
Abstract
The extracellular matrix (ECM) is one of the major components of tumors that plays multiple crucial roles, including mechanical support, modulation of the microenvironment, and a source of signaling molecules. The quantity and cross-linking status of ECM components are major factors determining tissue stiffness. During tumorigenesis, the interplay between cancer cells and the tumor microenvironment (TME) often results in the stiffness of the ECM, leading to aberrant mechanotransduction and further malignant transformation. Therefore, a comprehensive understanding of ECM dysregulation in the TME would contribute to the discovery of promising therapeutic targets for cancer treatment. Herein, we summarized the knowledge concerning the following: (1) major ECM constituents and their functions in both normal and malignant conditions; (2) the interplay between cancer cells and the ECM in the TME; (3) key receptors for mechanotransduction and their alteration during carcinogenesis; and (4) the current therapeutic strategies targeting aberrant ECM for cancer treatment.
Similar content being viewed by others
Introduction
Cancer is a leading cause of death which severely impedes the health career for extension of life expectancy in the world. The incidence and mortality of cancer are increasing year by year. According to the latest global cancer statistics in 2020, 19.3 million new cases were diagnosed and cancer contributed to 10.0 million deaths1. Therefore, cancer is becoming one of the most serious problems which threaten public health. The most striking attributes of cancer are uncontrolled proliferation, local invasion, and distant metastasis. Nowadays, the mainstream therapies for cancer treatment include surgery, chemotherapy, radiotherapy, targeted therapy, and immunotherapy. Cancer-related death is mainly caused by tumor recurrence and distant metastasis after systemic antitumor treatment. Although great advances have been achieved for cancer treatment in recent years, especially in the field of targeted therapy and immune therapy, the pursuit for converting this life-threatening disease into a manageable chronic condition has never stopped. The comprehensive understanding of cancer cells, as well as the microenvironment supporting the malignant behavior of cancer cells, are of equal importance for developing novel therapeutics against cancer.
The extracellular matrix (ECM), which comprises the interstitial elements within tissues or organs for all metazoan organisms, plays vital roles for all biological processes by providing architectural support, anchorage for cell adhesion, a reservoir for water, and various growth factors, as well as inductions for intracellular signaling pathways. According to a comprehensive study utilizing both proteomic analyses of the in vivo ECM composition and in silico prediction2, 278 genes were identified as core elements of the “matrisome” for humans, accounting for 1% of the entire proteome.
Ever since the identification and characterization of collagen, the most abundant component of ECM, in the 1930s, the complicated network of ECM started to be gradually revealed through modern biochemistry methods3. Most of the proteins in the ECM can be classified into two groups, fibrous proteins, and glycosaminoglycan. The former include collagen, fibronectin, elastin, and laminin, and the latter mainly consists of hyaluronic acid, chondroitin sulfate, keratan sulfate, and heparan sulfate. These molecules are crosslinked and distributed heavily in the ECM, forming the mesh structure for tissues. As early as the 1970s, the critical roles of ECM in the determination of cell morphology and responses to growth factors had been proven with solid empirical evidence4. Then the pursuit of discovering the intermembrane signal transducers linking ECM and intercellular signaling pathways produced many great works marked by the identification of integrins in 1980s5,6,7,8,9. Ever since then, the field of ECM–cell interaction developed rapidly, and the vast signaling network bridging extracellular environment and complicated cell behaviors started to reveal itself gradually due to the continuous efforts of researchers and technological advances. During the 2010s, the clinical application of Ibrutinib (a small molecular compound inhibiting integrin signaling) for the treatment of lymphoid leukemia and lymphoma was a hallmark event for the successful translation of biological knowledge to practical medicines in this research area10,11. At the same time, bioengineering of artificial and natural ECM materials also achieved great success in multiple branches of medicine, such as osteology, odontology, dermatology, and ophthalmology. For example, an artificial dermal regeneration template has been invented for the treatment of aplasia cutis congenital, a severer disorder characterized by the congenital absence of skin12.
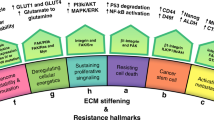
As one of the major components of the tumor microenvironment (TME), the dysregulation of ECM is a remarkable feature of cancer (Fig. 1). During the development of cancer, malignant cells contribute to ECM stiffness, and, in return, the stiffened ECM alters the characteristics of cancer cells. The communication between cancer cells and the ECM activates several vital pathways related to mechanotransduction. Therefore, a comprehensive understanding of the dysregulation of the ECM in the TME would contribute to the discovery of promising therapeutic targets for cancer treatment. In the present review, the structures and functions of multiple ECM components, such as collagen, fibronectin, elastin, and so on, were introduced. Then we summarized their alterations and the underlying mechanisms during matrix stiffness in cancer. Meanwhile, the downstream biological effects of matrix stiffness on both cancer cells and other cells in TME were also discussed. Subsequently, several pivotal receptors for ECM and their roles in malignant transformation were summarized. Afterward, both clinical and preclinical therapeutic applications of ECM-related signaling for cancer treatment were discussed in-depth based on our current knowledge from basic researches and clinical studies. Finally, the vision and several potential Gordian Knots for targeting ECM-related signaling for cancer treatment were summarized and discussed to call for more attention to this research field.

Schematic illustration of ECM components in normal tissue (left) and the TME (right). Matrix stiffness is mainly related to excessive collagen and HA within TME. Both cancer cells and fibroblasts contribute to the remodeling of the ECM during its stiffness, fundamentally influencing many critical biological processes during the development of cancer
Major ECM components: structure and function
Collagen
Collagen makes up most of the ECM, accounting for approximately 90% of the ECM and 30% of the total protein in humans13. Currently, 28 types of collagens have been identified, encoded by 43 genes14. All collagens are homotrimers or heterotrimers of three polypeptide chains (α chains), comprising numerous Gly-X-Y repeats, X and Y being frequently proline and 4-hydroxyproline, respectively15. Glycine provides conformational flexibility, while proline provides conformational rigidity. Therefore, the rod-shaped triple helix is stabilized by interchain hydrogen bonds and electrostatic interactions13. α Chains vary greatly in size (ranging from 662 up to 3152 amino acids for the human α1 (collagen type X) and α3 (collagen type VI) chains, respectively) as well as the frequencies of imperfections and interruptions of Gly-X-Y repeats, resulting in differences in plasticity, flexibility and recognition patterns among various types of collagens13.
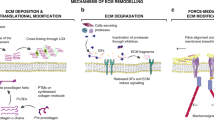
A more intensive collagen mesh is constructed based on the posttranscriptional product for collagen maturation. Collagen forms macromolecules by intermolecular cross-linking16. The cross-linked collagen provides tissue intensity and tenacity. For example, the mechanical properties of fibril-forming collagen are largely dependent on the extent of covalent cross-links within and between triple helixes, including disulfide bonds, the Nε(γ-glutamyl)lysine isopeptide, reducible and mature cross-links produced via the lysyl oxidase pathway, advanced glycation end products, hydroxylysine-methionine cross-links, and arginyl ketoimine adducts called arginoline17. Moreover, the existence of noncollagenous domains in collagens that can assemble mutually between collagens or even between collagens and other ECM proteins increases the complexity of such supramolecules18. Thus, collagens can form fibrils, beaded filaments, anchoring fibrils, and even networks19.
Collagens are long-lived proteins due to their high glycation level, but their degradation is still critical for both normal turnover and pathological destruction of connective tissue20. Matrix metalloproteinases (MMPs) participate in the physiological and pathological degradation of collagens. These zinc-dependent endopeptidases comprise a large family of 28 members21,22. For example, MMP-1, MMP-8, MMP-13, and MMP-14 cleave fibril-forming collagens I, II, and III, while MMP-2 and MMP-9 cleave denatured collagens and collagen IV14. Sheddases are another family of enzymes that digest collagens14,23. During the digestion of MMPs, signaling molecules such as endostatin and tumstatin are released from collagens, simultaneously leading to the alteration of mechanical characteristics as well as signaling transduction in the microenvironment24,25. As the major component of the ECM, the amount and posttranslational modifications of collagens often undergo tremendous alterations during the development of cancer, resulting in a fundamental influence on the behavior of cancer cells and other cells in the TME, features that would be discussed later in this review.
Fibronectin
Fibronectin is low in abundance but has diverse functions in the ECM. Soluble fibronectin is secreted by hepatocytes and into the circulation26,27, while the insoluble form is produced by a variety of mesenchymal cells, including fibroblast28 and endothelial cells. In the vasculature, vascular smooth muscle cells are a major producer of fibronectin29.
Fibronectin is a type of dimer proteoglycan, which is interwoven with two subunits via a disulfide bond at the C-terminus30,31. A fibronectin subunit weighs approximately between 220 and 250 kD32. There are several structural domains in both subunits, and the structural domains mainly consist of three structural modules, 12 repeat fibronectin type I, 2 repeat fibronectin type II, and 15–17 repeat fibronectin type III33,34,35. These structural domains constitute the functional domains of fibronectin, including a domain that weighs 70 kD at the N-terminus (fibronectin type I1–9), a central binding domain (CBD) that weighs 120 kD (fibronectin type III1–12), and a heparin-binding domain (HepII) (fibronectin type III12–14).
Fibronectin has profound effects on cell adhesion, migration36, proliferation37,38, blood coagulation39,40, vascularization41,42,43, clearance of bacteria by phagocytes44, and wound healing45,46, among others. In the ECM, fibronectin connects various structural proteins to form an integrated matrix, such as collagens47, fibrillin48, and tenascin-C49,50. For example, the antibody targeting the collagen-binding site in fibronectin could suppress the fibrillogenesis of collagen, suggesting that type I collagen cannot assemble without fibronectin51.
Other than binding to multiple structural proteins to reinforce the ECM, fibronectin directly interacts with many other proteins to exert regulatory functions52,53,54. First, fibronectin contains abundant arginine–glycine–asparagine (RGD) sequences that can recognize and bind to integrins on the cell membrane55. Therefore, fibronectin has a profound effect on intracellular signaling transduction by inducing integrin attachment. For example, the interaction of MMP-9-degraded fibronectin and integrin αvβ6 leads to aggressive migration and invasion via ERK1/2 and PI3K/AKT/Smad-1/5/8 pathways in breast cancer56. By contrast, many growth factors can directly interact with fibronectin. For example, insulin-like growth factor (IGF), fibroblast growth factor (FGF), transforming growth factor-beta (TGF-β), hepatocyte growth factor (HGF), and platelet-derived growth factor (PDGF) can interact with the fibronectin domain57,58,59,60. FGF, vascular endothelial growth factor (VEGF), and PDGF can bind to the heparin II domain in fibronectin58, and PDGF can attach to the fibronectin first type III repeat (FNIII1)57. Moreover, extra domain A (EDA) in fibronectin can increase VEGF-C expression in colorectal carcinoma61. Therefore, although fibronectin is low in abundance in the ECM, it plays a vital role during malignant transformation.
Elastin and laminin
Elastin is the primary component of elastic fibers and is mainly found in ligaments and vascular walls. Elastin maintains the tenacity and intensity of tissues along with collagen by rebelling against tissue deformation or rupture. Compared with collagen, elastin is highly resilient because of its amino constituents and dynamic three-dimensional (3D) structure. Glycine makes up one-third of the polypeptide, and proline accounts for approximately 10%, while hydroxyproline accounts for less than 1%. The β-turn in the polypeptide chain is produced based on the interaction of Gly4 (N–H) and Gly1 (C = O) or Leu5 (N–H) and Val2 (C = O), resulting in the resilience of elastin.
Laminin, together with collagen, makes up the constituents of the basement membrane. Therefore, laminin is involved in vascularization, especially in the process of vessel maturation62. During re-epithelialization in wound healing, laminin is upregulated to provide an interface for the adherence of epithelial cells to adhere and stretch63. Laminin is polymerized by three different chains, one α chain, one β chain, and one γ chain, which are encoded by separate genes64,65. Five forms of α chains (LAMA1-5) and three forms of β chains (LAMB1-3) and γ chains (LAMC1-3) can be found in laminins66. For example, laminin comprising α2β2γ1 is named laminin-221. The laminin network is constructed along with collagen type IV, fibronectin, and perlecan in basement membranes63,67.
Hyaluronic acid
Hyaluronic acid (HA), another primary component in ECM, is a high-molecular-weight glycosaminoglycan comprising disaccharide repeats of N-acetylglucosamine and glucuronic acid68. HA is synthesized by the alternative addition of glucuronic acid and N-acetylglucosamine to the growing chain, using their activated nucleotide sugars (uridine diphosphate glucose (UDP) and UDP-N-acetylglucosamine) as substrates. This reaction is mediated by hyaluronan synthase 1, 2, and 3 (HAS1–HAS3) localized on the cell membrane. The enzymatic degradation of HA is mediated by hyaluronidase, b-d-glucuronidase, and β-N-acetyl-hexosaminidase69. The number of repeated disaccharides in a completed HA molecule can reach 10,000 or even more. Its long polymer chains form random coils entangled in solution, and its numerous hydroxyls capture a huge quantity of water by forming hydrogen bonds70. Therefore, HA mechanically increases the elastoviscosity in the ECM.
In addition to its unique viscoelastic nature, HA functions as an important “reservoir” for water, buffering ion exchange, water, and osmotic balance within the ECM. Moreover, some substances and biomacromolecules are selectively permeable to HA due to their charged surface and selective domains. Therefore, HA can serve as a sieve: particles with a huge molecular size are hindered and immobilized, while smaller molecules tend to pass through HA more efficiently71. Additionally, HA can be recognized by various types of cells through membrane receptors such as CD44 and receptor for hyaluronan-mediated motility (RHAMM), as well as intracellular signaling transducers such as cell division cycle 37 (CDC37)72, P-3273, and hyaluronan binding protein 4 (HABP4)74. Such recognition plays vital role in many biological procedures, including cell mobility, invasion, proliferation, and inflammation.
Chondroitin sulfate, keratan sulfate, and heparan sulfate
Chondroitin sulfate, keratan sulfate, and heparan sulfate are heteropolysaccharides that also belong to the class of glycosaminoglycans75. Their molecular structures are similar to that of HA and comprise repeated disaccharides. The difference among HA, chondroitin sulfate, keratan sulfate, and heparan sulfate lies in the carbohydrate of the monomer and sulfate ester position. The carbohydrates that comprise chondroitin sulfate are [→4GlcAb1→3GalNAcb1→], and the sulfated site could be position 4 (CS A, chondroitin-4-sulfate) or position 6 (CS C, chondroitin-6-sulfate) of the repeating unit76,77. Keratan sulfate comprises [→3Galb1→4GlcNAcb1→], and sulfate esters can be found at C-6 in one carbohydrate or both monosaccharides with a hydroxyl group78. Heparan sulfate comprises [→4GlcAb1→4GlcNAca1→]. O-sulfation modification of heparan sulfate mainly occurs at C-2 of iduronic acid (IdoA) and C-6 of glucosamine, and sometimes at C-2 of GlcA and C-3 of glucosamine79. Various sulfate group sites lead to the heterogeneity of the glycosaminoglycan structures and functions75.
These three types of glycosaminoglycans described above have the following characteristics. First, glycosaminoglycans bind to proteins by covalent bonds80. For example, chondroitin sulfate can bind to matrix proteins, growth factors, cytokines, chemokines, and protease inhibitors81,82,83. Second, the sulfate substituent on the carbohydrate can mediate the coupling between glycosaminoglycans and metal ions, thus preventing the formation of peroxide catalyzed by metal ions. Finally, the hydrogen bond is formed when glycosaminoglycans interact with water, leading to the mechanical effect of viscoelasticity.
Fibroblasts
Stromal cells, including fibroblasts and pericytes, are the major source of ECM84. Fibroblasts are widely distributed in most connective tissues such as the bone marrow, lymph nodes, ovaries, and solid tumors85. Pericytes are specifically located surrounding the endothelial cells on the interior surface of blood vessels86,87.
Fibroblasts are one of the major cell types within the TME in terms of both number and function. Some studies have shown that fibroblasts account for 70 to 90% of the whole tumor volume of breast cancer and pancreatic cancer88,89,90. More importantly, fibroblasts play a central role in the formation and turnover of ECM for two reasons. First, fibroblasts directly produce structural macromolecules, such as collagen, fibronectin, and laminin91. Second, enzymes involved in the modification and degradation of these structural macromolecules are also secreted by fibroblasts such as lysyl hydroxylases and metalloproteinases92,93.
Fibroblasts are regulated by many signals, including cytokines, chemicals, and environmental signals, such as heat and mechanical forces, thus contributing to ECM remodeling. For example, TGF-β can enhance the production of both collagen and fibronectin, as well as procollagen lysyl hydroxylase 2 (LOX2), while tumor necrosis factor-alpha (TNF-α) can inhibit collagen synthesis in fibroblasts92,94. Moreover, TNF-α and interleukin (IL)-1 can induce the production of MMP-1, -3, and -9 by fibroblasts, leading to the degradation of collagen in the TME94. Interestingly, primary human dermal fibroblasts proliferate faster and produce more collagen on amine-rich (NH3) surfaces when cultured in vitro compared with surfaces coated with carboxyl acid (COOH) and hydrocarbon (CH3)95. Similarly, the collagen levels of human patellar tendon fibroblasts, cardiac fibroblasts, and periodontal ligament fibroblasts are all enhanced by repeated mechanical stretching96,97,98. Moreover, repeated mild heat shocks have been shown to increase dermal fibroblast activity and collagen production99. These lines of evidence suggest that fibroblasts are highly flexible cells that convert signals from multiple sources into changes in ECM components.
The ECM is an orchestration of many components, including but not limited to matrix proteins, glycosaminoglycans, growth factors, enzymes, and fibroblasts, whose balance is critical to maintaining tissue homeostasis. For cancer, which is a complicated disease involving active interaction between cells and their microenvironment, ECM stiffness is a distinctive feature and a promising therapeutic target (Fig. 1).
