the subject-level neuron- and glia-specific β values for each DNAm site and detect sites at which DNAm differs between subjects with SZ and NPC subjects. The cell-type proportions were refit in TCA version 1.1.0 and the cell-type-specific differential methylation analysis is done with default TCA settings and adjusted for age, race, and PMI assuming they affect tissue-level DNAm.
Relating DNA methylation and gene transcription to GWAS signal at the MAD1L1-containing SZ risk locus
Fine mapping
GWAS [3] have established signals of association between SNPs in a locus containing MAD1L1. FINEMAP, a software package that evaluates various potential causal variant configurations to produce posterior probabilities of association (PPA) that a given SNP or set of SNPs can account for the GWAS signal [35], was used to localize the GWAS signal to a set of plausible causal SNPs at this locus.
Colocalization analysis
SNPs that associate with methylation levels of cytosine in DNA, or mQTLs, at the MAD1L1 locus were identified using data from Jaffe et al. [7]. Likewise, PsychENCODE data [13] were used to identify SNPs that associate with the abundance of a gene transcript, or tQTLs. To assess for colocalization among GWAS signal, mQTLs, and tQTLs at the MAD1L1 locus, PPA was computed for each SNP with GWAS p value <5 × 10−15 (FINEMAP 0.002 < PPA < 0.253) using multi-trait colocalization (MOLOC, 78). A PPA >0.8 was considered evidence of colocalization (see Supplementary methods for details).
Results
SZ-associated differential DNA methylation was identified at many individual sites and genomic regions, including within MAD1L1
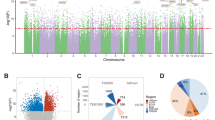
DNAm differed between subjects with SZ and NPC subjects at more sites than would be expected by chance (Fig. 1A). DNAm differed at 242 sites between subjects with SZ and NPC subjects with an FDR cutoff of q = 0.1 (Table 2). Of these 242 DMSs, DNAm differed at 101 sites with an FDR cutoff of q = 0.05. No global differences in DNAm were identified between SZ and NPC subjects (Supplementary Fig. 3). The sites at which DNAm differed between subjects with SZ and NPC subjects were broadly distributed across all autosomes (Fig. 1B).

A Probability plot showing that the analysis for sites at which DNAm differed between SZ and NPC subjects is enriched in small p values compared to what would be expected by chance. The y = x line represents the distribution of p values that would be expected by chance. B Manhattan plot showing that the DNAm differed between subjects with SZ and NPC subjects at many DNAm sites, and the sites were distributed across many autosomes. The horizontal lines represent FDR cutoff of q = 0.1 (bottom) and q = 0.05 (top). DNAm DNA methylation, SZ schizophrenia, NPC non-psychiatric comparison, FDR false discovery rate.
DNAm is known to differ markedly between neurons and glia (36), and detection of DNAm differences between groups in tissue with multiple cell types can be confounded by cell composition. In STG samples studied here, neuronal proportion did not differ between subjects with SZ and NPC subjects (SZ = 0.46 ± 0.05; NPC = 0.46 ± 0.04; p = 0.50) (Supplementary Table 2A), and we have previously shown that pyramidal neuron number in layer 3 of this brain region did not differ between subjects with SZ and NPC subjects (37). After adjusting for neuron proportion, DNAm differed at 256 sites between SZ and NPC subjects with the FDR cutoff of q = 0.1 (Supplementary Table 2B). Of these 256 sites, 210 were among the 242 detected prior to adjusting for neuron proportion thus suggesting that cell composition does not account for the majority of observed differences in DNAm.
Genomic regions in which DNAm at multiple contiguous sites differs between SZ and NPC subjects, or DMRs, may be more biologically meaningful or have different functional consequences than those of a single DMS. There were 44 genomic regions in which DNAm at two or more contiguous, measured sites differed between subjects with SZ and NPC subjects (Table 3).
Notably, three DMSs and one DMR were identified within the mitotic arrest deficient 1-like 1 (MAD1L1) gene. MAD1L1 is one of 130 genes thought likely to explain the association between SNPs at 270 GWAS loci and SZ [3]. The MAD1L1-associated differential methylation we identified was located in exon 6, and the methylation levels were lower in SZ subjects relative to NPC subjects.