Matrix stiffness in cancer: phenomena, mechanisms, and biological effects
Alterations of tissue stiffness in cancer
Stiffness is defined as the extent of deformation when the external force is applied to an object or material100,101. For most tissues without bones, their stiffness is largely dependent on the quantity and components of the ECM. Tumors frequently exhibit higher stiffness than normal tissues. For example, an elastography study of breast cancer demonstrated that the stiffness of tumor tissue is higher than that of normal tissue102. In another study, in vivo shear-wave elastography analysis of 337 breast cancer patients also revealed that tissue stiffness values are positively correlated with malignant phenotypes, including larger tumor sizes, higher histologic grades, and estrogen receptor (ER) status, with triple-negative breast cancer tissues ranking stiffest103. Similarly, a study of 373 patients with focal liver lesions showed that the mean stiffness values of hepatocellular carcinoma, intrahepatic cholangiocarcinoma, and metastasis were 34 (range: 4.4–188), 25 (range: 5.5–79), and 30 (range: 4.7–64), respectively, which are significantly higher than those of hemangioma (9.3, range: 3.1–41), focal nodular hyperplasia (10, range: 2.9–26) and cirrhotic nodules (11, range: 4.4–49)104. In addition to breast cancer and liver cancer, pancreatic tumors are also stiffer than normal pancreatic tissue105,106,107.
However, although tumors are macroscopically stiffer than normal tissues, Plodinec et al.108 observed the existence of dispersed softened regions within human breast cancer biopsies and breast cancer tissues in mouse mammary tumor virus-polyoma middle T antigen transgenic mice using an indentation-type atomic force microscopy method, and this finding might be related to metastatic spreading.
Mechanisms of matrix stiffness in cancer
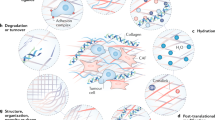
Matrix stiffness mainly depends on the ECM components and proportion, which is a cardinal phenomenon in many cancers accompanying TME sclerosis. Generally, overabundant collagen and HA are frequently observed throughout the tumor and are responsible for its stiffness109,110,111. However, solid tumors are 3D structures, whose periphery and interior parts show different mechanical characteristics. The interior parts of solid tumors mainly bear compressive stress from tumor cells and a stiffening matrix112, while the periphery of the tumor is subjected to tensile stress from the tumor mass and surrounding tissues112. Thus, the primary components responsible for stiffness of the periphery and interior of a tumor might be different. For example, excess collagen mostly contributes to the stiffness at the periphery of a tumor113. Superfluous HA mainly deposits at the interior of a tumor to counteract the compressive stresses exerted by the periphery layer of the tumor114.
Overall, the excessive intratumoral deposition of collagen and HA can be attributed to accelerated synthesis and slowed catabolism. First, some tumor cells can synthesize components of the ECM such as collagen and HA, exhibiting some characteristics of fibroblasts. For example, Fang et al.115 showed that type I collagen could be produced by not only fibroblasts but also by cancer cells in the lung and esophageal cancer. Similarly, HA-positive tumor cells can be identified in epithelial ovarian cancer116, breast cancer117, colorectal cancer118, prostate cancer119, and gastric cancer120. In addition to secreting components of the ECM, cancer cells can also produce enzymes involved in the maturation of ECM proteins. For example, IHC analysis revealed that gastric cancer cells could produce LOX to enhance collagen crosslinking121. It is worth mentioning that MMP-2 and MMP-9 are upregulated in human colorectal cancer122, and it might partly attribute to the compensatory responses to matrix stiffness so that the local matrix is degraded, which further enhances the motility of cancer cells.
Unfortunately, the production of ECM components and enzymes could be further accelerated when tumor cells receive external signals from growth factors123. For example, HAS mRNA transcription can be stimulated by epidermal growth factor (EGF), keratinocyte growth factor (KGF), and PDGF in keratinocytes124,125,126, and these growth factors are frequently overexpressed in cancer.
By contrast, some enzymes catalyzing the degradation of the ECM tend to be suppressed in the TME. For example, hyaluronidase activity decreases in ovarian cancer compared with that in normal ovarian tissue127. Moreover, MMP-28, namely epilysin, is significantly downregulated in lung squamous cell carcinoma and adenocarcinoma128.
In addition to the direct mechanisms described above, tumor cells could secrete growth factors to attract fibroblasts to migrate towards the TME and then transform normal fibroblasts into cancer-associated fibroblasts (CAFs) with a stronger ability to proliferate and promote ECM accumulation. In turn, a stiffened ECM accelerates the growth of tumor cells. Such communication between cancer cells and fibroblasts forms a positive loop feeding the rapid progression of this disease (Fig. 2). During the process of this bidirectional interaction, the TGF-β/Smad2/3 and C–X–C motif chemokine ligand 12 (CXCL12)/C–X–C motif chemokine receptor 4 (CXCR4) signaling pathways are most critical (Fig. 3). Specifically, TGF-β, which could be derived from cancer cells129, acts potently on fibroblasts to enhance the synthesis of collagen and fibronectin130 as well as chemokines related to tumor promotion, such as CXCR3, CXCR4, C–C motif chemokine receptor 9 (CCR9), CXCL10, CXCL12, C–C motif chemokine ligand 21 (CCL21), and CCL25131. These chemokines enhance tumor cell invasion and eventually the occurrence of organ-specific metastases132. Moreover, macrophages are lured into the TME by tumor cells133 and are further transformed towards the M2 type of macrophages134. Next, these M2 macrophages contribute to the activation of CAFs by secreting more TGF-β into the TME135,136, feeding more fuel into this positive loop and leading to malignant transformation (Fig. 2).

The signaling loop formed by cancer cells, macrophages and fibroblasts contributes to ECM stiffness, in which TGF-β plays a central role

Intracellular signaling network triggered by TGF-β and CXCL12, two critical factors inducing ECM stiffness
The biological effect of matrix stiffness on cancer cells
A stiffened ECM has fundamental influences on critical biological processes of cancer development, including uncontrolled proliferation, metastasis, angiogenesis, resistance against therapeutics, genome instability (GIN), and immunosuppressive TME (Fig. 4). The mechanisms by which ECM stiffness remodels these key processes will be discussed in-depth in this chapter.

Crucial biological processes affected by ECM stiffness for cancer
ECM and growth factors
ECM are important modifiers for the function of many growth factors. Firstly, accumulated ECM can function as a reservoir of growth factors, thus creating a niche with concentrated signaling molecules for the sustained malignant transformation. For instance, Somasundaram et al. proved that PDGF bound to collagen to accumulate in ECM137. Heparin-binding growth factor-1, a growth factor associated with angiogenesis, also binds to type I and type IV collagens138. What’s more, Paralkar et al.139 found that TGF-β bound to type IV collagen in the basement membrane. It is also reported that ECM contains IGFs140. Secondly, ECM facilitates the presentation of growth factors to their receptors. For example, glypican-3, a heparan sulfate proteoglycan, promotes the interaction between Wnts and Frizzled through complexing with Wnts, thus stimulating the growth of hepatocellular carcinoma cells both in vitro and in vivo141. Interestingly, glypican-3 also directly binds Frizzled through the glycosaminoglycan chains142. Furthermore, ECM degradation contributes to the release of growth factors and cytokines143,144. During tumorigenesis, MMP-2 and MMP-9 are upregulated in human colorectal cancer122, and growth factors released from ECM cleaved by MMPs would promote tumor progression. For example, the VEGF is released when heparan sulfate is degraded, and such process promotes angiogenesis in colorectal carcinoma145.
Effects on the proliferation of cancer cells
Tumor cells proliferate more slowly in a soft matrix, and matrix stiffness contributes to cancerous proliferation by multiple signaling pathways (Fig. 5)146,147,148,149. For example, the superfluous collagens bind to integrin on the cell membrane, leading to its allosteric alteration. In addition, the intracellular domain of the integrin β subunit recruits the cytoskeletal protein talin and other cytoskeletal linker proteins, leading to the formation of focal adhesions and activation of Src family kinases (SFKs). Thereafter, assembly of the actin cytoskeleton increases cytoskeletal tension, influencing the myocardin-related transcription factor (MRTF)/serum response factor (SRF) complex. With the help of MRTF/SRF, the signals from the cytoskeleton are transmitted into the nucleus. In addition, activated focal adhesion kinase (FAK) enhances the activity of PI3K100,150. Downstream proteins, such as AP-1 (oncogene c-Jun/c-Fos) are activated via Rac/PAK/MEK/ERK, and target of rapamycin is inhibited, eventually contributing to the proliferation of tumor cells. Another important pathway involved in cancer cell proliferation on hard surfaces is the Hippo pathway. This pathway comprises three components, mammalian Ste20-like kinases 1/2 (MST1/2), large tumor suppressor 1/2 (LATS1/2), and yes-associated transcriptional regulator/tafazzin (YAP/TAZ). When matrix stiffness occurs, the activated integrin linked kinase (ILK)-integrin signaling enhances the phosphorylation of myosin phosphatase target subunit 1 and inhibits its activity151, leading to the suppression of a signaling cascade comprising Merlin, MST1/2, and LATS1/2151. The blockade of upstream signals results in the translocation of YAP/TAZ from the cytoplasm to the nucleus152, where they initiate the transcription of genes involved in cell proliferation, such as cyclin D1 and forkhead box M1153. In a prospective cohort study in 528 patients with chronic hepatitis B patients, those with a higher liver stiffness (≥10 kPa) showed a significantly higher possibility of developing hepatocellular carcinoma than those showing lower liver stiffness (<10 kPa)154.

Schematic summary of key membrane receptors recognizing ECM components and their downstream signaling networks
Effects on the mobility of cancer cells
Cancer cells also tend to exhibit higher mobility on a stiffened ECM for many reasons. First, the stiffness of ECM can directly activate several signaling transduction pathways involved in cell migration (Fig. 5). For example, Rho/Rho-associated coiled-coil containing protein kinase signaling is activated when cytoskeletal tension is increased in a stiffened matrix155,156. Simultaneously, collagen-induced integrin clustering induces the recruitment of focal adhesion signaling molecules, such as FAK, Src, paxillin, Rac, Rho, and Ras, eventually promoting the contraction and progression of cancer cells157,158. As mentioned previously, PI3K is also activated when matrix stiffness occurs150,159,160, and the downstream molecules AP-1 promotes the migration and invasion of tumor cells. Moreover, the stiffness of the ECM can regulate the expression level of key elements of signaling pathways, indirectly leading to their dysregulation. Gkretsi et al.161 reported that Ras suppressor-1 (RSU-1), a cell-ECM protein, is overexpressed in breast cancer cells embedded in stiffer 3D collagen I gels, and silencing RSU-1 led to the inhibition of urokinase plasminogen activator (UPA) and MMP-13, resulting in reduced invasion activity in breast cancer cells.
Effects on tumor vascularity
Vascularization is an indicator of tumor development and progression162. During angiogenesis, vascular loops are formed which link the parental vessels and neovessels, and the matrix of basement membrane is synthesized to reinforce the elasticity and tenacity of blood vessel163. Basement membrane, mainly composed of collagen, laminin, fibronectin, often exhibit several abnormal characteristics in tumors, such as disconnection with endothelial cells, disorder of layer and structure, and extensively infiltration into tumor stroma164. Vascularization decreases with the increasing matrix density in both collagen and fibrin matrix165,166,167,168. Matrix stiffness also interrupts endothelial cell–cell junction so that the integrity of barrier breaks down and leads to the leaky vascular169. Mechanically, several mechanosensors are in the employ of endothelial cells to convert the mechanical clues into cells so that intracellular biochemical signaling cascades can be activated, such as integrins, actin cytoskeleton, cell-cell adhesion receptors, and other membrane proteins such as ion channels and G-protein-coupled receptors. Taking the mechanical sensor complex of endothelial cells as an example, which is composed of platelet and endothelial cell adhesion molecule 1 (PECAM1), vascular endothelial cadherin (VE–cadherin), and VEGF receptor (VEGFR), PECAM1 functions as a direct mechanosensory, and VE–cadherin serves as an adapter, and VEGFR activates intracellular signaling which consumes energy from GTPase170,171,172. In addition, upregulation of MMP activity in stiffened TME also augments vascular hyperplasia, intrusion, and neovascular branching169.
ECM also affects vascularization indirectly through hypoxia. Overgrowth of cancer cells, as well as a structural and functional abnormality of ECM both, contributes to the hypoxia of solid tumor173. In turn, the hypoxia circumstance affects the vascularization via multiple mechanisms, the most well understood of which is the activation of hypoxia-inducible factors (HIFs)174. Overexpression of HIF-1α has been reported in many malignancies175,176. In addition, multiple studies have demonstrated the involvement of HIF-1/VEGF signaling in breast cancer177, gastric cancer178, bladder cancer179, ovarian cancer180, and so on.
Effects on the therapeutic efficacy of chemotherapies, radiotherapies, and targeted therapies
The stiffened matrix in tumors also decreases drug sensitivity, which can partially explain the poor therapeutic efficacy of chemotherapies and immune therapies in many circumstances181. First, a stiffened matrix forms physical barriers for drug infiltration into tumor tissue, and researchers have shown that decreasing HA deposited in tumor tissues benefited systemic chemotherapy in colorectal cancer patients with liver metastasis182. Second, besides the physical barrier, a stiff matrix compresses micro blood vessels, making it difficult for drugs to access core tumor tissues via the vasculature. Third, ECM stiffness induces hypoxia in the tumor microenvironment183,184, further inducing neovascular chaos, resulting in disorganized and perforated intratumoral microvessels. This leaky vasculature negatively influences the transport efficiency of chemotherapeutic drugs185. Finally, ECM stiffness is involved in the transformation of tumor cells to cancer stem cells (CSCs), which have the ability to proliferate in a hypoxic environment. Many lines of evidence have shown that CSCs are more resistant to anti-cancer drugs than bulk cells186,187,188.
Matrix stiffness also plays a role in radiation resistance for tumors189,190,191,192. β1 integrins are upregulated in several types of cancer, such as lung cancer193 and colorectal carcinoma191, and several studies have demonstrated that the upregulation of β1 integrins contribute to the survival of tumor cells in pancreatic, prostate, glioblastoma, melanoma, and colorectal carcinoma after the treatment of radiation190,193,194,195. Mechanically, the downstream signals of β1 integrins would be activated in response of radiation, such as ILK, FAK, paxillin194, c-Jun N2-terminal kinase (JNK), PI3K, and AKT/protein kinase B (PKB)196. The activation of PI3K/AKT leads to the radiation resistance197, and inhibition of integrin signaling attenuates the insensitivity of cancer cells exposed to radiation189,196.
Effects on genome stability
Recently, some researchers have reported that GIN could also be enhanced by matrix stiffness, whose detailed mechanisms remain under investigation. Some researchers speculate that matrix stiffness would promote cell mitosis, during which spontaneous mutations would accumulate accompanied by fast DNA replication198,199. In addition, matrix stiffness increases the probability of nucleus envelope rupture200. Nucleus envelope rupture would cause the leakage of nuclear contents into the cytoplasm, such as nucleic acid and nuclease, ultimately causing DNA damage and GIN201. Finally, the pore size of the matrix is smaller under stiffer conditions202 due to the overabundant deposition of matrix proteins such as collagen203. While cancer cells invade, they need to squeeze through smaller pores and undergo more physical damage204. Such squeezing movement would isolate some mobilizable nuclear proteins away from DNA205, such as DNA repair proteins (e.g., BRCA1), thus increasing the possibility of GIN.
Effects on infiltrated immune cells and immune therapies
During the development of cancer, immune cells infiltrate into TME and play either anti-tumor and pro-tumor roles. For example, CD8+ T cells, CD4+ Th1 cells, dendritic cells (DC), natural killer (NK) cells, and M1 macrophages mainly exhibited inhibitory effects on cancer progression, while regulatory T cells (Tregs), CD4+ Th2 cells, myeloid-derived suppressor cells (MDSCs) and M2 macrophages mainly show pro-tumor effects206. These cells are constantly influenced by the physical, chemical, and biological signals emitted by ECM in TME.
T cells, especially CD8+ T cells, are the major players in the immune response against cancerous cells, and they are also the primary targets for checkpoint inhibitor (CPI) therapy currently. The intratumoral ECM contributed to both exclusion and inactivation of T cells in TME by multiple mechanisms. Firstly, the center-axial infiltration of T cells towards chemotaxis gradients is interrupted by the haptotaxis signals from altered ECM207,208,209. Instead of entering TME, some T cells migrate along ECM-rich encapsulation of tumors due to the gradients of substrate rigidity and adhesion molecules such as aligned collagen fibers210,211. Such an accumulation of T cells on the periphery of cancerous tissues are reported in many kinds of cancers such as colorectal cancer and lung cancer212,213. Secondly, ECM-rich tumors exhibit poor diffusion and hypoxia. The retarded exchange of substances enhances glycolytic metabolism and acidification, which suppresses the activation of T lymphocytes through the specific interaction between V-domain immunoglobulin suppressor of T cell activation and co-inhibitory receptor P-selectin glycoprotein ligand-1 in acidic TME214. Moreover, the hypoxic microenvironment increases the production of immunosuppressive factors like TGF-β in favor of Treg differentiation, and it also induces neovascularization with abnormal structure and reduced expression of surface glycoproteins and cell adhesion molecules which are critical for the extravasation of T lymphocytes215. Thereby, poor diffusion due to stiffened ECM contributes remarkably to the suppression of anti-tumor responses mediated by T lymphocytes in TME. Thirdly, ECM proteins are directly involved in the regulation of T lymphocytes. For example, CD8+ T cells are suppressed by collagen through leukocyte associated Ig-like receptor-1 (LAIR-1)/SH2-containing inositol phosphatase signaling axis in murine cancer models216, and high molecular weight HA could enhance the activity of Tregs in vitro217. Lung cancer cells also express more PD-L1 in response to ECM stiffness to shut down the anti-tumor immune response mediated by CD8+ T cells218. Fourthly, stiffened ECM impairs the efficacy of immune therapies. For example, two important obstacles for the application of chimeric antigen receptor (CAR)-engineered T cells therapy in non-hematopoietic malignancies are the low infiltration rate and the immunosuppressive TME, both of which are directly associated with intratumoral ECM accumulation219. Similarly, CPI therapies mediated by blocking antibodies are also less penetrable for tumors with high rigidity220.
Tumor-associated macrophages are the most frequent immune cells found in the TME221. ECM components are able to regulate the polarization of macrophages. For example, collagen and HA are able to drive M2 polarization in vitro222,223, while fibronectin enhances cytotoxicity of macrophages against tumor cells, resembling the M1 polarization224. Other than macrophages, NK cells have also been shown to be functionally suppressed by transmembrane collagens like Collagen type XVII via LAIR-1225.