SZ-associated differential DNA methylation at some individual sites was specific to neurons or glia
Cell-type deconvolution identified nine DMSs in neurons (Fig. 2A, C) and two DMSs in glia (Fig. 2B, C) with an FDR cutoff of q = 0.1. One of the sites of SZ-associated differential methylation identified within MAD1L1 in bulk tissue analysis was determined to be due to neuron-specific DNAm alterations by cell-type deconvolution. (Fig. 2B, C). All 11 sites for which DNAm differed between SZ and NPC subjects in a cell-type-specific manner were also identified as being differentially methylated in the bulk tissue analysis (Table 2).

A Manhattan plot showing neuron-specific DNAm differences between SZ and NPC subjects at nine sites. B Manhattan plot showing glia-specific DNAm differences between SZ and NPC subjects at two sites. C Box plots of DNAm (β value) at sites of cell-type-specific differences in DNAm between SZ and NPC subjects. DNAm DNA methylation, SZ schizophrenia, NPC non-psychiatric comparison.
Brain methylation and transcript quantitative trait loci for MAD1L1 and schizophrenia GWAS signals are colocalized at the MAD1L1-containing locus
Based on the fine-mapping of the GWAS signal at the MAD1L1-containing locus (Fig. 3A), no single SNP stood out as the causal variant (all had PPA <0.5); moreover, there was no support for more than one causal variant at this locus either (Fig. 3B and Supplementary Table 3A). Many mQTLs and tQTLs fell within this locus (Fig. 3C), with the tQTLs affecting the expression of three transcripts mapping onto two genes (Fig. 3C).

A Negative log (base 10) p values for association at chromosome 7p22.3 from a genome-wide association study (GWAS) of schizophrenia [3]. The purple diamond represents SNP rs12668848 (p = 1.110 × 10−18). Not shown: insertion/deletion variants. B Mirror plot of fine-mapping posterior probability (PPA; upper plot) and SZ associations (from A) at chromosome 7p22.3 (lower plot). In the upper plot, the largest PPA was 0.254 (purple diamond). The remaining points are PPA computed on SZ-GWAS SNPs with association p < 5 × 10−15. The lower plot shows SNPs with SZ association p <5 × 10−12. The green points represent SNPs used for fine mapping because they have SZ association p < 5 × 10−15; SNPs not shown have negligible PPA. C Mirror plot of transcript quantitative trait loci (tQTL; upper plots) and methylation quantitative trait loci (mQTL; lower plots) for MAD1L1 (left) and MRM2 (right). The tQTLs and mQTLs were obtained from PsychENCODE [76] and Jaffe et al. [7], respectively. For MAD1L1 (upper left), 95 and 165, out of a total of 262 tQTL SNPs mapped to transcripts ENST00000437877 (teal points) and ENST00000450235 (red points), respectively. For MRM2 (upper right), 154 and 545, out of a total of 699 tQTL SNPs mapped to transcripts ENST00000467199 (black points) and ENST00000480040 (green points), respectively. In the lower half of each plot, mQTLs are depicted that show 11,368 mQTL SNPs mapping to 280 CpG sites associated with MAD1L1 (yellow points; lower left) and 129 mQTL SNPs mapping to 4 CpG sites associated with MRM2 (blue points; lower right).
Joint analysis of GWAS and tQTL data for three transcripts yielded PPA = 0.95, 0.74, and 0.29 for ENST00000437877 (MAD1L1), ENST00000450235 (MAD1L1), and ENST00000486040 (MRM2), respectively (Supplementary Table 3B). By the convention that PPA >0.8 is sufficient evidence for colocalization, ENST00000437877 (MAD1L1) was colocalized at rs58120505. Joint analysis of GWAS, tQTL, and mQTL data also provided compelling evidence of colocalization for two MAD1L1 transcripts (Supplementary Table 3C); for ENST00000437877, again the greatest evidence accrued at rs58120505. Diagnostics for these colocalizations were imperfect (Supplementary Fig. 4), however; this might be explained by an imperfect match between the etiological effect of the causal genetic variant and the tissue used to produce the tQTL and mQTL resources.
Our results comported with a recent study by Perzel Mandell et al. [36], who used whole-genome bisulfite sequencing to assess DNAm in 183 subjects (344 samples) of human postmortem brain tissue, as well as characterize the genome-wide genetic variation of all subjects. In the Perzel Mandell study, two brain regions were characterized, the hippocampus and the dorsolateral prefrontal cotex. Using the signal from a GWAS study of SZ [35], they selected an index SNP to represent the GWAS signal in each locus (usually the SNP with the smallest p value). They found that these index SNPs were highly likely to be mQTLs. Their index SNP for the GWAS signal around MAD1L1, rs10650434, was no exception; it was a significant mQTL, associating with almost 2,000 CpG sites in the locus regardless of a brain region, although the strongest mQTL signals, as judged by p value, were for CpG sites within a few kilobases (kb) of the SNP itself. Notably, the index SNP, rs10650434, lies within 5 kb of the SNP we colocalized, rs58120505, and alleles of the two SNPs are in almost perfect linkage disequilibrium (r2 = 0.992), according to genotypes from 498 samples of European ancestry reported in the 1000 Genomes Project [37].
Discussion
In the STG of SZ subjects, we identified differences in DNAm levels relative to NPC subjects at 242 individual sites and 44 genomic regions with multiple sites. Notably, we identified SZ-associated differential methylation in MAD1L1, a gene contained within one of the 270 SZ risk loci identified in the largest GWAS study to date and one of 130 genes thought highly likely to explain the association between GWAS loci and SZ [3]. The MAD1L1- associated differential methylation we identified was characterized by lower DNAm in SZ subjects relative to NPC subjects, a difference we determined to be driven by neuron-specific alterations in DNAm. This finding is consistent with studies in the prefrontal cortex that also identified genome-wide significant DMRs in MAD1L1 [7]. Using publicly available data, we identified brain mQTLs and tQTLs for MAD1L1 and found evidence for colocalization with the GWAS signal at the MAD1L1-containing locus.
Our findings, and those of Perzel Mandell et al. [36], implicate MAD1L1 methylation in SZ etiology and/or pathophysiology, and suggest alterations in MAD1L1 methylation and transcription may mediate SZ risk at the MAD1L1-containing locus. Despite pointing to a potential molecular mechanism by which SZ risk SNPs at the MAD1L1-containing locus confer risk, the biological mechanisms affected by MAD1L1-associated SZ risk variants and differential methylation that might be relevant to conferring SZ risk remain unclear. MAD1L1 is expressed in many human tissues [38, 39] and is known to have a role in regulating the spindle assembly checkpoint during mitosis [40]. Genetic mutations that disrupt MAD1L1 expression are associated with aneuploidy and multiple cancers [38, 39]. During development, MAD1L1 is most strongly expressed in differentiating cells and is critical for the transition from proliferation to terminal differentiation in a broad range of cell types [41, 42]. Given MAD1L1 is expressed in both neurons and glia of most brain regions [43,44,45], the differentiation of neurons and glia may be disrupted if MAD1L1 expression is affected by SZ-associated differential methylation during neurodevelopment. Such a disruption would be predicted to alter the delicate balance of the various neuronal and glia subtypes and thus brain circuitry, perhaps giving rise to the dysfunctional brain circuits that are associated with the clinical features of SZ [46].
Alternatively, MAD1L1 may act post-neurodevelopment as its expression in terminally differentiated cells, including post-mitotic neurons and glia, suggests a function in addition to those related to development. Studies have found that its expression in terminally differentiated cells may be necessary for maintaining the differentiated state [47,48,49]. Indeed, even modest decreases in MAD1L1 expression lead to dedifferentiation in some cell types [48]. Some evidence points to a role for dedifferentiation of post-mitotic neurons in the cognitive decline and behavioral changes associated with normal brain aging in humans [50,51,52], and a similar mechanism could conceivably contribute to SZ etiology and/or pathophysiology. That said, these proposed mechanisms are conjecture and critical next steps should focus on understanding MAD1L1 in the brain, generally, and translating MAD1L1-associated SNPs and differential methylation into molecular mechanisms for SZ, specifically.