Receptors for ECM in normal and cancer cells
Many receptor proteins play an important role in the interaction between ECM and cells, such as integrin, discoidin domain receptors (DDRs), CD44, RHAMM, LAIR-1, and the mannose receptor family, including urokinase plasminogen activator receptor-associated protein226. Next, we will focus mainly on integrin, DDRs, CD44, and RHAMM, which are frequently discussed in the context of cancer (Fig. 5).
Integrin
Integrins are transmembrane heterodimers comprising α subunits and β subunits. In mammals, 18 α subunits and 8 β subunits combine into 24 integrin heterodimers. Among the 24 integrins, four (α1β1, α2β1, α10β1, and α11β1) can bind collagen226. Moreover, integrins can bind to various proteins that contain the RGD sequence, such as fibronectin, fibrinogen, laminin, and vitronectin227,228,229,230. Other than functioning as an anchor, integrins serve as switching points that connect the ECM to the intracellular actin cytoskeleton. Specifically, integrins perceive the ECM mechanical force and then transfer such signals to intracellular proteins such as FAK and Src tyrosine kinases, a process called mechanotransduction. In addition to outside-in signal transduction, integrins also transmit signals from the inside to the outside of the cell when intracellular stimulating molecules bind to β subunits, further influencing the affinity between integrins and the ECM so that cell adhesion, migration, and ECM characteristics might change. For example, Pollan et al.231 reported that the adhesion of prostatic cancer cells could be attenuated by silencing CUB domain-containing protein-1 (CDCP1) due to the reduction of inside-out signaling mediated by integrin β1 subunit. Interestingly, the metastatic adhesion of circulating cancer cells may be upregulated by the inside-out signaling via FAK/integrin232.
Much research has shown that several integrin proteins are highly expressed in solid tumors and are involved in tumor progression. For example, integrin αvβ3 is upregulated in prostate cancer and promotes cell migration via activation of the PI3K/AKT pathway233. Similarly, immunohistochemistry (IHC) analysis conducted by Desgrosellier JS revealed that the positive rate of integrin αvβ3 was significantly higher in metastasis than in primary tumors of pancreatic and breast cancer, and integrin αvβ3 enhanced tumor migration and metastasis by the recruitment of Src kinase234. Furthermore, several studies have demonstrated that the upregulation of integrin αvβ3 is correlated with a poor prognosis for patients with oral squamous carcinoma235,236,237,238, breast cancer239, gastric cancer240,241, colorectal cancer242, pancreatic ductal adenocarcinoma243, and cervical squamous carcinoma244. Other than integrin αvβ3, integrin αvβ6 is overexpressed in oral squamous carcinoma235,237, breast carcinoma239,245, gastric cancer240,246, pancreatic ductal adenocarcinoma243,247, ovarian cancer248, colorectal cancer242,249,250, cholangiocarcinoma251,252, and non-small cell lung cancer253.
Discoidin domain receptors
DDRs can spontaneously bind to collagen and are not regulated by intracellular or extracellular signals. The structure of DDRs includes collagen-binding the discoidin domain at the N-terminus, extracellular juxtamembrane domain, transmembrane domain, intracellular juxtamembrane domain, and tyrosine kinase domain at the C-terminus254. There are two types of DDRs, namely, DDR1 and DDR2. DDR1 is commonly expressed in epithelial cells, and DDR2 is generally present in mesenchymal cells such as fibroblasts255. Specifically, DDR1 interacts with collagen type I and IV, while DDR2 binds to collagen type I, II, and X. When the collagen-binding discoidin domain interacts with collagen, the conformation of DDRs changes and the phosphorylation of the tyrosine kinase domain leads to the recruitment of adapter proteins (e.g., ShcA and Nck2) to the cytoplasmic domain of DDRs256. Both integrin and DDRs can sense ECM stiffness and then transmit this signal into cells. However, ECM cell signal transduction mediated by DDRs is unidirectional, while the one mediated by integrin is bidirectional.
Although heterogeneity remains regarding the expression of DDRs in multiple cancers, many studies have reported that DDRs are overexpressed in cancers. For example, DDR1 overexpression has been observed in breast cancer257,258,259,260, nonsmall cell lung carcinomas261,262,263,264, glioblastoma265, ovarian tumor266,267,268,269, endometrial tumors270, esophageal carcinoma271, head and neck squamous cell carcinomas260, hepatocellular carcinoma272, cholangiocarcinoma273, and prostate cancer274. Similarly, DDR2 overexpression is reported in nasopharyngeal carcinoma275, cholangiocarcinoma273, thyroid cancer276, Hodgkin’s lymphoma277,278, and acute myelocytic leukemia279. In addition, DDR1 overexpression is significantly correlated with a poor prognosis in pancreatic ductal adenocarcinoma280, gastric cancer281, and nonsmall cell lung cancer263,282, while increased DDR2 levels could function as an independent indicator of a worse clinical outcome in breast cancer283.
CD44
CD44 mainly functions as a receptor for HA, collagen, fibronectin, and growth factors. CD44 comprises an extracellular domain, a transmembrane domain, and a cytoplasmic domain284, whose isoform heterogeneity is mainly due to the alternative splicing of premRNA and posttranscriptional modifications such as glycosylation (N- and O-glycosylations). HA–CD44 interaction activates multiple cell receptors, such as c-MET, EGF receptor (EGFR), erb-b2 receptor tyrosine kinase 2 (ErbB2), and TGF-β, which then promotes oncogenic pathways. In addition to membrane receptors, the HA–CD44 interaction also activates intracellular signal transducers, such as Grb2, Gab-1, Src, and Rac GTPase families. Thus, many aspects of malignant transformation, such as uncontrolled proliferation, migration and drug resistance could be induced by the HA-CD44 interaction284,285. In addition, the binding of lymphocytes to fibronectin is also mediated by CD44286, which is pivotal for the infiltration of lymphocytes into the TME. A phase I clinical trial demonstrated that recombinant fibronectin CH296 (FN-CH296) stimulates T cells to achieve strong tumor inhibitory effects in patients with advanced cancer287.
Overexpression of CD44 standard (CD44s) and CD44 variant (CD44v) isoforms is widely reported in many types of cancer288. In gastric cancer, Yansu Chen et al.289 performed a meta-analysis comprising 2403 cases and identified that higher CD44 expression is correlated with a poor overall survival rate and serves as an independent risk factor. A similar observation regarding the prognostic value of CD44 is also reported in other types of cancer, including renal cell carcinoma290,291,292,293,294,295, prostate cancer296,297,298, pancreatic cancer299,300,301, lung cancer302,303,304,305,306,307, breast cancer308, colorectal cancer309,310,311,312,313,314,315,316,317,318, and hepatocellular carcinoma319,320,321,322. Recently, CD44s and CD44v isoforms have been identified as surface biomarkers for CSCs in pancreatic cancer323, salivary gland tumor324, laryngeal and nasopharyngeal carcinoma325,326,327, head and neck malignancy328,329,330,331,332,333,334,335, gastric cancer336,337,338,339,340,341, colon cancer312,342,343,344,345,346, glioma347,348,349, lung cancer306,350,351, breast cancer352, ovarian cancer353, prostate cancer296,354,355,356, and leukemia/lymphoma357.
Receptor for hyaluronan-mediated motility
RHAMM is a unique ECM receptor which lacks a transmembrane domain, and it exhibits both intracellular (cytoplasmic and nuclear) and extracellular (membrane-bound or soluble) localizations358,359,360,361. RHAMM exhibits highly diverse functions in different subcellular compartments. On cell membrane, HA is the major ligand for membrane-bound RHAMM362. RHAMM couples with integral cell surface receptor proteins such as CD44 and growth factor receptors, and HA-RHAMM–CD44 coupling is necessary for the activation of Src/Ras/ERK and FAK/Ras/ERK signaling pathways mediated by CD44363,364,365,366. Antibodies blocking RHAMM-HA recognition would completely inhibit HA-mediated locomotion, while antibodies blocking CD44-HA recognition failed to change locomotion, suggesting that RHAMM plays a central role for cell motility along HA fibers367,368. Moreover, intracellular RHAMM forms direct interaction with MEK/ERK366, and it also localizes to multiple subcellular structures including actin filaments, podosomes, the centrosome, microtubules and the mitotic spindle364,369. During cell migration, spectrin-α (an actin-associated protein) and RHAMM interact in a complex at the nodes of the actin net to coordinate microtubule polarization370. In the centrosome, RHAMM interacts with dynein and maintains spindle pole stability369. In the nucleus RHAMM is able to regulate HA-induced activation of the Aurora A kinase (AURKA) by associating with TPX2 (TPX2 microtubule nucleation factor), a critical protein for AURKA recruiting and activating371. During mitosis, RHAMM regulates mitotic spindle formation through interacting with tubulin, ERK and TPX2 to recruit and activate AURKA360,369,372,373. In mammary epithelium, RHAMM works in concert with TPX2, BRCA1, and AURKA to regulate the apicobasal polarization374.
As a dual oncogenic protein promoting proliferation and migration both on cell membrane and intracellularly, RHAMM is overexpressed and correlated with poor prognosis in many kinds of solid tumors, including but not limited to breast cancer375,376,377, colorectal cancer378,379, stomach cancer380, prostate cancer381,382, hepatocellular carcinoma383,384, pancreatic ductal adenocarcinoma385, lung cancer386,387, bladder cancer388, oral squamous cell carcinoma389, and head and neck cancers390. Recently, Choi et al.391 reported that RHAMMB isoform was crucial for in vivo metastatic capacity of mouse and human pancreatic cancer while RHAMMA, carrying an extra 15-amino acid-stretch, did not promote metastasis in spontaneous and experimental metastasis mouse models.
Matrix components as therapeutic targets for cancer
Therapies targeting collagen
Collagen is one of the most fundamental components in the ECM, the breaking of which could facilitate the penetration of many conventional chemotherapeutic agents and nanoparticles through the barrier of the stiffened matrix in the TME. To alleviate the excessive deposition of collagen in solid tumors with TME sclerosis, several therapeutic strategies have been developed, mostly focusing on the synthesis, degradation, and cross-linking of collagen (Table 1).
Considering TGF-β’s crucial role during collagen synthesis, TGF-β signaling is the most promising target to inhibit collagen synthesis392,393. For example, an anticoccidial named halofuginone has been shown to reduce collagen synthesis by inhibiting TGF-β signaling in animal models of pancreatic cancer394, lung cancer395, melanoma396, and breast cancer397, and tumor migration396,397. Moreover, the therapeutic effect of fresolimumab, a monoclonal antibody targeting TGF-β, is currently actively evaluated in several clinical trials (clinicaltrials.gov identifier: NCT01401062 and NCT02581787) to treat cancer398. However, a treatment target involving TGF-β should be regarded cautiously because of its extensive roles in both inflammatory and tumorigenesis399. In addition to therapies targeting TGF-β, some pilot studies have also found that the classic anti-hypertensive drug losartan contributes to the inhibition of collagen synthesis in both animal models400,401 and clinical trials (clinicaltrials.gov identifier: NCT01821729). Although its molecular mechanism still requires further investigation, its safety profile makes losartan a promising choice for designing new therapies targeting collagen synthesis in cancer.
Collagenases can degrade collagen, which could attenuate the stiffness of the matrix and simultaneously contribute to more efficient drug delivery into solid tumors402. Due to their chemical nature as proteins, several strategies have been developed to overcome their problem of large molecular sizes to achieve effective transport of collagenases into tumors, such as the oncolytic herpes simplex virus vector403 and collagozome (a liposomal formulation of collagenase type I)404. For example, Zinger A et al. found that the tumor size was reduced by 87% when mice bearing pancreatic tumor xenografts were sequentially treated with collagozome and paclitaxel compared with mice treated with empty liposomes and paclitaxel404. More importantly, the researchers found no evidence of the existence of tumor cells in the circulatory system, suggesting that the process of ECM degradation did not trigger tumor metastasis404. Interestingly, a hormone named relaxin could improve the penetration of antitumor drugs by indirectly promoting the synthesis of collagenase in osteosarcoma tumor models405. In addition to collagenases, a few studies showed that strategies aimed at regulating the quantity or activity of MMPs could also be helpful for cancer treatment, such as marimastat (BB-2516), prinomastat (AG3340), tanomastat (BAY 12-9566), and neovastat406,407.
However, two major concerns exist regarding the application of collagenase during cancer therapy. First, the process of collagen degradation might lead to the release of growth factors and cytokines anchored in collagens, which would initiate a cascade of inflammatory signals and tumor progression408. Second, the breakdown of collagen might facilitate tumor migration and invasion409,410. Therefore, the best time point for the application of such treatment should be considered cautiously and validated experimentally. Theoretically, collagenase-based therapies should be applied to early detected cancer with obvious matrix stiffness that has shown no signs of invasion or metastasis.
Inhibition of collagen cross-linking is another strategy to target ECM stiffness in cancer. For example, studies have found that LOXs are frequently upregulated in many cancers, including thyroid cancer and colorectal cancer411,412,413. Inhibition of LOXs has been shown to enhance chemotherapeutic drug delivery in mouse models of pancreatic cancer414 and breast cancer415. Theoretically, LOX inhibition might be beneficial during the development phase of cirrhotic cancer. However, it might not work for tumors with an existing mature collagen mesh, greatly limiting its application.
Therapies targeting TGFβR
TGF-β receptor (TGFβR) is a tetramer which consists of two different transmembrane kinase, namely type I receptor (TGFβR1) and type II receptor (TGFβR2), and both of them have the ability to lead to the phosphorylation of serines, threonines, and tyrosines416. When TGF-β binds to TGFβR, the seine at the C-terminal of the adapters such as Smad2 and Smad3 phosphorylates. These adapters, together with Smad4, translocate into the cell nucleus, finally binding to transcription factors so that the transcription of target genes is activated or suppressed416,417,418. TGF-β plays a dual role in cancer. It is a tumor suppressor during the initiating stage early stage of cancer, while it functions as an oncoprotein in advanced stages of cancer419. Mechanically, the direct effect when TGF-β binds to TGFβR is proapoptotic420, so TGFβR is downregulated or mutant in various types of cancer. However, tumor cells themselves overexpress TGF-β, which is excreted into TME and targets nonparenchymal cells421 like fibroblasts and Treg cells. TGF-β promotes fibroblasts to produce ECM components, and enhances the differentiation and function of Treg cells to induce immunosuppressive TME422.
Therapies targeting TGF-β have been discussed in the section “Therapies Targeting Collagen” earlier in this review. Small molecules which target TGFβR are widely used in experiments of cancer therapies423 (Table 1). SB-431542 and SB-505124 have been shown to suppress proliferation, motility, and vascularization in mice models of glioma and renal carcinoma424,425,426. SB-431542 also enhances the activity of DC and CD8+ T cells423,427. The fatal weakness of these two inhibitors lies in their instability and low specificity which causes severe systematic toxicity. Ki26894 has been reported to suppress bone metastasis in mice models of breast cancer and gastric cancer428,429,430. Other small molecules that inhibit TGFβR, such as LY-2109761431,432,433,434,435, SD-093436, SD-208436, and LY-580276436, have also been tested in various kinds of cancer.
In consideration of the complex role of TGF-β and TGFβR in tumors, the therapeutic strategy targeting them should be really cautious. Agonist of TGFβR directly inhibits the growth of tumor cells, but also promotes stromal cells to produce ECM components and contributes to immunosuppression. As for advanced tumors, inhibition of TGFβR suppresses the metastasis and invasion of tumor. Therefore, a comprehensive understanding of tumor traits, disease stage, and TME are prerequisites when applying a therapeutic strategy targeting TGF-β signaling.
Therapies targeting AT1R
Angiotensin II type 1 receptor (AT1R) and Angiotensin II type 2 receptor (AT2R) are both receptors for Angiotensin II (AngII)437. They belong to the family of G-protein-coupled receptors, which are seven-span transmembrane proteins. AT1R is considered as the leading receptor for AngII to exert vasoconstriction functions, while AT2R tends to be a counter-regulatory factor. The downstream signaling of AT1R, directly or indirectly, includes MAPK, c-Src, Tyk2, Pyk2, Jak2, Ras, AKT, receptor tyrosine kinases, and redox-sensitive transcription factors such as nuclear factor kappa B (NFκB) and HIF-1α438,439,440,441. The phosphorylation of tyrosine in growth factor receptors, integrins, and adhesion-associated adapter proteins such as paxillin, tensin, and Grb2 all promotes the function of AngII, eventually enhancing the phosphorylation of MEK and ERK1/2 induced by EGFR442,443,444,445,446.
AT1R plays important roles in promoting cell proliferation, angiogenesis, and inflammation in TME. Firstly, AT1R activates EGFR in breast cancer447 and prostate cancer448, contributing to the activation of ERK and signal transducer and activator of transcription 3 (STAT3) phosphorylation, and protein kinase C (PKC) activation, thus promoting the proliferation of cancer cells. Secondly, the activating of EGFR by AT1R leads to the increased expression of VEGF in both cancer cells and endothelial cells, and intratumoral endothelial cells are activated in either paracrine or autocrine manner, which contributes to angiogenesis in TME449,450. Lastly, AT1R promotes the transcription of cytokines and chemokines, such as IL-6, IL-12, IL-8, and monocyte chemoattractant protein-1 through activating NFκB and AP-1451, thus resulting in inflammation. Dysregulations of AT1R and AT2R has been reported in the breast in situ carcinoma452, invasive breast carcinoma453, skin squamous cell carcinoma454, cervical cancer455, ovarian cancer456, and prostate cancer448.
Angiotensin receptor blockers (ARBs) are widely used as traditional antihypertension drugs, and recent research revealed that they could suppress growth and metastasis of cancer (Table 1). Candesartan, a long-acting angiotensin receptor antagonist, inhibits lung metastasis in mice intravenously injected with 3LL cells457. Moreover, tumor growth and angiogenesis are inhibited by candesartan in mouse melanoma model457,458 and xenograft models of human prostate and ovarian cancer cells448,456. Losartan, another angiotensin receptor blocker, is able to inhibit the release of growth factors like VEGF and suppresses tumor growth of glioma cells both in vivo and in vitro459.