This study is the first to identify SZ-associated differential methylation in the STG. Others have previously reported DNAm differences between subjects with SZ and NPC subjects in the prefrontal cortex [7, 53,54,55,56], striatum [55], hippocampus [55, 57], and cerebellum [55] thus suggesting that altered DNAm in multiple brain regions contributes to SZ neurobiology. Our findings add to the growing body of literature that implicates altered epigenetic pathways, including DNAm as well as histone modifications [58,59,60], in SZ neurobiology. Though most often studied separately, there is extensive crosstalk between DNAm and histone modification pathways [61,62,63]. This crosstalk drives the establishment of composite epigenetic signatures that depend on epigenetic regulatory enzymes (e.g., DNA methyltransferases, histone methyltransferases, etc.) with protein domains that specifically recognize methylated DNA and/or modified histones and thus allow for linking of DNAm and histone modifications at appropriate sites in the genome. SETD1A, a gene in which loss-of-function mutations confer a large increase in risk for SZ [64], is an example of an enzyme linking DNAm and histone modification. SETD1A methylates histones after it is localized to unmethylated DNA via an interaction with CXXC-finger protein-1 [65]. This body of literature suggests that a more complete understanding of how these epigenetic pathways and their interactions are altered in SZ is likely to be fruitful in identifying molecular mechanisms contributing to SZ. Epigenetic editing technologies that use highly specific DNA-targeting tools (e.g., CRISPR) to methylate DNA or modify histones in a locus-specific manner will be valuable in dissecting these molecular mechanisms in cell culture and animal models [66, 67].
Though our findings for MAD1L1 strongly implicate genetic variation as a causal mechanism for its SZ-associated differential methylation, our findings of other DMSs in this study, like those of all postmortem brain studies, are only correlative and cannot establish causal relationships. The SZ-associated differences in DNAm that we identified in the STG are likely a combination of genetic and environmental factors [68,69,70].
Though differential methylation may be associated with SZ risk factors, it may be the result of exposure to antipsychotics or other confounds. Studies of peripheral tissues indicate that antipsychotics do alter DNAm [71]. However, DNAm alterations are already present in subjects with only brief (<16 weeks) antipsychotic treatment [72], thus suggesting that much SZ-associated differential methylation is intrinsic to the illness. Some studies have even found that SZ-associated DNAm alterations in peripheral tissues are normalized by treatment with antipsychotics [73], raising the possibility that the therapeutic effects of antipsychotics are mediated, in part, by DNAm changes. Such findings also make it likely that antipsychotics mask some SZ-associated differential methylation from being detected in studies.
An additional potential confound particularly relevant in studies of DNAm in subjects with SZ is cigarette smoking. Cigarette smoking is much more common among individuals with SZ than the general population and is known to induce robust DNAm changes in peripheral tissues [74]. Cigarette smoking does affect DNAm in the brain, however, none of the DMSs or regions identified in this study have been found to be among the sites most strongly affected by cigarette smoking [75].
This study lays the groundwork for more detailed investigations of SZ-associated differential methylation in the STG. Future studies should focus on identifying the biological mechanisms by which altered DNAm, especially within MAD1L1, contributes to SZ etiology and pathophysiology. To this end, studies that use epigenetic editing technology to recapitulate SZ-associated differential methylation in cell cultures and animal models will be useful.
References
Hilker R, Helenius D, Fagerlund B, Skytthe A, Christensen K, Werge TM, et al. Heritability of schizophrenia and schizophrenia spectrum based on the nationwide Danish Twin Register. Biol Psychiatry. 2018;83:492–8. https://doi.org/10.1016/j.biopsych.2017.08.017.
Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Pardiñas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet. 2018;50:381–9. https://doi.org/10.1038/s41588-018-0059-2.
Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landén M, et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci. 2016;19:1433–41. https://doi.org/10.1038/nn.4402.
Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49:27–35. https://doi.org/10.1038/ng.3725.
Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM, et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci. 2016;19:48–54.
Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 2016;19:40–47.
Boks MP, Houtepen LC, Xu Z, He Y, Ursini G, Maihofer AX, et al. Genetic vulnerability to DUSP22 promoter hypermethylation is involved in the relation between in utero famine exposure and schizophrenia. NPJ Schizophr. 2018;4:16. https://doi.org/10.1038/s41537-018-0058-4.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92.
Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, Dsouza C, Fouse SD, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–7. https://doi.org/10.1038/nature09165.
Wagner JR, Busche S, Ge B, Kwan T, Pastinen T, Blanchette M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014;15:R37. https://doi.org/10.1186/gb-2014-15-2-r37.