Therapies targeting HA
Similar to collagen, two types of therapeutic strategies targeting HA are under investigation, including the inhibition of HA synthesis and enhancement of HA degradation (Table 1).
4-Methylumbelliferone (4-MU) is an inhibitor of HAS. 4-MU has been shown to suppress the activation of CSCs and attenuate chemoresistance in animal models of ovarian cancer460. Moreover, Kohli et al.461 demonstrated that liposomes containing 4-MU could potently suppress HA synthesis, eventually facilitating the penetration of more liposome drugs into breast cancer xenografts.
Hyaluronidase has exhibited beneficial effects for diseases such as bladder cancer, brain cancer, and gastrointestinal cancer by degrading HA within the TME402,462. A few clinical trials are currently evaluating the therapeutic effects of combining hyaluronidase and chemotherapeutic agents such as gemcitabine and fluorouracil463,464. Currently, the long-term effects of hyaluronidase on cancer therapy remain under investigation.
Therapies targeting fibronectin
The researches regarding the application of fibronectin in cancer treatment are mainly focused on its application as a target for precise drug delivery (Table 1). EDA and extra domain B (EDB) of fibronectin are frequently upregulated in tumor neovasculature465,466,467. Therefore, several targeted cancer therapies have been developed targeting EDB. For example, the murine monoclonal antibody against the cryptic domain adjacent to human fibronectin EDB, BC-1 was fused with murine IL12 (huBC-1-mIL-12) and showed inhibitory effects on various kinds of cancer xenografts in immunocompetent severe combined immune deficiency mice, including colon cancer, skin tumor, and prostate cancer468,469. A clinical trial of huBC-1-mIL-12 was conducted, and 46% of patients were in stable condition after 6 or more cycles of treatments470. Another antibody that targets EDB, L19, was fused with IL-2 (L19-IL-2) and significantly improved the tumor-inhibitory efficiency of IL-2 in tumor-bearing mice471,472. Patients who received L19-IL-2 treatment showed stable condition without treatment-related death during its clinical trial in renal cell carcinoma and melanoma patients473,474. Other than cytokines, EDB also serves as the delivery target for antibody-mediated radioisotopes. A fusion protein of L19 and small immunoprotein (SIP) marked with 131I not only slowed tumor growth, but also prolonged the survival of mice bearing F9 teratocarcinoma and head and neck carcinoma xenografts475,476. In addition, 131I-labeled L19-SIP could be applied to visualize the tumor lesions in lymphoma477 and prostate cancer478 patients. However, no curative effect was observed in these patients. Moreover, EDB binding peptides have also been applied for the delivery of chemotherapeutic agents. Saw et al.479 developed APTEDB, a novel class of high-affinity peptides targeting EDB, and doxorubicin-containing APTEDB liposomes reduced 55% of tumor size while the free doxorubicin reduced 20% of tumor size in tumor allograft mice model. Similarly, APTEDB-decorated nanoparticles encapsulating paclitaxel has been applied for the inhibition of neovasculature in a mice model of glioma tumor, and such modification significantly enhanced the intratumoral accumulation of paclitaxel and prolonged the survival time480.
Therapies targeting sensors of matrix stiffness
Integrin is a promising drug target due to its crucial role in both mechanotransduction and other oncogenic processes for malignancy transformation (Table 1). Integrin α11β1, α5β1, α9β1, and αvβ3 are widely expressed by tumor cells and tumor stromal cells, including fibroblasts, endothelial cells, and tumor-associated macrophages, substantially influencing the characteristics of the TME481,482,483,484. Many preclinical studies have demonstrated that the inhibition of integrin could strongly suppress disease progression485. For example, Vitaxin, a humanized monoclonal antibody targeting integrin αvβ3, showed therapeutic potential in breast, lung, and colon cancer patients by preventing intratumoral angiogenesis during clinical trials486. Similarly, volociximab, an antibody that binds specifically to integrin α5β1, also exhibited remarkable therapeutic efficacy in clinical trials involving ovarian cancer, peritoneal cancer, pancreatic cancer and renal cancer patients487,488,489,490. In addition, Paolillo et al.491 found that 1a-RGD, a small-molecule integrin antagonist that targets the RGD–integrin interaction, could augment detachment-mediated anoikis while suppressing cell migration in glioma cancer cell lines. Cilengitide, a specific peptide antagonist targeting the binding between integrin αvβ3 and RGD, shows a good safety profile and clinical improvement for patients with head and neck tumors492,493,494,495. However, it should be cautioned that the binding of cilengitide with integrin is accompanied by conformation alteration, leading to adverse effects such as agonist-like activities496.
Emerging studies have demonstrated the role of DDR1 in cancer progression and metastasis497,498,499 (Table 1). Aguilera et al.500 knocked down DDR1 by siRNA and found that migration was inhibited in pancreatic ductal adenocarcinoma cells. The combination of DDR1 inhibitors and classical chemotherapeutic drugs has been reported to reduce the tumor burden in both orthotopic xenografts and autochthonous pancreatic cancer models500. Moreover, an in vivo study showed that the knockdown of DDR1 suppressed tumor growth and multiorgan metastasis in breast cancer mouse models497. Similarly, in a KRAS-mutant lung adenocarcinoma mouse model, inhibition of DDR1 attenuated tumor aggression501. The signal transduction triggered by DDRs could be blocked by tyrosine kinase inhibitors (TKIs), such as imatinib, nilotinib, and dasatinib, some of which have been applied as a cancer treatment for more than a decade502,503,504,505,506. For example, nilotinib, a second-generation TKI, suppresses tumor metastasis of colorectal cancer cells by inhibiting the DDR signaling pathway in an intrasplenic tumor mouse model507. Moreover, lung squamous cell carcinoma patients with a DDR2 mutation were more sensitive to dasatinib than those with wild-type DDR2508. Other than TKIs, 3-(2-(pyrazolo[1,5-a]pyrimidin-6-yl) ethynyl)benzamides have been identified as selective DDR1 inhibitors with a relatively low IC50 and could potently attenuate cancer invasion, adhesion, and tumorigenesis in vitro509.
Considering the importance of the CD44-HA and RHAMM-HA interactions in tumor cells, they might be promising therapeutic targets for cancer treatment (Table 1). Efforts have been made by many research groups to evaluate the antitumor effect of CD44 antibodies. For example, bivatuzumab (e.g., the first humanized monoclonal antibody against CD44v6 underwent clinical trials), displayed a moderate antitumor effect in patients with advanced squamous cell carcinoma of the head and neck or esophagus510. Subsequently, more CD44 antibodies entered clinical trials, such as RO5429083 (clinicaltrials.gov identifier: NCT01358903 and NCT01641250). Moreover, another siRNA-based strategy has been developed to inhibit the mRNA transcription of CD44 or CD44v511,512,513,514,515,516. However, due to the lack of a comprehensive understanding of all CD44 isoforms and the consequences of knocking down a mixture of CD44 isoforms, some challenges persist for the clinical applications of a siRNA-based strategy targeting CD44. Recently, verbascoside has been identified as small molecular-weight inhibitors for CD44 dimerization, and it showed inhibitory effects on the growth of intracranial tumors in a mouse model of glioma517. DNA aptamer targeting the HA-binding domain of CD44 also exhibited suppressive effects on the invasiveness of breast cancer cell line MDA-MB-231518. Several strategies targeting RHAMM-HA interaction are also under preclinical studies in multiple kinds of cancer. For example, small interfering RNA-mediated suppression of RHAMM has been shown to sensitize lung adenocarcinoma A549 cells to radiotherapy519. A soluble peptide containing the HA-binding domain of RHAMM inhibited both proliferation and migration of multiple glioma cell lines368. Several shorter blocking peptides (7 to 15mer) for RHAMM-HA interactions have been screened out, but their therapeutic efficacy has not been evaluated in the cancer models yet520,521.
Other therapeutic strategies alleviating matrix stiffness in cancer
A few studies have reported that tranilast522, pirfenidone523, fasudil524, metformin525, and dexamethasone526 could alleviate matrix stiffness in tumors. In addition, traditional drugs, such as hydroxychloroquine527, retinoic acid receptor agonists527, and FAK inhibitors399, have the potential to attenuate matrix stiffness in tumors. More mechanistic studies are urgently required to exploit these drugs for cancer therapy.
Cell–ECM interaction as therapeutic targets in leukemia—chronic lymphocytic leukemia as an example
In the previous paragraphs of this review, we mainly focused on the ECM of solid tumors. However, cell–ECM interactions also play nonnegligible roles in leukemia which are often referred to as “liquid cancer”. Even though ECM molecules are relatively sparse in blood, intensive but temporary cell-ECM interactions occur within the bone marrow and peripheral lymphatic organs where hematopoietic cells receive signals for proliferation and differentiation. Similar to solid tumors, collagens, proteoglycans, and glycoproteins form ECM in the bone marrow and lymph nodes. However, unlike solid tumors in which most cancer cells are permanently embedded in stiffened ECM, leukemia cells only form temporary connections with ECM in structured niches within bone marrow and lymphoid organs.
Leukemia can be classified as acute or chronic, according to the degree of cell differentiation, and as myelogenous or lymphocytic, according to the predominant type of cell involved. Generally, leukemia can be categorized as acute lymphocytic leukemia, chronic lymphocytic leukemia (CLL), acute myelogenous leukemia (AML), and chronic myelogenous leukemia (CML). In this section, CLL would be the most frequently used example since remarkable breakthroughs have been made in managing this disease by targeting cell-ECM interactions in the past decade.
In the cortex of lymph nodes, a network of fibroblastic reticular cells (FRCs) secretes type III collagen that produces reticular fibers which are highly stretchable, which allows lymph nodes to enlarge rapidly to accommodate fast-dividing lymphocytes in the circumstances of infections or leukemia. On the apical surface of FRC (the side that faces the cavity where lymphocytes reside), integral membrane proteins (such as vascular cell adhesion molecule, VCAM-1) and other macromolecules tethered to FRC membranes (such as HA) provide abundant anchorage points for lymphocytes and antigen-presenting cells528,529,530. CLL cells actively proliferate inside the lymphoid tissues, but they would stop proliferation during their circulation in blood531. Therefore, lymphadenopathy is typically observed in CLL patients. Bruton tyrosine kinase (BTK), a key element of B-cell antigen receptor (BCR) signalosome, plays vital roles for CLL homing and retention in lymph nodes by controlling integrin α4β1-mediated adhesion to fibronectin and VCAM-1, as well as chemotaxis signals mediated by CXCL12-, CXCL13-, and CCL19-induced signaling532. CLL patient cells expressed higher BTK mRNA compared to normal B cells533. BTK inhibitor Ibrutinib (PCI-32765) treatment can achieve rapid (within days) and sustained reduction of lymphadenopathy accompanied by transient lymphocytosis due to early exiting of CLL cells from lymph nodes534,535. Ibrutinib has been approved for patients with previously treated mantle cell lymphoma, CLL, and several other B-cell-related diseases, and more BTK inhibitors are in the pipeline536.
Other than BCR signaling, cell-HA interactions in the bone marrow and lymph nodes are also critical for hematological malignancies. For example, CD44v6 expression in diffuse large B-cell lymphoma (DLBCL) correlates with advanced disease stage, and coexpression of any of the CD44 isoforms with RHAMM could identify a subgroup of DLBCL patients with a very poor prognosis independent of the International Prognostic Index537. RHAMM-HA interaction promotes directional cell locomotion538,539,540. For example, B cells could mobilize along HA molecules through RHAMM-HA interaction upon activation by chemokines such as CCL21 and IL-8, which is critical for proper B-cell positioning within the lymph nodes541,542. RHAMM is highly expressed on B-cell CLL (B-CLL) cells and considered as a promising tumor-associated antigen543,544,545. RHAMM-R3 peptide vaccination could trigger an anticancer immune response in CLL patients546. On the other hand, the N-linked glycosylated CD44 variant (CD44v6) forms high-avidity interaction with HA under the stimulation of the CD40 ligand, thus producing an adhesive force to stop CLL mobility, thus retaining CLL cells in specialized compartments of lymph node where they would encounter intensive survival and proliferation-inducing signals542. Moreover, engagement of CD44 would activate the intercellular PI3K/AKT and MAPK/ERK pathways, and increase the expression level of myeloid cell leukemia sequence 1 protein to suppress spontaneous and drug-induced apoptosis in CLL cells547. These biological functions are in consistent with the observations that CD44 is highly expressed and negatively correlated with prognosis in CLL548,549. Zhang et al.550 reported that a humanized monoclonal antibody specific for CD44 (RG7356) was directly cytotoxic for CLL cells both in vitro and in vivo, but had little effects on normal B cells.
Other than CLL, CD44-HA interaction plays similar tissue homing and pro-survival roles in AML and CML. Several CD44 antibodies have been shown to suppress AML cells, such as RG7356, HI44a, A3D8, and ARH460-16-2551,552,553,554. In the Phase I clinical study of RG7356 in AML patients, one complete response (CR) with incomplete platelet recovery (CRp) and one partial response (PR) were recorded, suggesting its potential as an effective therapy for AML551. Inhibition of HA synthesis with 4-MU enhanced the anti-proliferative effect of imatinib and doxorubicin in CML555,556.
The success of Ibrutinib and RG7356 in hematological malignancies suggests that cell–ECM interactions, as well as the signaling network involved in the tissue homing process, are highly workable and druggable targets for combating hematological malignancies.
Conclusion and outlook
Cancer is a complex systematic disease involving constant interactions between cancer cells, the ECM, and other cell types present in the TME. Therefore, ECM stiffness plays a vital role during the process of cancer progression, making it a promising therapeutic target for cancer management.
Firstly, ECM serves as a promising and direct therapeutic target for cancer treatment. One of the most successful cases is the application of TKIs in non-small cell lung cancer with EGFR mutation557 and chronic myeloid leukemia558, which is partially mediated by the blockage of the signal transduction stimulated by DDRs. Therefore, it is urgent for us to identify more cancer-specific ECM targets with the potential of inhibiting cell proliferation, migration, and angiogenesis in the future, thus effectively suppress cancer progression without hurting healthy tissues.
Secondly, intratumoral ECM could be the antigens for the design of tumor vaccine and CAR therapy. Lin et al. showed that vaccination targeting EDA domain of fibronectin enhanced macrophages infiltration, inhibited tumor vasculature, decreased pulmonary metastases559 in a polyoma middle T oncoprotein mouse breast cancer model560. Zhang et al.561 applied CAR macrophage therapy which recognized tumor antigen ErbB2 so that CD147 signaling was simulated and MMPs was increased in a breast cancer mice model. Such CAR therapy softened the ECM, enhanced T cells infiltration, and inhibited tumor growth in mice. However, tumor vaccine and CAR therapy targeting ECM components is relatively more difficult for clinical translation, compared to directly targeting ECM components and related signaling pathways with small molecular compounds or antibodies. The main challenge lies in the scarcity of effective and specific ECM targets in cancer. Another problem is how to accurately predict the host responses to tumor vaccine and CAR therapy in real patients. The complex immune responses in human and animal models in vivo have not been not fully elucidated, which also raises new cautions regarding the safety and workability of translating knowledge obtained in animal models to human patients.
Thirdly, researches of tumor ECM contribute to the development of effective tools for tumor diagnosis and imaging. Traditional imaging heavily relies on radionuclide and single fluorescence, which are limited by the adverse reactions and the accessibility of imaging medium into dense solid tumors. Santimaria et al.562 labeled L19, a monoclonal antibody targeting EDB fibronectin, with 123I for imaging of colorectal cancer, lung cancer, and brain cancer. Novel imaging materials and methods are springing up these days, and they are safer and more convenient for tumor imaging with lower toxicity. For instance, gold nanoparticles (AuNPs), whose diameter ranges from 1 to 100 nm, possess the traits of sensitivity, specificity, and low clearance, and therefore are designed for coupling tumor-specific RNA563,564. Researchers applied second-harmonic generation (SHG) and two-photon excited fluorescence for high contrast imaging of tumors in the deep positions of the body based on the SHG signature of ECM components565. In addition, radiomics based on machine learning algorithms are more and more capable of identifying early malignancies, whose combination with detection reagents targeting intratumoral ECM might be a powerful method for the early detection of cancer. What’s more, noninvasive liquid biopsy testing ECM components or fragments could be another direction strategy for cancer at its early stage. Moon et al.566 performed liquid biopsy of fibronectin on circulating extracellular vesicles for early detection of breast cancer, and the area under the curves reached 0.70.
Fourthly, ECM normalization can serve as a powerful adjuvant for conventional chemotherapy and immunotherapy. After applying the nanoparticles loaded with pheophorbide a (PPa, a photosensitiser that induced immunogenetic cell death) and NLG919 (an inhibitor of indoleamine 2,3-dioxygenase 1 which enhanced the proliferation of cytotoxic T lymphocytes and inhibited regulatory T cells) targeting MMP-2/9 in ECM, the effect of immunotherapy would be greatly enhanced567. The advances of nanotechnology provide opportunities for the spatial and temporal regulation of drug release in TME. Several clinical trials of nanoparticles loaded with albumin-bound (NAB)-paclitaxel is ongoing in early breast cancer568,569, advanced biliary tract cancer570,571, pancreatic cancer572, and so on. Nanoparticle loaded with multiple drugs targeting ECM is now a research hotspot that is promising to achieve low toxicity and high efficiency for their precisely controlled combination of different therapeutic agents. However, some challenges also remain for clinical translation up to now. The most obvious problem lies in the complexity of chemical composition during the in vivo metabolism of such nanoparticles loaded with multiple drugs. Moreover, drug delivery efficiency, tumor-targeting efficacy, drug toxicity, and controllable drug release in time and in place are all issues that need to be considered.