Javitt DC, Sweet RA. Auditory dysfunction in schizophrenia: integrating clinical and basic features. Nat Rev Neurosci. 2015;16:535–50.
Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, et al. Comprehensive functional genomic resource and integrative model for the human brain. Science. 2018;362:eaat8464. https://doi.org/10.1126/science.aat8464.
Giambartolomei C, Liu JZ, Zhang W, Hauberg M, Shi H, Boocock J, et al. A Bayesian framework for multiple trait colocalization from summary association statistics. Bioinformatics. 2018;34:2538–45. https://doi.org/10.1093/bioinformatics/bty147.
Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73.
Sweet RA, Henteleff RA, Zhang W, Sampson AR, Lewis DA. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology. 2009;34:374–89.
American Psychiatric Association. DSM-IV: Diagnostic and Statistical Manual of Mental Health Disorders. 4th ed. Arlington, VA: American Psychiatric Association; 1994.
McKinney B, Ding Y, Lewis DA, Sweet RA. DNA methylation as a putative mechanism for reduced dendritic spine density in the superior temporal gyrus of subjects with schizophrenia. Transl Psychiatry. 2017;7:e1032–e1032.
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38.
Nestler EJ, Peña CJ, Kundakovic M, Mitchell A, Akbarian S. Epigenetic basis of mental illness. Neuroscientist. 2016;22:447–63.
Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016;17:208–208.
Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 2016;8:389–99.
Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–8.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinforma (Oxf, Engl). 2014;30:1363–9.
Cox TF, Cox MAA. Multidimensional scaling. Boca Raton, FL: Chapman & Hall/CRC; 2001.
Xing J, Watkins WS, Shlien A, Walker E, Huff CD, Witherspoon DJ, et al. Toward a more uniform sampling of human genetic diversity: a survey of worldwide populations by high-density genotyping. Genomics. 2010;96:199–210.
Horvath S. Erratum to: DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115.
Akbarian S, Beeri MS, Haroutunian V. Epigenetic determinants of healthy and diseased brain aging and cognition. JAMA Neurol. 2013;70:711–8.
McKinney BC, Lin C-W, Rahman T, Oh H, Lewis DA, Tseng G, et al. DNA methylation in the human frontal cortex reveals a putative mechanism for age-by-disease interactions. Transl Psychiatry. 2019;9:39–39.
Lewis DA. The human brain revisited: opportunities and challenges in postmortem studies of psychiatric disorders. Neuropsychopharmacology. 2002;26:143–54.
McCullumsmith RE, Hammond JH, Shan D, Meador-Woodruff JH. Postmortem brain: an underutilized substrate for studying severe mental illness. Neuropsychopharmacology. 2014;39:65–87.
Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, Lord RV, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6. https://doi.org/10.1186/1756-8935-8-6.
Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013;8:290–302.
Rahmani E, Schweiger R, Rhead B, Criswell LA, Barcellos LF, Eskin E, et al. Cell-type-specific resolution epigenetics without the need for cell sorting or single-cell biology. Nat Commun. 2019;10:3417. https://doi.org/10.1038/s41467-019-11052-9.
Benner C, Spencer CCA, Havulinna AS, Salomaa V, Ripatti S, Pirinen M. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 2016;32:1493–501. https://doi.org/10.1093/bioinformatics/btw018.
Perzel Mandell KA, Eagles NJ, Wilton R, Price AJ, Semick SA, Collado-Torres L, et al. Genome-wide sequencing-based identification of methylation quantitative trait loci and their role in schizophrenia risk. Nat Commun. 2021;12:5251.
Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Tsukasaki K, Miller CW, Greenspun E, Eshaghian S, Kawabata H, Fujimoto T, et al. Mutations in the mitotic check point gene, MAD1L1, in human cancers. Oncogene. 2001;20:3301–5. https://doi.org/10.1038/sj.onc.1204421.
Sun Q, Zhang X, Liu T, Liu X, Geng J, He X, et al. Increased expression of mitotic arrest deficient-like 1 (MAD1L1) is associated with poor prognosis and insensitive to taxol treatment in breast cancer. Breast Cancer Res Treat. 2013;140:323–30. https://doi.org/10.1007/s10549-013-2633-8.