Lastly, there are still Gordian Knots for developing cancer treatment targeting ECM in the field of both scientific research and clinical practices. Currently, there is a lack of materials that can accurately simulate the ECM in vitro. Although Matrigel is widely applied in 3D culture and Gelatin Methacryloyl is frequently used in 3D print, the physical and biological properties, as well as the interaction between different components of ECM is very difficult for an in vitro system to compare with. In addition, ECM stiffness is rarely fully simulated in mice models of solid tumors. Mice have the advantages of quick reproduction, stable genetic background, and low cost. However, it is not very accurate to compare ECM characteristics in tumor-bearing mice whether subcutaneously or orthotopically with real human tumors, due to the great differences in scales and histology. For example, a large proportion of liver cancer is accompanied by cirrhosis, but the subcutaneous and orthotopic liver cancer models, which are commonly used in our drug evaluation, cannot reflect the characteristics of cirrhosis. Dong et al.573 either mixed cell suspension with collagen or induced liver cirrhosis with CCl4 before the inoculation of xenografts, and the growth of the tumor and the signal transduction pattern altered, resembling human liver cancer specimens accompanied with cirrhosis. Furthermore, systematic assessment is still lacking for the comparison regarding the differences of specific ECM components in different cancer, which is actually the fundamental step for developing strategies of specific therapies and sensitive detection. Last but not least, drug libraries are absent which target ECM components, probably due to the lack of accurate in vitro and in vivo models.
Overall, ECM components contribute greatly to the microenvironment of almost every single cell in the human body, and its dysregulation is closely related to the development and progression of many diseases such as cancer. Over the past two decades, there have been many achievements for the successful application of our knowledge regarding ECM dysregulation in the design of anticancer therapy. With the advances and interdisciplinary integration in cell biology, oncology, material science, and nanotechnology, more and more potent anticancer strategies targeting ECM components and ECM-associated signaling pathways would be translated from basic research to clinical usage, eventually improving the life quality of cancer patients.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. https://doi.org/10.3322/caac.21660 (2021).
Naba, A. et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell Proteom. 11, M111 014647 (2012).
Piez, K. A. History of extracellular matrix: a personal view. Matrix Biol. 16, 85–92 (1997).
Gospodarowicz, D., Greenburg, G. & Birdwell, C. R. Determination of cellular shape by the extracellular matrix and its correlation with the control of cellular growth. Cancer Res. 38, 4155–4171 (1978).
Tamkun, J. W. et al. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell 46, 271–282 (1986).
Pytela, R., Pierschbacher, M. D., Ginsberg, M. H., Plow, E. F. & Ruoslahti, E. Platelet membrane glycoprotein IIb/IIIa: member of a family of Arg-Gly-Asp–specific adhesion receptors. Science 231, 1559–1562 (1986).
Kishimoto, T. K., O’Connor, K., Lee, A., Roberts, T. M. & Springer, T. A. Cloning of the beta subunit of the leukocyte adhesion proteins: homology to an extracellular matrix receptor defines a novel supergene family. Cell 48, 681–690 (1987).
Argraves, W. S. et al. Amino acid sequence of the human fibronectin receptor. J. Cell Biol. 105, 1183–1190 (1987).
Hynes, R. O. The emergence of integrins: a personal and historical perspective. Matrix Biol. 23, 333–340 (2004).
Wang, M. L. et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 369, 507–516 (2013).
Cameron, F. & Sanford, M. Ibrutinib: first global approval. Drugs 74, 263–271 (2014).
Trah, J. et al. Integra((R))-dermal regeneration template and split-thickness skin grafting: a therapy approach to correct aplasia cutis congenita and epidermolysis bullosa in Carmi syndrome. Dermatol. Ther. 8, 313–321 (2018).
van der Rest, M. & Garrone, R. Collagen family of proteins. FASEB J. 5, 2814–2823 (1991).
Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 3, a004978 (2011).
Xu, S. et al. The role of collagen in cancer: from bench to bedside. J. Transl. Med. 17, 309 (2019).
Tjin, G. et al. Lysyl oxidases regulate fibrillar collagen remodelling in idiopathic pulmonary fibrosis. Dis. Model. Mech. 10, 1301–1312 (2017).
Eyre, D. R., Weis, M. A. & Wu, J. J. Maturation of collagen Ketoimine cross-links by an alternative mechanism to pyridinoline formation in cartilage. J. Biol. Chem. 285, 16675–16682 (2010).
Gordon, M. K. & Hahn, R. A. Collagens. Cell Tissue Res. 339, 247–257 (2010).
Burgeson, R. E. & Nimni, M. E. Collagen types. Molecular structure and tissue distribution. Clin. Orthop. Relat. Res. 250–272, https://europepmc.org/article/med/1516320 (1992).
Panwar, P. et al. Aging-associated modifications of collagen affect its degradation by matrix metalloproteinases. Matrix Biol. 65, 30–44 (2018).
Cui, N., Hu, M. & Khalil, R. A. Biochemical and biological attributes of matrix metalloproteinases. Prog. Mol. Biol. Transl. Sci. 147, 1–73 (2017).
Visse, R. & Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 92, 827–839 (2003).
Murphy, G. Regulation of the proteolytic disintegrin metalloproteinases, the ‘Sheddases’. Semin. Cell Dev. Biol. 20, 138–145 (2009).
Hamano, Y. et al. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell 3, 589–601 (2003).
Heljasvaara, R. et al. Generation of biologically active endostatin fragments from human collagen XVIII by distinct matrix metalloproteases. Exp. Cell Res. 307, 292–304 (2005).
To, W. S. & Midwood, K. S. Plasma and cellular fibronectin: distinct and independent functions during tissue repair. Fibrogenes. Tissue Repair 4, 21 (2011).
Maurer, L. M., Ma, W. & Mosher, D. F. Dynamic structure of plasma fibronectin. Crit. Rev. Biochem. Mol. Biol. 51, 213–227 (2015).
Adachi, Y. et al. Fibronectin production by cultured human lung fibroblasts in three-dimensional collagen gel culture. Vitr. Cell Dev. Biol. Anim. 34, 203–210 (1998).
Okazaki, J., Mawatari, K., Liu, B. & Kent, K. C. The effect of protein kinase C and its alpha subtype on human vascular smooth muscle cell proliferation, migration and fibronectin production. Surgery 128, 192–197 (2000).
Mao, Y. & Schwarzbauer, J. E. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 24, 389–399 (2005).
Erickson, H. P. Stretching fibronectin. J. Muscle Res. Cell Motil. 23, 575–580 (2002).
White, E. S. & Muro, A. F. Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 63, 538–546 (2011).
Skorstengaard, K., Jensen, M. S., Petersen, T. E. & Magnusson, S. Purification and complete primary structures of the heparin-, cell-, and DNA-binding domains of bovine plasma fibronectin. Eur. J. Biochem. 154, 15–29 (1986).
Pankov, R. & Yamada, K. M. Fibronectin at a glance. J. Cell Sci. 115, 3861–3863 (2002).
Hynes, R. O. in Fibronectins 113–175 (Springer New York, NY, 1990).
Xiao, J. et al. Expression of fibronectin in esophageal squamous cell carcinoma and its role in migration. BMC Cancer 18, 976 (2018).
Dong, Y. et al. Fibronectin type III domain-containing protein 5 promotes proliferation and differentiation of goat adipose-derived stem cells. Res Vet. Sci. 125, 351–359 (2019).
Zhang, W. H., Li, X. L., Guo, Y. & Zhang, Y. Proliferation and osteogenic activity of fibroblasts induced with fibronectin. Braz. J. Med Biol. Res. 50, e6272 (2017).
Wang, Y. & Ni, H. Fibronectin maintains the balance between hemostasis and thrombosis. Cell Mol. Life Sci. 73, 3265–3277 (2016).
Cho, J. & Mosher, D. F. Role of fibronectin assembly in platelet thrombus formation. J. Thromb. Haemost. 4, 1461–1469 (2006).
Astrof, S. & Hynes, R. O. Fibronectins in vascular morphogenesis. Angiogenesis 12, 165–175 (2009).
Kumra, H. et al. Roles of fibronectin isoforms in neonatal vascular development and matrix integrity. PLoS Biol. 16, e2004812 (2018).
Lugano, R. et al. CD93 promotes beta1 integrin activation and fibronectin fibrillogenesis during tumor angiogenesis. J. Clin. Investig. 128, 3280–3297 (2018).
Czop, J. K. Phagocytosis of particulate activators of the alternative complement pathway: effects of fibronectin. Adv. Immunol. 38, 361–398 (1986).
Grinnell, F. Fibronectin and wound healing. J. Cell Biochem. 26, 107–116 (1984).
Valenick, L. V., Hsia, H. C. & Schwarzbauer, J. E. Fibronectin fragmentation promotes alpha4beta1 integrin-mediated contraction of a fibrin-fibronectin provisional matrix. Exp. Cell Res. 309, 48–55 (2005).
Kadler, K. E., Hill, A. & Canty-Laird, E. G. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 20, 495–501 (2008).
Sabatier, L. et al. Fibrillin assembly requires fibronectin. Mol. Biol. Cell 20, 846–858 (2009).
Chung, C. Y. & Erickson, H. P. Glycosaminoglycans modulate fibronectin matrix assembly and are essential for matrix incorporation of tenascin-C. J. Cell Sci. 110, 1413–1419 (1997). Pt 12.
Chiquet-Ehrismann, R. & Tucker, R. P. Tenascins and the importance of adhesion modulation. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a004960 (2011).
McDonald, J. A., Kelley, D. G. & Broekelmann, T. J. Role of fibronectin in collagen deposition: Fab’ to the gelatin-binding domain of fibronectin inhibits both fibronectin and collagen organization in fibroblast extracellular matrix. J. Cell Biol. 92, 485–492 (1982).
Zollinger, A. J. & Smith, M. L. Fibronectin, the extracellular glue. Matrix Biol. 60-61, 27–37 (2017).
Zhu, J. & Clark, R. A. F. Fibronectin at select sites binds multiple growth factors and enhances their activity: expansion of the collaborative ECM-GF paradigm. J. Investig. Dermatol 134, 895–901 (2014).
Dallas, S. L. et al. Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. J. Biol. Chem. 280, 18871–18880 (2005).
Oxford, J. T., Reeck, J. C. & Hardy, M. J. Extracellular matrix in development and disease. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20010205 (2019).
Li, W., Liu, Z., Zhao, C. & Zhai, L. Binding of MMP-9-degraded fibronectin to beta6 integrin promotes invasion via the FAK-Src-related Erk1/2 and PI3K/Akt/Smad-1/5/8 pathways in breast cancer. Oncol. Rep. 34, 1345–1352 (2015).
Lin, F. et al. Fibronectin growth factor-binding domains are required for fibroblast survival. J. Investig. Dermatol. 131, 84–98 (2011).
Martino, M. M. & Hubbell, J. A. The 12th-14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 24, 4711–4721 (2010).
Wijelath, E. S. et al. Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circ. Res. 99, 853–860 (2006).
Hynes, R. O. The extracellular matrix: not just pretty fibrils. Science 326, 1216–1219 (2009).
Xiang, L. et al. The extra domain A of fibronectin increases VEGF-C expression in colorectal carcinoma involving the PI3K/AKT signaling pathway. PLoS ONE 7, e35378 (2012).
Hallmann, R. et al. Expression and function of laminins in the embryonic and mature vasculature. Physiol. Rev. 85, 979–1000 (2005).
Iorio, V., Troughton, L. D. & Hamill, K. J. Laminins: roles and utility in wound repair. Adv. Wound Care 4, 250–263 (2015).
Aumailley, M. et al. A simplified laminin nomenclature. Matrix Biol. 24, 326–332 (2005).
Colognato, H. & Yurchenco, P. D. Form and function: the laminin family of heterotrimers. Dev. Dyn. 218, 213–234 (2000).
McLean, W. H. et al. An unusual N-terminal deletion of the laminin alpha3a isoform leads to the chronic granulation tissue disorder laryngo-onycho-cutaneous syndrome. Hum. Mol. Genet. 12, 2395–2409 (2003).
Durbeej, M. Laminins. Cell Tissue Res. 339, 259–268 (2010).
Balazs, E. A., Laurent, T. C. & Jeanloz, R. W. Nomenclature of hyaluronic acid. Biochem. J. 235, 903 (1986).
Stern, R. Hyaluronan catabolism: a new metabolic pathway. Eur. J. Cell Biol. 83, 317–325 (2004).
Scott, J. E. Supramolecular organization of extracellular matrix glycosaminoglycans, in vitro and in the tissues. FASEB J. 6, 2639–2645 (1992).
Henry, C. B. & Duling, B. R. Permeation of the luminal capillary glycocalyx is determined by hyaluronan. Am. J. Physiol. 277, H508–514 (1999).
Grammatikakis, N. et al. A novel glycosaminoglycan-binding protein is the vertebrate homologue of the cell cycle control protein, Cdc37. J. Biol. Chem. 270, 16198–16205 (1995).
Deb, T. B. & Datta, K. Molecular cloning of human fibroblast hyaluronic acid-binding protein confirms its identity with P-32, a protein co-purified with splicing factor SF2. Hyaluronic acid-binding protein as P-32 protein, co-purified with splicing factor SF2. J. Biol. Chem. 271, 2206–2212 (1996).
Huang, L., Grammatikakis, N., Yoneda, M., Banerjee, S. D. & Toole, B. P. Molecular characterization of a novel intracellular hyaluronan-binding protein. J. Biol. Chem. 275, 29829–29839 (2000).
Volpi, N. Therapeutic applications of glycosaminoglycans. Curr. Med. Chem. 13, 1799–1810 (2006).
Kinoshita-Toyoda, A. et al. Structural determination of five novel tetrasaccharides containing 3-O-sulfated D-glucuronic acid and two rare oligosaccharides containing a beta-D-glucose branch isolated from squid cartilage chondroitin sulfate E. Biochemistry 43, 11063–11074 (2004).
Bishnoi, M., Jain, A., Hurkat, P. & Jain, S. K. Chondroitin sulphate: a focus on osteoarthritis. Glycoconj. J. 33, 693–705 (2016).
Funderburgh, J. L. Keratan sulfate: structure, biosynthesis, and function. Glycobiology 10, 951–958 (2000).
Li, J. P. & Kusche-Gullberg, M. Heparan sulfate: biosynthesis, structure, and function. Int. Rev. Cell Mol. Biol. 325, 215–273 (2016).
Laremore, T. N., Zhang, F., Dordick, J. S., Liu, J. & Linhardt, R. J. Recent progress and applications in glycosaminoglycan and heparin research. Curr. Opin. Chem. Biol. 13, 633–640 (2009).
Nandini, C. D. & Sugahara, K. Role of the sulfation pattern of chondroitin sulfate in its biological activities and in the binding of growth factors. Adv. Pharm. 53, 253–279 (2006).
Jackson, R. L., Busch, S. J. & Cardin, A. D. Glycosaminoglycans: molecular properties, protein interactions, and role in physiological processes. Physiol. Rev. 71, 481–539 (1991).
Mizumoto, S., Yamada, S. & Sugahara, K. Molecular interactions between chondroitin-dermatan sulfate and growth factors/receptors/matrix proteins. Curr. Opin. Struct. Biol. 34, 35–42 (2015).
Dick, M. K., Miao, J. H. & Limaiem, F. in StatPearls (2020).
Kalluri, R. & Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401 (2006).
In Mesenchymal Stem Cells and Skeletal Regeneration (eds Elena A. Jones, Xuebin Yang, Peter Giannoudis, & Dennis McGonagle) 49–66 (Academic Press, 2013).
Jones, E. A., Yang, X., Giannoudis, P. & McGonagle, D. in Mesenchymal Stem Cells and Skeletal Regeneration (eds Elena A. Jones, Xuebin Yang, Peter Giannoudis, & Dennis McGonagle) 33–38 (Academic Press, 2013).
Nazemi, M. & Rainero, E. Cross-talk between the tumor microenvironment, extracellular matrix, and cell metabolism in cancer. Front. Oncol. 10, 239 (2020).
Louault, K. et al. Interactions between cancer-associated fibroblasts and tumor cells promote MCL-1 dependency in estrogen receptor-positive breast cancers. Oncogene 38, 3261–3273 (2019).
Ricci, F., Kern, S. E., Hruban, R. H. & Iacobuzio-Donahue, C. A. Stromal responses to carcinomas of the pancreas: juxtatumoral gene expression conforms to the infiltrating pattern and not the biologic subtype. Cancer Biol. Ther. 4, 302–307 (2005).
Lynch, M. D. & Watt, F. M. Fibroblast heterogeneity: implications for human disease. J. Clin. Investig. 128, 26–35 (2018).
Voloshenyuk, T. G., Landesman, E. S., Khoutorova, E., Hart, A. D. & Gardner, J. D. Induction of cardiac fibroblast lysyl oxidase by TGF-beta1 requires PI3K/Akt, Smad3, and MAPK signaling. Cytokine 55, 90–97 (2011).
Brilla, C. G., Maisch, B., Zhou, G. & Weber, K. T. Hormonal regulation of cardiac fibroblast function. Eur. Heart J. 16, 45–50 (1995). Suppl C.
Siwik, D. A., Chang, D. L. & Colucci, W. S. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ. Res. 86, 1259–1265 (2000).
Bachhuka, A., Hayball, J., Smith, L. E. & Vasilev, K. Effect of surface chemical functionalities on collagen deposition by primary human dermal fibroblasts. ACS Appl. Mater. Interfaces 7, 23767–23775 (2015).
Balestrini, J. L. & Billiar, K. L. Equibiaxial cyclic stretch stimulates fibroblasts to rapidly remodel fibrin. J. Biomech. 39, 2983–2990 (2006).
Husse, B., Briest, W., Homagk, L., Isenberg, G. & Gekle, M. Cyclical mechanical stretch modulates expression of collagen I and collagen III by PKC and tyrosine kinase in cardiac fibroblasts. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1898–1907 (2007).
Yang, G., Crawford, R. C. & Wang, J. H. Proliferation and collagen production of human patellar tendon fibroblasts in response to cyclic uniaxial stretching in serum-free conditions. J. Biomech. 37, 1543–1550 (2004).
Mayes, A. E. & Holyoak, C. D. Repeat mild heat shock increases dermal fibroblast activity and collagen production. Rejuvenation Res. 11, 461–465 (2008).
Gkretsi, V. & Stylianopoulos, T. Cell adhesion and matrix stiffness: coordinating cancer cell invasion and metastasis. Front. Oncol. 8, 145 (2018).
Kalli, M. & Stylianopoulos, T. Defining the role of solid stress and matrix stiffness in cancer cell proliferation and metastasis. Front. Oncol. 8, 55 (2018).