Amon A. The spindle checkpoint. Curr Opin Genet Dev. 1999;9:69–75. https://doi.org/10.1016/S0959-437X(99)80010-0.
Hurlin PJ, Foley KP, Ayer DE, Eisenman RN, Hanahan D, Arbeit JM. Regulation of Myc and Mad during epidermal differentiation and HPV-associated tumorigenesis. Oncogene. 1995;11:2487–501.
Ohta Y, Hamada Y, Saitoh N, Katsuoka K. Effect of the transcriptional repressor Mad1 on proliferation of human melanoma cells. Exp Dermatol. 2002;11:439–47. https://doi.org/10.1034/j.1600-0625.2002.110507.x.
Cichon S, Mühleisen TW, Degenhardt FA, Mattheisen M, Miró X, Strohmaier J, et al. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am J Hum Genet. 2011;88:372–81. https://doi.org/10.1016/j.ajhg.2011.01.017.
Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–9. https://doi.org/10.1038/nature11405.
GTEx Analysis Release V6p (dbGaP Accession phs000424.v6.p1). 2017.
Lewis DA, Sweet RA. Schizophrenia from a neural circuitry perspective: advancing toward rational pharmacological therapies. J Clin Invest. 2009;119:706–16.
Foley KP, Eisenman RN. Two MAD tails: what the recent knockouts of Mad1 and Mxi1 tell us about the MYC/MAX/MAD network. Biochim Biophys Acta. 1999;1423:M37–47.
Han S, Park K, Kim HY, Lee MS, Kim HJ, Kim YD, et al. Clinical implication of altered expression of Mad1 protein in human breast carcinoma. 2000;88:1623–32. https://doi.org/10.1002/(SICI)1097-0142(20000401)88:7<1623::AID-CNCR17>3.0.CO;2-W.
Pan L, Zhang X, Suo X, Wang F, Niu Z, Dong Z. Expression and mutation analysis of the Myc antagonist gene Mad1 in acute leukemia. Blood. 2006;108:4331. https://doi.org/10.1182/blood.v108.11.4331.4331.
Park DC, Polk TA, Park R, Minear M, Savage A, Smith MR. Aging reduces neural specialization in ventral visual cortex. Proc Natl Acad Sci USA. 2004;101:13091–5. https://doi.org/10.1073/pnas.0405148101.
Oh G, Ebrahimi S, Wang SC, Cortese R, Kaminsky ZA, Gottesman II. et al. Epigenetic assimilation in the aging human brain. Genome Biol. 2016;17:76–76.
Koen JD, Rugg MD. Neural dedifferentiation in the aging brain. Trends Cogn Sci. 2019;23:547–59.
Wockner LF, Noble EP, Lawford BR, Young RM, Morris CP, Whitehall VL, et al. Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl Psychiatry. 2014;4:e339.
Numata S, Ye T, Herman M, Lipska BK. DNA methylation changes in the postmortem dorsolateral prefrontal cortex of patients with schizophrenia. Front Genet. 2014;5:280.
Viana J, Hannon E, Dempster E, Pidsley R, Macdonald R, Knox O, et al. Schizophrenia-associated methylomic variation: molecular signatures of disease and polygenic risk burden across multiple brain regions. Hum Mol Genet. 2017;26:210–25.
Pai S, Li P, Killinger B, Marshall L, Jia P, Liao J, et al. Differential methylation of enhancer at IGF2 is associated with abnormal dopamine synthesis in major psychosis. Nat Commun. 2019;10:2046. https://doi.org/10.1038/s41467-019-09786-7.
Ruzicka WB, Subburaju S, Benes FM. Circuit- and diagnosis-specific DNA methylation changes at gamma-aminobutyric acid-related genes in postmortem human hippocampus in schizophrenia and bipolar disorder. JAMA Psychiatry. 2015;72:541–51.
O’dushlaine C, Rossin L, Lee PH, Duncan L, Parikshak NN, Newhouse S, et al. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015;18:199–209. https://doi.org/10.1038/nn.3922.
Chase KA, Rosen C, Rubin LH, Feiner B, Bodapati AS, Gin H, et al. Evidence of a sex-dependent restrictive epigenome in schizophrenia. J Psychiatr Res. 2015;65:87–94. https://doi.org/10.1016/j.jpsychires.2015.04.005.