Liu, T., Babaniyi, O. A., Hall, T. J., Barbone, P. E. & Oberai, A. A. Noninvasive in-vivo quantification of mechanical heterogeneity of invasive breast carcinomas. PLoS ONE 10, e0130258 (2015).
Chang, J. M. et al. Stiffness of tumours measured by shear-wave elastography correlated with subtypes of breast cancer. Eur. Radiol. 23, 2450–2458 (2013).
Lu, Q. et al. Hepatocellular carcinoma: stiffness value and ratio to discriminate malignant from benign focal liver lesions. Radiology 275, 880–888 (2015).
Rice, A. J. et al. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 6, e352 (2017).
Erkan, M. et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat. Rev. Gastroenterol. Hepatol. 9, 454–467 (2012).
Whatcott, C. J. et al. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin. Cancer Res. 21, 3561–3568 (2015).
Plodinec, M. et al. The nanomechanical signature of breast cancer. Nat. Nanotechnol. 7, 757–765 (2012).
Insua-Rodriguez, J. & Oskarsson, T. The extracellular matrix in breast cancer. Adv. Drug Deliv. Rev. 97, 41–55 (2016).
Kalluri, R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 3, 422–433 (2003).
Giussani, M., Merlino, G., Cappelletti, V., Tagliabue, E. & Daidone, M. G. Tumor-extracellular matrix interactions: Identification of tools associated with breast cancer progression. Semin. Cancer Biol. 35, 3–10 (2015).
Mohammadi, H. & Sahai, E. Mechanisms and impact of altered tumour mechanics. Nat. Cell Biol. 20, 766–774 (2018).
Stylianopoulos, T. et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc. Natl Acad. Sci. USA 109, 15101–15108 (2012).
Stylianopoulos, T. et al. Coevolution of solid stress and interstitial fluid pressure in tumors during progression: implications for vascular collapse. Cancer Res. 73, 3833–3841 (2013).
Fang, S. et al. Clinical significance and biological role of cancer-derived Type I collagen in lung and esophageal cancers. Thorac. Cancer 10, 277–288 (2019).
Anttila, M. A. et al. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res. 60, 150–155 (2000).
Auvinen, P. et al. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am. J. Pathol. 156, 529–536 (2000).
Ropponen, K. et al. Tumor cell-associated hyaluronan as an unfavorable prognostic factor in colorectal cancer. Cancer Res. 58, 342–347 (1998).
Lipponen, P. et al. High stromal hyaluronan level is associated with poor differentiation and metastasis in prostate cancer. Eur. J. Cancer 37, 849–856 (2001).
Setala, L. P. et al. Hyaluronan expression in gastric cancer cells is associated with local and nodal spread and reduced survival rate. Br. J. Cancer 79, 1133–1138 (1999).
Zhao, L. et al. LOX inhibition downregulates MMP-2 and MMP-9 in gastric cancer tissues and cells. J. Cancer 10, 6481–6490 (2019).
Zucker, S. & Vacirca, J. Role of matrix metalloproteinases (MMPs) in colorectal cancer. Cancer Metastasis Rev. 23, 101–117 (2004).
Jacobson, A., Brinck, J., Briskin, M. J., Spicer, A. P. & Heldin, P. Expression of human hyaluronan synthases in response to external stimuli. Biochem. J. 348, 29–35 (2000). Pt 1.
Pienimaki, J. P. et al. Epidermal growth factor activates hyaluronan synthase 2 in epidermal keratinocytes and increases pericellular and intracellular hyaluronan. J. Biol. Chem. 276, 20428–20435 (2001).
Karvinen, S. et al. Keratinocyte growth factor stimulates migration and hyaluronan synthesis in the epidermis by activation of keratinocyte hyaluronan synthases 2 and 3. J. Biol. Chem. 278, 49495–49504 (2003).
Pasonen-Seppanen, S. et al. EGF upregulates, whereas TGF-beta downregulates, the hyaluronan synthases Has2 and Has3 in organotypic keratinocyte cultures: correlations with epidermal proliferation and differentiation. J. Investig. Dermatol. 120, 1038–1044 (2003).
Hiltunen, E. L. et al. Elevated hyaluronan concentration without hyaluronidase activation in malignant epithelial ovarian tumors. Cancer Res. 62, 6410–6413 (2002).
Gobin, E. et al. A pan-cancer perspective of matrix metalloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer 19, 581 (2019).
Terra, M. et al. Tumor-derived TGFbeta alters the ability of plasmacytoid dendritic cells to respond to innate immune signaling. Cancer Res. 78, 3014–3026 (2018).
Ignotz, R. A. & Massague, J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 261, 4337–4345 (1986).
Kuzet, S. E. & Gaggioli, C. Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res. 365, 607–619 (2016).
Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 4, 540–550 (2004).
Donzelli, S. et al. Expression of ID4 protein in breast cancer cells induces reprogramming of tumour-associated macrophages. Breast Cancer Res. 20, 59 (2018).
Tan, B. et al. Inhibition of Rspo-Lgr4 facilitates checkpoint blockade therapy by switching macrophage polarization. Cancer Res. 78, 4929–4942 (2018).
Purcell, J. W. et al. LRRC15 is a novel mesenchymal protein and stromal target for antibody-drug conjugates. Cancer Res. 78, 4059–4072 (2018).
Zhao, P. et al. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem. Sci. 9, 2674–2689 (2018).
Somasundaram, R. & Schuppan, D. Type I, II, III, IV, V, and VI collagens serve as extracellular ligands for the isoforms of platelet-derived growth factor (AA, BB, and AB). J. Biol. Chem. 271, 26884–26891 (1996).
Thompson, J. A. et al. Site-directed neovessel formation in vivo. Science 241, 1349–1352 (1988).
Paralkar, V. M., Vukicevic, S. & Reddi, A. H. Transforming growth factor beta type 1 binds to collagen IV of basement membrane matrix: implications for development. Dev. Biol. 143, 303–308 (1991).
Jones, J. I., Gockerman, A., Busby, W. H. Jr, Camacho-Hubner, C. & Clemmons, D. R. Extracellular matrix contains insulin-like growth factor binding protein-5: potentiation of the effects of IGF-I. J. Cell Biol. 121, 679–687 (1993).
Capurro, M. I., Xiang, Y. Y., Lobe, C. & Filmus, J. Glypican-3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res. 65, 6245–6254, (2005).
Capurro, M., Martin, T., Shi, W. & Filmus, J. Glypican-3 binds to Frizzled and plays a direct role in the stimulation of canonical Wnt signaling. J. Cell Sci. 127, 1565–1575, (2014).
Imai, K., Hiramatsu, A., Fukushima, D., Pierschbacher, M. D. & Okada, Y. Degradation of decorin by matrix metalloproteinases: identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem. J. 322, 809–814 (1997). (Pt 3).
Maeda, S., Dean, D. D., Gomez, R., Schwartz, Z. & Boyan, B. D. The first stage of transforming growth factor beta1 activation is release of the large latent complex from the extracellular matrix of growth plate chondrocytes by matrix vesicle stromelysin-1 (MMP-3). Calcif. Tissue Int. 70, 54–65 (2002).
Hawinkels, L. J. et al. VEGF release by MMP-9 mediated heparan sulphate cleavage induces colorectal cancer angiogenesis. Eur. J. Cancer 44, 1904–1913 (2008).
Alonso-Nocelo, M. et al. Matrix stiffness and tumor-associated macrophages modulate epithelial to mesenchymal transition of human adenocarcinoma cells. Biofabrication 10, 035004 (2018).
Tasdemir, N. et al. Comprehensive phenotypic characterization of human invasive lobular carcinoma cell lines in 2D and 3D cultures. Cancer Res. 78, 6209–6222 (2018).
Zhang, M. et al. Soft fibrin matrix downregulates DAB2IP to promote Nanog-dependent growth of colon tumor-repopulating cells. Cell Death Dis. 10, 151 (2019).
Zhang, R. et al. Increased matrix stiffness promotes tumor progression of residual hepatocellular carcinoma after insufficient heat treatment. Cancer Sci. 108, 1778–1786 (2017).
Levental, K. R. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906 (2009).
Serrano, I., McDonald, P. C., Lock, F., Muller, W. J. & Dedhar, S. Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nat. Commun. 4, 2976 (2013).
Sabra, H. et al. beta1 integrin-dependent Rac/group I PAK signaling mediates YAP activation of Yes-associated protein 1 (YAP1) via NF2/merlin. J. Biol. Chem. 292, 19179–19197 (2017).
Mizuno, T. et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 31, 5117–5122 (2012).
Fung, J., Lai, C. L., Seto, W. K., Wong, D. K. & Yuen, M. F. Prognostic significance of liver stiffness for hepatocellular carcinoma and mortality in HBeAg-negative chronic hepatitis B. J. Viral Hepat. 18, 738–744 (2011).
Paszek, M. J. et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254 (2005).
Samuel, M. S. et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 19, 776–791 (2011).
Shi, Q. & Boettiger, D. A novel mode for integrin-mediated signaling: tethering is required for phosphorylation of FAK Y397. Mol. Biol. Cell 14, 4306–4315 (2003).
Lawson, C. D. & Burridge, K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 5, e27958 (2014).
Friedland, J. C., Lee, M. H. & Boettiger, D. Mechanically activated integrin switch controls alpha5beta1 function. Science 323, 642–644 (2009).
Rubashkin, M. G. et al. Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylinositol (3,4,5)-triphosphate. Cancer Res. 74, 4597–4611 (2014).
Gkretsi, V., Stylianou, A., Louca, M. & Stylianopoulos, T. Identification of Ras suppressor-1 (RSU-1) as a potential breast cancer metastasis biomarker using a three-dimensional in vitro approach. Oncotarget 8, 27364–27379 (2017).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Folkman, J. How is blood vessel growth regulated in normal and neoplastic tissue? G.H.A. Clowes memorial Award lecture. Cancer Res. 46, 467–473 (1986).
Baluk, P., Morikawa, S., Haskell, A., Mancuso, M. & McDonald, D. M. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 163, 1801–1815 (2003).
Ghajar, C. M., Blevins, K. S., Hughes, C. C., George, S. C. & Putnam, A. J. Mesenchymal stem cells enhance angiogenesis in mechanically viable prevascularized tissues via early matrix metalloproteinase upregulation. Tissue Eng. 12, 2875–2888 (2006).
Shen, C. J. et al. Decreased cell adhesion promotes angiogenesis in a Pyk2-dependent manner. Exp. Cell Res. 317, 1860–1871 (2011).
Cross, V. L. et al. Dense type I collagen matrices that support cellular remodeling and microfabrication for studies of tumor angiogenesis and vasculogenesis in vitro. Biomaterials 31, 8596–8607 (2010).
Edgar, L. T., Underwood, C. J., Guilkey, J. E., Hoying, J. B. & Weiss, J. A. Extracellular matrix density regulates the rate of neovessel growth and branching in sprouting angiogenesis. PLoS ONE 9, e85178 (2014).
Bordeleau, F. et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl Acad. Sci. USA 114, 492–497 (2017).
Kutys, M. L. & Chen, C. S. Forces and mechanotransduction in 3D vascular biology. Curr. Opin. Cell Biol. 42, 73–79 (2016).
Hahn, C. & Schwartz, M. A. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 10, 53–62 (2009).
Lakshmikanthan, S. et al. Rap1 promotes endothelial mechanosensing complex formation, NO release and normal endothelial function. EMBO Rep. 16, 628–637 (2015).
Vaupel, P., Mayer, A. & Hockel, M. Tumor hypoxia and malignant progression. Methods Enzymol. 381, 335–354 (2004).
Semenza, G. L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 9, 47–71 (2014).
Zhong, H. et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 59, 5830–5835 (1999).
Talks, K. L. et al. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 157, 411–421 (2000).
Pakravan, K. et al. MicroRNA-100 shuttled by mesenchymal stem cell-derived exosomes suppresses in vitro angiogenesis through modulating the mTOR/HIF-1alpha/VEGF signaling axis in breast cancer cells. Cell Oncol. 40, 457–470 (2017).
Oh, S. Y. et al. Clinicopathologic significance of HIF-1alpha, p53, and VEGF expression and preoperative serum VEGF level in gastric cancer. BMC Cancer 8, 123 (2008).
Chen, M. C., Lee, C. F., Huang, W. H. & Chou, T. C. Magnolol suppresses hypoxia-induced angiogenesis via inhibition of HIF-1alpha/VEGF signaling pathway in human bladder cancer cells. Biochem. Pharm. 85, 1278–1287 (2013).
Wong, C., Wellman, T. L. & Lounsbury, K. M. VEGF and HIF-1alpha expression are increased in advanced stages of epithelial ovarian cancer. Gynecol. Oncol. 91, 513–517 (2003).
Netti, P. A., Berk, D. A., Swartz, M. A., Grodzinsky, A. J. & Jain, R. K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 60, 2497–2503 (2000).
Rahbari, N. N. et al. Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases. Sci. Transl. Med. 8, 360ra135 (2016).
Najafi, M., Farhood, B. & Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell Biochem. 120, 2782–2790 (2019).
Piersma, B., Hayward, M. K. & Weaver, V. M. Fibrosis and cancer: a strained relationship. Biochim. Biophys. Acta Rev. Cancer 1873, 188356 (2020).
Ramjiawan, R. R., Griffioen, A. W. & Duda, D. G. Anti-angiogenesis for cancer revisited: is there a role for combinations with immunotherapy? Angiogenesis 20, 185–204 (2017).
Shibue, T. & Weinberg, R. A. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 14, 611–629 (2017).
Reya, T., Morrison, S. J., Clarke, M. F. & Weissman, I. L. Stem cells, cancer, and cancer stem cells. Nature 414, 105–111 (2001).
Al-Hajj, M., Becker, M. W., Wicha, M., Weissman, I. & Clarke, M. F. Therapeutic implications of cancer stem cells. Curr. Opin. Genet. Dev. 14, 43–47 (2004).
Abdollahi, A. et al. Inhibition of alpha(v)beta3 integrin survival signaling enhances antiangiogenic and antitumor effects of radiotherapy. Clin. Cancer Res. 11, 6270–6279 (2005).
Cordes, N., Seidler, J., Durzok, R., Geinitz, H. & Brakebusch, C. beta1-integrin-mediated signaling essentially contributes to cell survival after radiation-induced genotoxic injury. Oncogene 25, 1378–1390 (2006).
Meineke, V. et al. Ionizing radiation modulates cell surface integrin expression and adhesion of COLO-320 cells to collagen and fibronectin in vitro. Strahlenther. Onkol. 178, 709–714 (2002).
Onoda, J. M., Piechocki, M. P. & Honn, K. V. Radiation-induced increase in expression of the alpha IIb beta 3 integrin in melanoma cells: effects on metastatic potential. Radiat. Res. 130, 281–288 (1992).
Cordes, N., Blaese, M. A., Meineke, V. & Van Beuningen, D. Ionizing radiation induces up-regulation of functional beta1-integrin in human lung tumour cell lines in vitro. Int. J. Radiat. Biol. 78, 347–357 (2002).
Cordes, N. et al. Human pancreatic tumor cells are sensitized to ionizing radiation by knockdown of caveolin-1. Oncogene 26, 6851–6862 (2007).
Cordes, N., Hansmeier, B., Beinke, C., Meineke, V. & van Beuningen, D. Irradiation differentially affects substratum-dependent survival, adhesion, and invasion of glioblastoma cell lines. Br. J. Cancer 89, 2122–2132 (2003).
Park, C. C., Zhang, H. J., Yao, E. S., Park, C. J. & Bissell, M. J. Beta1 integrin inhibition dramatically enhances radiotherapy efficacy in human breast cancer xenografts. Cancer Res. 68, 4398–4405 (2008).
Johnson, G. E., Ivanov, V. N. & Hei, T. K. Radiosensitization of melanoma cells through combined inhibition of protein regulators of cell survival. Apoptosis 13, 790–802 (2008).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
Seshadri, R., Kutlaca, R. J., Trainor, K., Matthews, C. & Morley, A. A. Mutation rate of normal and malignant human lymphocytes. Cancer Res. 47, 407–409 (1987).
Tamiello, C. et al. Soft substrates normalize nuclear morphology and prevent nuclear rupture in fibroblasts from a laminopathy patient with compound heterozygous LMNA mutations. Nucleus 4, 61–73 (2013).
Maciejowski, J., Li, Y., Bosco, N., Campbell, P. J. & de Lange, T. Chromothripsis and kataegis induced by telomere crisis. Cell 163, 1641–1654 (2015).
Yang, Y. L., Leone, L. M. & Kaufman, L. J. Elastic moduli of collagen gels can be predicted from two-dimensional confocal microscopy. Biophys. J. 97, 2051–2060 (2009).
Swift, J. et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341, 1240104 (2013).
Harada, T. et al. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 204, 669–682 (2014).
Irianto, J. et al. Nuclear constriction segregates mobile nuclear proteins away from chromatin. Mol. Biol. Cell 27, 4011–4020 (2016).
Caruana, I., Simula, L., Locatelli, F. & Campello, S. T lymphocytes against solid malignancies: winning ways to defeat tumours. Cell Stress 2, 200–212 (2018).
Luo, X. et al. Lymphocytes perform reverse adhesive haptotaxis mediated by LFA-1 integrins. J. Cell Sci. https://doi.org/10.1242/jcs.242883 (2020).
Ferrero, E. et al. Tumor-driven matrix invasion by infiltrating lymphocytes: involvement of the alpha1 integrin I-domain. Eur. J. Immunol. 28, 2530–2536 (1998).
Hauzenberger, D., Klominek, J. & Sundqvist, K. G. Functional specialization of fibronectin-binding beta 1-integrins in T lymphocyte migration. J. Immunol. 153, 960–971 (1994).
Pruitt, H. C. et al. Collagen fiber structure guides 3D motility of cytotoxic T lymphocytes. Matrix Biol. 85-86, 147–159 (2020).
Bougherara, H. et al. Real-Time Imaging of Resident T Cells in Human Lung and Ovarian Carcinomas Reveals How Different Tumor Microenvironments Control T Lymphocyte Migration. Front Immunol. 6, 500 (2015).
Funada, Y. et al. Prognostic significance of CD8+ T cell and macrophage peritumoral infiltration in colorectal cancer. Oncol. Rep. 10, 309–313 (2003).
Salmon, H. et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 122, 899–910 (2012).