Fullard JF, Halene TB, Giambartolomei C, Haroutunian V, Akbarian S, Roussos P. Understanding the genetic liability to schizophrenia through the neuroepigenome. Schizophr Res. 2016;177:115–24.
Ooi SKT, Qiu C, Bernstein E, Li K, Jia D, Yang Z, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. 2007;448:714–7. https://doi.org/10.1038/nature05987.
Hashimoto H, Vertino PM, Cheng X. Molecular coupling of DNA methylation and histone methylation. Epigenomics. 2010;2:657–69.
Du J, Johnson LM, Jacobsen SE, Patel DJ. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015;16:519–32. https://doi.org/10.1038/nrm4043.
Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci. 2016;19:571–7. https://doi.org/10.1038/nn.4267.
Tate CM, Lee JH, Skalnik DG. CXXC finger protein 1 restricts the Setd1A histone H3K4 methyltransferase complex to euchromatin. FEBS J. 2010;277:210–23. https://doi.org/10.1111/j.1742-4658.2009.07475.x.
Stricker SH, Köferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet. 2017;18:51–66.
Holtzman L, Gersbach CA. Editing the epigenome: reshaping the genomic landscape. Annu Rev Genomics Hum Genet. 2018;19:43–71. https://doi.org/10.1146/annurev-genom-083117-021632.
Van Os J, Rutten BPF, Poulton R. Gene-environment interactions in schizophrenia: review of epidemiological findings and future directions. Schizophr Bull. 2008;34:1066–82.
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. 2008;105:17046–9. https://doi.org/10.1073/pnas.0806560105.
Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5:5592. https://doi.org/10.1038/ncomms6592.
Castellani CA, Melka MG, Diehl EJ, Laufer BI, O'Reilly RL, Singh SM. DNA methylation in psychosis: insights into etiology and treatment. Epigenomics. 2015;7:67–74.
Nishioka M, Bundo M, Koike S, Takizawa R, Kakiuchi C, Araki T, et al. Comprehensive DNA methylation analysis of peripheral blood cells derived from patients with first-episode schizophrenia. J Hum Genet. 2013;58:91–97.
Abdolmaleky HM, Pajouhanfar S, Faghankhani M, Joghataei MT, Mostafavi A, Thiagalingam S. Antipsychotic drugs attenuate aberrant DNA methylation of DTNBP1 (dysbindin) promoter in saliva and post-mortem brain of patients with schizophrenia and Psychotic bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2015;168:687–96. https://doi.org/10.1002/ajmg.b.32361.
Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9:436–47.
Markunas CA, Semick SA, Quach BC, Tao R, Deep-Soboslay A, Carnes MU, et al. Genome-wide DNA methylation differences in nucleus accumbens of smokers vs. nonsmokers. Neuropsychopharmacology. 2021;46:554–60.
Psych EC, Akbarian S, Liu C, Knowles JA, Vaccarino FM, Farnham PJ, et al. The PsychENCODE project. Nat Neurosci. 2015;18:1707–12.
Funding
This work was supported by NIH Grants K23 MH112798 (BCM), R37 MH057881 (BD), and R01 MH071533, MH116046, and AG027224 (RAS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, or the United States Government.
Author information
Authors and Affiliations
Contributions
BCM, LLM, BD, and RAS designed the study; BCM and CMH performed the experiments; LLM, YW, LK, BD, JW, and YD analyzed the results; all authors contributed to interpretation of the results; BCM wrote the manuscript with significant contributions from LLM, BD, and RAS; all authors provided feedback on drafts of the manuscript and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
DAL receives investigator-initiated research support from Pfizer and Merck, and serves as a paid consultant on basic science research for Astellas. No other authors report any conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
McKinney, B.C., McClain, L.L., Hensler, C.M. et al. Schizophrenia-associated differential DNA methylation in brain is distributed across the genome and annotated to MAD1L1, a locus at which DNA methylation and transcription phenotypes share genetic variation with schizophrenia risk. Transl Psychiatry 12, 340 (2022). https://doi.org/10.1038/s41398-022-02071-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-02071-0
This article is cited by
-
Genome-wide DNA methylation and transcriptome analyses reveal the key gene for wool type variation in sheep
Journal of Animal Science and Biotechnology (2023)