Johnston, R. J. et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 574, 565–570 (2019).
Castermans, K. & Griffioen, A. W. Tumor blood vessels, a difficult hurdle for infiltrating leukocytes. Biochim. Biophys. Acta 1776, 160–174 (2007).
Peng, D. H. et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8(+) T cell exhaustion. Nat. Commun. 11, 4520 (2020).
Bollyky, P. L. et al. Intact extracellular matrix and the maintenance of immune tolerance: high molecular weight hyaluronan promotes persistence of induced CD4+CD25+ regulatory T cells. J. Leukoc. Biol. 86, 567–572 (2009).
Miyazawa, A. et al. Regulation of PD-L1 expression by matrix stiffness in lung cancer cells. Biochem. Biophys. Res. Commun. 495, 2344–2349 (2018).
Long, K. B. et al. CAR T cell therapy of non-hematopoietic malignancies: detours on the road to clinical success. Front. Immunol. 9, 2740 (2018).
Jain, R. K. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 9, 253–266 (1990).
Lewis, C. E. & Pollard, J. W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 66, 605–612 (2006).
Kaplan, G. In vitro differentiation of human monocytes. Monocytes cultured on glass are cytotoxic to tumor cells but monocytes cultured on collagen are not. J. Exp. Med.157, 2061–2072 (1983).
Kim, H., Cha, J., Jang, M. & Kim, P. Hyaluronic acid-based extracellular matrix triggers spontaneous M2-like polarity of monocyte/macrophage. Biomater. Sci. 7, 2264–2271 (2019).
Perri, R. T. et al. Fibronectin enhances in vitro monocyte-macrophage-mediated tumoricidal activity. Blood 60, 430–435 (1982).
Rygiel, T. P., Stolte, E. H., de Ruiter, T., van de Weijer, M. L. & Meyaard, L. Tumor-expressed collagens can modulate immune cell function through the inhibitory collagen receptor LAIR-1. Mol. Immunol. 49, 402–406 (2011).
Leitinger, B. Transmembrane collagen receptors. Annu. Rev. Cell Dev. Biol. 27, 265–290 (2011).
Takagi, J., Strokovich, K., Springer, T. A. & Walz, T. Structure of integrin alpha5beta1 in complex with fibronectin. EMBO J. 22, 4607–4615 (2003).
Huang, J. et al. Platelet integrin alphaIIbbeta3: signal transduction, regulation, and its therapeutic targeting. J. Hematol. Oncol. 12, 26 (2019).
Beaulieu, J. F. Integrin α6β4 in colorectal cancer: expression, regulation, functional alterations and use as a biomarker. Cancers 12, 41 (2019).
Choi, Y., Kim, E., Lee, Y., Han, M. H. & Kang, I. C. Site-specific inhibition of integrin alpha v beta 3-vitronectin association by a ser-asp-val sequence through an Arg-Gly-Asp-binding site of the integrin. Proteomics 10, 72–80 (2010).
Pollan, S. G. et al. Regulation of inside-out beta1-integrin activation by CDCP1. Oncogene 37, 2817–2836 (2018).
Downey-Biechler, C., Craig, D. H., More, S. K. & Basson, M. D. Inside-out signaling through FAK-integrin axis may regulate circulating cancer cell metastatic adhesion. Proc. Natl Acad. Sci. USA 116, 19795–19796 (2019).
Zheng, D. Q., Woodard, A. S., Tallini, G. & Languino, L. R. Substrate specificity of alpha(v)beta(3) integrin-mediated cell migration and phosphatidylinositol 3-kinase/AKT pathway activation. J. Biol. Chem. 275, 24565–24574 (2000).
Desgrosellier, J. S. et al. An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat. Med. 15, 1163–1169 (2009).
Thomas, G. J., Nystrom, M. L. & Marshall, J. F. Alphavbeta6 integrin in wound healing and cancer of the oral cavity. J. Oral. Pathol. Med. 35, 1–10 (2006).
Regezi, J. A., Ramos, D. M., Pytela, R., Dekker, N. P. & Jordan, R. C. Tenascin and beta 6 integrin are overexpressed in floor of mouth in situ carcinomas and invasive squamous cell carcinomas. Oral. Oncol. 38, 332–336 (2002).
Jones, J., Watt, F. M. & Speight, P. M. Changes in the expression of alpha v integrins in oral squamous cell carcinomas. J. Oral. Pathol. Med. 26, 63–68 (1997).
Li, H. X. et al. Expression of alphavbeta6 integrin and collagen fibre in oral squamous cell carcinoma: association with clinical outcomes and prognostic implications. J. Oral. Pathol. Med. 42, 547–556 (2013).
Allen, M. D. et al. Altered microenvironment promotes progression of preinvasive breast cancer: myoepithelial expression of alphavbeta6 integrin in DCIS identifies high-risk patients and predicts recurrence. Clin. Cancer Res. 20, 344–357 (2014).
Kawashima, A. et al. Expression of alphav integrin family in gastric carcinomas: increased alphavbeta6 is associated with lymph node metastasis. Pathol. Res. Pract. 199, 57–64 (2003).
Zhang, Z. Y. et al. Integrin alphanvbeta6 acts as a prognostic indicator in gastric carcinoma. Clin. Oncol. 20, 61–66 (2008).
Bates, R. C. et al. Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J. Clin. Investig. 115, 339–347 (2005).
Sipos, B. et al. Immunohistochemical screening for beta6-integrin subunit expression in adenocarcinomas using a novel monoclonal antibody reveals strong up-regulation in pancreatic ductal adenocarcinomas in vivo and in vitro. Histopathology 45, 226–236 (2004).
Hazelbag, S. et al. Overexpression of the alpha v beta 6 integrin in cervical squamous cell carcinoma is a prognostic factor for decreased survival. J. Pathol. 212, 316–324 (2007).
Saha, A. et al. High-resolution in vivo imaging of breast cancer by targeting the pro-invasive integrin alphavbeta6. J. Pathol. 222, 52–63 (2010).
Zhuang, Z. et al. Clinical significance of integrin alphavbeta6 expression effects on gastric carcinoma invasiveness and progression via cancer-associated fibroblasts. Med. Oncol. 30, 580 (2013).
Li, Z. et al. Integrin beta6 acts as an unfavorable prognostic indicator and promotes cellular malignant behaviors via ERK-ETS1 pathway in pancreatic ductal adenocarcinoma (PDAC). Tumour Biol. 37, 5117–5131 (2016).
Ahmed, N., Riley, C., Rice, G. E., Quinn, M. A. & Baker, M. S. Alpha(v)beta(6) integrin-A marker for the malignant potential of epithelial ovarian cancer. J. Histochem Cytochem 50, 1371–1380 (2002).
Wang, B. et al. SDF-1/CXCR4 axis promotes directional migration of colorectal cancer cells through upregulation of integrin alphavbeta6. Carcinogenesis 35, 282–291 (2014).
Yang, G. Y. et al. Integrin alpha v beta 6 mediates the potential for colon cancer cells to colonize in and metastasize to the liver. Cancer Sci. 99, 879–887 (2008).
Li, Z. et al. Integrin beta6 serves as an immunohistochemical marker for lymph node metastasis and promotes cell invasiveness in cholangiocarcinoma. Sci. Rep. 6, 30081 (2016).
Patsenker, E. et al. The alphavbeta6 integrin is a highly specific immunohistochemical marker for cholangiocarcinoma. J. Hepatol. 52, 362–369 (2010).
Elayadi, A. N. et al. A peptide selected by biopanning identifies the integrin alphavbeta6 as a prognostic biomarker for nonsmall cell lung cancer. Cancer Res. 67, 5889–5895 (2007).
Itoh, Y. Discoidin domain receptors: Microenvironment sensors that promote cellular migration and invasion. Cell Adhes. Migr. 12, 378–385 (2018).
Leitinger, B. Discoidin domain receptor functions in physiological and pathological conditions. Int. Rev. Cell Mol. Biol. 310, 39–87 (2014).
Vogel, W., Gish, G. D., Alves, F. & Pawson, T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1, 13–23 (1997).
Barker, K. T. et al. Expression patterns of the novel receptor-like tyrosine kinase, DDR, in human breast tumours. Oncogene 10, 569–575 (1995).
Turashvili, G. et al. Novel markers for differentiation of lobular and ductal invasive breast carcinomas by laser microdissection and microarray analysis. BMC Cancer 7, 55 (2007).
Turashvili, G. et al. Novel immunohistochemical markers for the differentiation of lobular and ductal invasive breast carcinomas. Biomed. Pap. 151, 59–64 (2007).
Hidalgo-Carcedo, C. et al. Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat. Cell Biol. 13, 49–58 (2011).
Ford, C. E. et al. Expression and mutation analysis of the discoidin domain receptors 1 and 2 in non-small cell lung carcinoma. Br. J. Cancer 96, 808–814 (2007).
Rikova, K. et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 131, 1190–1203 (2007).
Yang, S. H. et al. Discoidin domain receptor 1 is associated with poor prognosis of non-small cell lung carcinomas. Oncol. Rep. 24, 311–319 (2010).
Valencia, K. et al. Inhibition of collagen receptor discoidin domain receptor-1 (DDR1) reduces cell survival, homing, and colonization in lung cancer bone metastasis. Clin. Cancer Res. 18, 969–980 (2012).
Ram, R. et al. Discoidin domain receptor-1a (DDR1a) promotes glioma cell invasion and adhesion in association with matrix metalloproteinase-2. J. Neurooncol. 76, 239–248 (2006).
Laval, S. et al. Isolation and characterization of an epithelial-specific receptor tyrosine kinase from an ovarian cancer cell line. Cell Growth Differ. 5, 1173–1183 (1994).
Alves, F. et al. Distinct structural characteristics of discoidin I subfamily receptor tyrosine kinases and complementary expression in human cancer. Oncogene 10, 609–618 (1995).
Heinzelmann-Schwarz, V. A. et al. Overexpression of the cell adhesion molecules DDR1, Claudin 3, and Ep-CAM in metaplastic ovarian epithelium and ovarian cancer. Clin. Cancer Res. 10, 4427–4436 (2004).
Quan, J., Yahata, T., Adachi, S., Yoshihara, K. & Tanaka, K. Identification of receptor tyrosine kinase, discoidin domain receptor 1 (DDR1), as a potential biomarker for serous ovarian cancer. Int. J. Mol. Sci. 12, 971–982 (2011).
Colas, E. et al. Molecular markers of endometrial carcinoma detected in uterine aspirates. Int J. Cancer 129, 2435–2444 (2011).
Nemoto, T., Ohashi, K., Akashi, T., Johnson, J. D. & Hirokawa, K. Overexpression of protein tyrosine kinases in human esophageal cancer. Pathobiology 65, 195–203 (1997).
Shen, Q. et al. Role of microRNA-199a-5p and discoidin domain receptor 1 in human hepatocellular carcinoma invasion. Mol. Cancer 9, 227 (2010).
Gu, T. L. et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS ONE 6, e15640 (2011).
Shimada, K. et al. Prostate cancer antigen-1 contributes to cell survival and invasion though discoidin receptor 1 in human prostate cancer. Cancer Sci. 99, 39–45 (2008).
Chua, H. H. et al. Upregulation of discoidin domain receptor 2 in nasopharyngeal carcinoma. Head Neck 30, 427–436 (2008).
Rodrigues, R. et al. Comparative genomic hybridization, BRAF, RAS, RET, and oligo-array analysis in aneuploid papillary thyroid carcinomas. Oncol. Rep. 18, 917–926 (2007).
Renne, C., Willenbrock, K., Kuppers, R., Hansmann, M. L. & Brauninger, A. Autocrine- and paracrine-activated receptor tyrosine kinases in classic Hodgkin lymphoma. Blood 105, 4051–4059 (2005).
Willenbrock, K. et al. Common features and differences in the transcriptome of large cell anaplastic lymphoma and classical Hodgkin’s lymphoma. Haematologica 91, 596–604 (2006).
Tomasson, M. H. et al. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 111, 4797–4808 (2008).
Huo, Y. et al. High expression of DDR1 is associated with the poor prognosis in Chinese patients with pancreatic ductal adenocarcinoma. J. Exp. Clin. Cancer Res. 34, 88 (2015).
Yuge, R. et al. Silencing of discoidin domain receptor-1 (DDR1) concurrently inhibits multiple steps of metastasis cascade in gastric cancer. Transl. Oncol. 11, 575–584 (2018).
Miao, L. et al. Discoidin domain receptor 1 is associated with poor prognosis of non-small cell lung cancer and promotes cell invasion via epithelial-to-mesenchymal transition. Med. Oncol. 30, 626 (2013).
Ren, T., Zhang, J., Zhang, J., Liu, X. & Yao, L. Increased expression of discoidin domain receptor 2 (DDR2): a novel independent prognostic marker of worse outcome in breast cancer patients. Med. Oncol. 30, 397 (2013).
Misra, S. et al. Hyaluronan-CD44 interactions as potential targets for cancer therapy. FEBS J. 278, 1429–1443 (2011).
Toole, B. P. Hyaluronan-CD44 interactions in cancer: paradoxes and possibilities. Clin. Cancer Res. 15, 7462–7468 (2009).
Jalkanen, S. & Jalkanen, M. Lymphocyte CD44 binds the COOH-terminal heparin-binding domain of fibronectin. J. Cell Biol. 116, 817–825 (1992).
Ishikawa, T. et al. Phase I clinical trial of fibronectin CH296-stimulated T cell therapy in patients with advanced cancer. PLoS ONE 9, e83786 (2014).
Gupta, A. et al. Promising noninvasive cellular phenotype in prostate cancer cells knockdown of matrix metalloproteinase 9. Sci. World J. 2013, 493689 (2013).
Chen, Y., Fu, Z., Xu, S., Xu, Y. & Xu, P. The prognostic value of CD44 expression in gastric cancer: a meta-analysis. Biomed. Pharmacother. 68, 693–697 (2014).
Paradis, V. et al. CD44 is an independent prognostic factor in conventional renal cell carcinomas. J. Urol. 161, 1984–1987 (1999).
Lim, S. D., Young, A. N., Paner, G. P. & Amin, M. B. Prognostic role of CD44 cell adhesion molecule expression in primary and metastatic renal cell carcinoma: a clinicopathologic study of 125 cases. Virchows Arch. 452, 49–55 (2008).
Lucin, K., Matusan, K., Dordevic, G. & Stipic, D. Prognostic significance of CD44 molecule in renal cell carcinoma. Croat. Med. J. 45, 703–708 (2004).
Yildiz, E. et al. Prognostic value of the expression of Ki-67, CD44 and vascular endothelial growth factor, and microvessel invasion, in renal cell carcinoma. BJU Int. 93, 1087–1093 (2004).
Rioux-Leclercq, N. et al. Clinical significance of cell proliferation, microvessel density, and CD44 adhesion molecule expression in renal cell carcinoma. Hum. Pathol. 32, 1209–1215 (2001).
Li, N. et al. Analysis of CD44 isoform v10 expression and its prognostic value in renal cell carcinoma. BJU Int. 85, 514–518 (2000).
Ni, J. et al. CD44 variant 6 is associated with prostate cancer metastasis and chemo-/radioresistance. Prostate 74, 602–617 (2014).
Tei, H., Miyake, H., Harada, K. & Fujisawa, M. Expression profile of CD44s, CD44v6, and CD44v10 in localized prostate cancer: effect on prognostic outcomes following radical prostatectomy. Urol. Oncol. 32, 694–700 (2014).
Gunia, S., May, M., Koch, S., Dietel, M. & Erbersdobler, A. Expression of CD44s in incidental prostate cancer is more strongly associated with Gleason scores on subsequent radical prostatectomies than conventional prognostic parameters. Pathobiology 76, 286–292 (2009).
Hou, Y. C., Chao, Y. J., Tung, H. L., Wang, H. C. & Shan, Y. S. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer 120, 2766–2777 (2014).
Zhou, G. et al. Expression of CD44v6 and integrin-beta1 for the prognosis evaluation of pancreatic cancer patients after cryosurgery. Diagn. Pathol. 8, 146 (2013).
Li, Z. et al. CD44v/CD44s expression patterns are associated with the survival of pancreatic carcinoma patients. Diagn. Pathol. 9, 79 (2014).
Ko, Y. H. et al. Prognostic significance of CD44s expression in resected non-small cell lung cancer. BMC Cancer 11, 340 (2011).
Nguyen, V. N., Mirejovsky, T., Melinova, L. & Mandys, V. CD44 and its v6 spliced variant in lung carcinomas: relation to NCAM, CEA, EMA and UP1 and prognostic significance. Neoplasma 47, 400–408 (2000).
Yang, S. Z., Ji, W. H., Mao, W. M. & Ling, Z. Q. Elevated levels of preoperative circulating CD44(+) lymphocytes and neutrophils predict poor survival for non-small cell lung cancer patients. Clin. Chim. Acta 439, 172–177 (2015).
Hirata, T., Fukuse, T., Naiki, H., Hitomi, S. & Wada, H. Expression of CD44 variant exon 6 in stage I non-small cell lung carcinoma as a prognostic factor. Cancer Res 58, 1108–1110 (1998).
Roudi, R. et al. Clinical significance of putative cancer stem cell marker CD44 in different histological subtypes of lung cancer. Cancer Biomark. 14, 457–467 (2014).
Situ, D. et al. Expression and prognostic relevance of CD44v6 in stage I non-small cell lung carcinoma. J. Cancer Res. Clin. Oncol. 136, 1213–1219 (2010).
Kong, Y. et al. Breast cancer stem cell markers CD44 and ALDH1A1 in serum: distribution and prognostic value in patients with primary breast cancer. J. Cancer 9, 3728–3735 (2018).
Fang, Y. J. et al. Impact of ERbeta and CD44 expression on the prognosis of patients with stage II colon cancer. Tumour Biol. 33, 1907–1914 (2012).
Kunimura, T., Yoshida, T., Sugiyama, T. & Morohoshi, T. The relationships between loss of standard CD44 expression and lymph node, liver metastasis in T3 colorectal carcinoma. J. Gastrointest. Cancer 40, 115–118 (2009).
Olschewski, H. [Appetite depressant drugs and the risk of primary pulmonary hypertension]. Pneumologie 51, 575–576 (1997).
Ozawa, M. et al. Prognostic significance of CD44 variant 2 upregulation in colorectal cancer. Br. J. Cancer 111, 365–374 (2014).
Saigusa, S. et al. Clinical significance of LGR5 and CD44 expression in locally advanced rectal cancer after preoperative chemoradiotherapy. Int J. Oncol. 41, 1643–1652 (2012).
Garouniatis, A. et al. FAK, CD44v6, c-Met and EGFR in colorectal cancer parameters: tumour progression, metastasis, patient survival and receptor crosstalk. Int J. Colorectal Dis. 28, 9–18 (2013).
Saito, S. et al. CD44v6 expression is related to mesenchymal phenotype and poor prognosis in patients with colorectal cancer. Oncol. Rep. 29, 1570–1578 (2013).
Li, X. D., Ji, M., Wu, J., Jiang, J. T. & Wu, C. P. Clinical significance of CD44 variants expression in colorectal cancer. Tumori 99, 88–92 (2013).
Fan, C. W. et al. Prognostic significance of relevant markers of cancer stem cells in colorectal cancer—a meta analysis. Hepatogastroenterology 59, 1421–1427 (2012).
Katoh, S. et al. Cancer stem cell marker in circulating tumor cells: expression of CD44 variant exon 9 is strongly correlated to treatment refractoriness, recurrence and prognosis of human colorectal cancer. Anticancer Res. 35, 239–244 (2015).
Hu, S. et al. IMP3 combined with CD44s, a novel predictor for prognosis of patients with hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 140, 883–893 (2014).
Zhou, Z. J. et al. Overexpression of HnRNP A1 promotes tumor invasion through regulating CD44v6 and indicates poor prognosis for hepatocellular carcinoma. Int J. Cancer 132, 1080–1089 (2013).
Mima, K. et al. CD44s regulates the TGF-beta-mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res. 72, 3414–3423 (2012).
Yang, G. H. et al. Osteopontin combined with CD44, a novel prognostic biomarker for patients with hepatocellular carcinoma undergoing curative resection. Oncologist 13, 1155–1165 (2008).
Li, L. et al. Antibody against CD44s inhibits pancreatic tumor initiation and postradiation recurrence in mice. Gastroenterology 146, 1108–1118 (2014).
Shen, S. et al. Tumor-initiating cells are enriched in CD44(hi) population in murine salivary gland tumor. PLoS ONE 6, e23282 (2011).
Janisiewicz, A. M. et al. CD44(+) cells have cancer stem cell-like properties in nasopharyngeal carcinoma. Int. Forum Allergy Rhinol. 2, 465–470 (2012).
Su, J. et al. Identification of cancer stem-like CD44+ cells in human nasopharyngeal carcinoma cell line. Arch. Med. Res. 42, 15–21 (2011).
Cain, J. W. et al. Identification of CD44 as a surface biomarker for drug resistance by surface proteome signature technology. Mol. Cancer Res. 9, 637–647 (2011).
Prince, M. E. et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl Acad. Sci. USA 104, 973–978 (2007).
Faber, A. et al. Interaction of a CD44+ head and neck squamous cell carcinoma cell line with a stromal cell-derived factor-1-expressing supportive niche: An in vitro model. Oncol. Lett. 7, 82–86 (2014).
Chikamatsu, K., Takahashi, G., Sakakura, K., Ferrone, S. & Masuyama, K. Immunoregulatory properties of CD44+ cancer stem-like cells in squamous cell carcinoma of the head and neck. Head Neck 33, 208–215 (2011).
Pries, R., Witrkopf, N., Trenkle, T., Nitsch, S. M. & Wollenberg, B. Potential stem cell marker CD44 is constitutively expressed in permanent cell lines of head and neck cancer. Vivo 22, 89–92 (2008).
Faber, A. et al. CD44 as a stem cell marker in head and neck squamous cell carcinoma. Oncol. Rep. 26, 321–326 (2011).
Baumann, M. & Krause, M. CD44: a cancer stem cell-related biomarker with predictive potential for radiotherapy. Clin. Cancer Res. 16, 5091–5093 (2010).
Bourguignon, L. Y., Shiina, M. & Li, J. J. Hyaluronan-CD44 interaction promotes oncogenic signaling, microRNA functions, chemoresistance, and radiation resistance in cancer stem cells leading to tumor progression. Adv. Cancer Res. 123, 255–275 (2014).
Bourguignon, L. Y., Wong, G., Earle, C. & Chen, L. Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J. Biol. Chem. 287, 32800–32824 (2012).
Hirata, K. et al. CD44 variant 9 expression in primary early gastric cancer as a predictive marker for recurrence. Br. J. Cancer 109, 379–386 (2013).
Chen, W. et al. Identification of CD44+ cancer stem cells in human gastric cancer. Hepatogastroenterology 60, 949–954 (2013).
Yoon, C. et al. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin. Cancer Res. 20, 3974–3988 (2014).
Lau, W. M. et al. CD44v8-10 is a cancer-specific marker for gastric cancer stem cells. Cancer Res. 74, 2630–2641 (2014).
Sun, M., Zhou, W., Zhang, Y. Y., Wang, D. L. & Wu, X. L. CD44(+) gastric cancer cells with stemness properties are chemoradioresistant and highly invasive. Oncol. Lett. 5, 1793–1798 (2013).
Takaishi, S. et al. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 27, 1006–1020 (2009).
Ohata, H. et al. Induction of the stem-like cell regulator CD44 by Rho kinase inhibition contributes to the maintenance of colon cancer-initiating cells. Cancer Res. 72, 5101–5110 (2012).
Todaro, M. et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 14, 342–356 (2014).
Kimura, Y. et al. CD44variant exon 9 plays an important role in colon cancer initiating cells. Oncotarget 4, 785–791 (2013).
Du, L. et al. CD44 is of functional importance for colorectal cancer stem cells. Clin. Cancer Res.14, 6751–6760 (2008).
Su, Y. J., Lai, H. M., Chang, Y. W., Chen, G. Y. & Lee, J. L. Direct reprogramming of stem cell properties in colon cancer cells by CD44. EMBO J. 30, 3186–3199 (2011).
Anido, J. et al. TGF-beta receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell 18, 655–668 (2010).
Pietras, A. et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 14, 357–369 (2014).
Fu, J. et al. TGM2 inhibition attenuates ID1 expression in CD44-high glioma-initiating cells. Neuro Oncol. 15, 1353–1365 (2013).
Shi, Y., Liu, C., Liu, X., Tang, D. G. & Wang, J. The microRNA miR-34a inhibits non-small cell lung cancer (NSCLC) growth and the CD44hi stem-like NSCLC cells. PLoS One 9, e90022 (2014).
Leung, E. L. et al. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One 5, e14062 (2010).
Yae, T. et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun. 3, 883 (2012).
Meng, E. et al. CD44+/CD24- ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin. Exp. Metastasis 29, 939–948 (2012).
Liu, C. et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med 17, 211–215 (2011).
Palapattu, G. S. et al. Selective expression of CD44, a putative prostate cancer stem cell marker, in neuroendocrine tumor cells of human prostate cancer. Prostate 69, 787–798 (2009).
Patrawala, L. et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 25, 1696–1708 (2006).
Miyatake, Y., Sheehy, N., Ikeshita, S., Hall, W. W. & Kasahara, M. Anchorage-dependent multicellular aggregate formation induces CD44 high cancer stem cell-like ATL cells in an NF-kappaB- and vimentin-dependent manner. Cancer Lett. 357, 355–363 (2015).
Turley, E. A. & Torrance, J. Localization of hyaluronate and hyaluronate-binding protein on motile and non-motile fibroblasts. Exp. Cell Res 161, 17–28 (1985).
Maxwell, C. A., McCarthy, J. & Turley, E. Cell-surface and mitotic-spindle RHAMM: moonlighting or dual oncogenic functions? J. Cell Sci. 121, 925–932 (2008).
Assmann, V., Jenkinson, D., Marshall, J. F. & Hart, I. R. The intracellular hyaluronan receptor RHAMM/IHABP interacts with microtubules and actin filaments. J. Cell Sci. 112, 3943–3954 (1999). Pt 22.
Assmann, V., Marshall, J. F., Fieber, C., Hofmann, M. & Hart, I. R. The human hyaluronan receptor RHAMM is expressed as an intracellular protein in breast cancer cells. J. Cell Sci. 111, 1685–1694 (1998). Pt 12.
Hardwick, C. et al. Molecular cloning of a novel hyaluronan receptor that mediates tumor cell motility. J. Cell Biol. 117, 1343–1350 (1992).
Misra, S., Hascall, V. C., Markwald, R. R. & Ghatak, S. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front. Immunol. 6, 201 (2015).
Turley, E. A., Noble, P. W. & Bourguignon, L. Y. Signaling properties of hyaluronan receptors. J. Biol. Chem. 277, 4589–4592 (2002).
Tolg, C. et al. Rhamm-/- fibroblasts are defective in CD44-mediated ERK1,2 motogenic signaling, leading to defective skin wound repair. J. Cell Biol. 175, 1017–1028 (2006).
Zhang, S. et al. The hyaluronan receptor RHAMM regulates extracellular-regulated kinase. J. Biol. Chem. 273, 11342–11348 (1998).
Turley, E. A., Austen, L., Moore, D. & Hoare, K. Ras-transformed cells express both CD44 and RHAMM hyaluronan receptors: only RHAMM is essential for hyaluronan-promoted locomotion. Exp. Cell Res. 207, 277–282 (1993).
Akiyama, Y. et al. Hyaluronate receptors mediating glioma cell migration and proliferation. J. Neurooncol. 53, 115–127 (2001).
Maxwell, C. A. et al. RHAMM is a centrosomal protein that interacts with dynein and maintains spindle pole stability. Mol. Biol. Cell 14, 2262–2276 (2003).
Silverman-Gavrila, R. V., Silverman-Gavrila, L. B., Bilal, K. H. & Bendeck, M. P. Spectrin alpha is important for rear polarization of the microtubule organizing center during migration and spindle pole assembly during division of neointimal smooth muscle cells. Cytoskeleton 72, 157–170 (2015).
Hatano, H. et al. RHAMM/ERK interaction induces proliferative activities of cementifying fibroma cells through a mechanism based on the CD44-EGFR. Lab Investig. 91, 379–391 (2011).
Jiang, J., Mohan, P. & Maxwell, C. A. The cytoskeletal protein RHAMM and ERK1/2 activity maintain the pluripotency of murine embryonic stem cells. PLoS ONE 8, e73548 (2013).
Chen, H. et al. Spatial regulation of Aurora A activity during mitotic spindle assembly requires RHAMM to correctly localize TPX2. Cell Cycle 13, 2248–2261 (2014).
Maxwell, C. A. et al. Interplay between BRCA1 and RHAMM regulates epithelial apicobasal polarization and may influence risk of breast cancer. PLoS Biol. 9, e1001199 (2011).
Wang, C. et al. The overexpression of RHAMM, a hyaluronan-binding protein that regulates ras signaling, correlates with overexpression of mitogen-activated protein kinase and is a significant parameter in breast cancer progression. Clin. Cancer Res. 4, 567–576 (1998).
Wang, Z. et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl Acad. Sci. USA 111, E89–98 (2014).
Hamilton, S. R. et al. The hyaluronan receptors CD44 and Rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J. Biol. Chem. 282, 16667–16680 (2007).
Lugli, A. et al. Overexpression of the receptor for hyaluronic acid mediated motility is an independent adverse prognostic factor in colorectal cancer. Mod. Pathol. 19, 1302–1309 (2006).
Zlobec, I., Baker, K., Terracciano, L. M. & Lugli, A. RHAMM, p21 combined phenotype identifies microsatellite instability-high colorectal cancers with a highly adverse prognosis. Clin. Cancer Res. 14, 3798–3806 (2008).
Li, H. et al. Expression of hyaluronan receptors CD44 and RHAMM in stomach cancers: relevance with tumor progression. Int J. Oncol. 17, 927–932 (2000).
Gust, K. M. et al. RHAMM (CD168) is overexpressed at the protein level and may constitute an immunogenic antigen in advanced prostate cancer disease. Neoplasia 11, 956–963 (2009).
Korkes, F. et al. Hyaluronan-mediated motility receptor (RHAMM) immunohistochemical expression and androgen deprivation in normal peritumoral, hyperplasic and neoplastic prostate tissue. BJU Int. 113, 822–829 (2014).
Liu, Y. C. et al. Hepatitis B virus X protein induces RHAMM-dependent motility in hepatocellular carcinoma cells via PI3K-Akt-Oct-1 signaling. Mol. Cancer Res. 18, 375–389 (2020).
He, X. et al. Upregulation of hyaluronan-mediated motility receptor in hepatocellular carcinoma predicts poor survival. Oncol. Lett. 10, 3639–3646 (2015).
Cheng, X. B., Sato, N., Kohi, S., Koga, A. & Hirata, K. Receptor for hyaluronic acid-mediated motility is associated with poor survival in pancreatic ductal adenocarcinoma. J. Cancer 6, 1093–1098 (2015).
Wang, D. et al. Expression of the receptor for hyaluronic acid mediated motility (RHAMM) is associated with poor prognosis and metastasis in non-small cell lung carcinoma. Oncotarget 7, 39957–39969 (2016).
Song, J. M. et al. Hyaluronan-CD44/RHAMM interaction-dependent cell proliferation and survival in lung cancer cells. Mol. Carcinog. 58, 321–333 (2019).
Niedworok, C. et al. The impact of the receptor of hyaluronan-mediated motility (RHAMM) on human urothelial transitional cell cancer of the bladder. PLoS ONE 8, e75681 (2013).
Shigeishi, H. et al. Overexpression of the receptor for hyaluronan-mediated motility, correlates with expression of microtubule-associated protein in human oral squamous cell carcinomas. Int. J. Oncol. 34, 1565–1571 (2009).
Shigeishi, H., Higashikawa, K. & Takechi, M. Role of receptor for hyaluronan-mediated motility (RHAMM) in human head and neck cancers. J. Cancer Res. Clin. Oncol. 140, 1629–1640 (2014).
Choi, S. et al. Function and clinical relevance of RHAMM isoforms in pancreatic tumor progression. Mol. Cancer 18, 92 (2019).
Liu, J. et al. TGF-beta blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc. Natl Acad. Sci. USA 109, 16618–16623 (2012).
Chauhan, V. P. et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 4, 2516 (2013).
Zion, O. et al. Inhibition of transforming growth factor beta signaling by halofuginone as a modality for pancreas fibrosis prevention. Pancrease 38, 427–435 (2009).
Lin, R. et al. Inhibition of TGF-beta signaling with halofuginone can enhance the antitumor effect of irradiation in Lewis lung cancer. Onco Targets Ther. 8, 3549–3559 (2015).
Juarez, P. et al. Halofuginone inhibits the establishment and progression of melanoma bone metastases. Cancer Res. 72, 6247–6256 (2012).
Juarez, P. et al. Halofuginone inhibits TGF-beta/BMP signaling and in combination with zoledronic acid enhances inhibition of breast cancer bone metastasis. Oncotarget 8, 86447–86462 (2017).
Morris, J. C. et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 9, e90353 (2014).
Lampi, M. C. & Reinhart-King, C. A. Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials.Sci Transl Med. 10, eaao0475 (2018).
Diop-Frimpong, B., Chauhan, V. P., Krane, S., Boucher, Y. & Jain, R. K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl Acad. Sci. USA 108, 2909–2914 (2011).
Zhao, Y. et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc. Natl Acad. Sci. USA 116, 2210–2219 (2019).
Dolor, A. & Szoka, F. C. Jr Digesting a path forward: the utility of collagenase tumor treatment for improved drug delivery. Mol. Pharm. 15, 2069–2083 (2018).
McKee, T. D. et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res 66, 2509–2513 (2006).
Zinger, A. et al. Collagenase nanoparticles enhance the penetration of drugs into pancreatic tumors. ACS Nano 13, 11008–11021 (2019).
Eikenes, L., Tufto, I., Schnell, E. A., Bjorkoy, A. & De Lange Davies, C. Effect of collagenase and hyaluronidase on free and anomalous diffusion in multicellular spheroids and xenografts. Anticancer Res 30, 359–368 (2010).
Coussens, L. M., Fingleton, B. & Matrisian, L. M. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392 (2002).
Overall, C. M. & Lopez-Otin, C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat. Rev. Cancer 2, 657–672 (2002).
Parks, W. C., Wilson, C. L. & Lopez-Boado, Y. S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 4, 617–629 (2004).
Fang, M., Yuan, J., Peng, C. & Li, Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. 35, 2871–2882 (2014).
Page-McCaw, A., Ewald, A. J. & Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 8, 221–233 (2007).
Boufraqech, M. et al. miR30a inhibits LOX expression and anaplastic thyroid cancer progression. Cancer Res. 75, 367–377 (2015).
Minna, E. et al. Cancer associated fibroblasts and senescent thyroid cells in the invasive front of thyroid carcinoma. Cancers https://doi.org/10.3390/cancers12010112 (2020).
Liu, X. et al. Identification and characterization of the promoter of cancer-related gene LOXL2. Exp. Cell Res. 387, 111786 (2020).
Miller, B. W. et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol. Med. 7, 1063–1076 (2015).
Kanapathipillai, M. et al. Inhibition of mammary tumor growth using lysyl oxidase-targeting nanoparticles to modify extracellular matrix. Nano Lett. 12, 3213–3217 (2012).
Derynck, R. & Budi, E. H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. https://doi.org/10.1126/scisignal.aav5183 (2019).
Hata, A. & Chen, Y. G. TGF-beta signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a022061 (2016).
Hill, C. S. Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. https://doi.org/10.1101/cshperspect.a022079 (2016).
Chen, Y. et al. Transforming growth factor beta signaling pathway: a promising therapeutic target for cancer. J. Cell Physiol. 235, 1903–1914 (2020).
Tucker, R. F., Shipley, G. D., Moses, H. L. & Holley, R. W. Growth inhibitor from BSC-1 cells closely related to platelet type beta transforming growth factor. Science 226, 705–707 (1984).
Reiss, M. TGF-beta and cancer. Microbes Infect. 1, 1327–1347 (1999).
Wan, Y. Y. & Flavell, R. A. ‘Yin-Yang’ functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol. Rev. 220, 199–213 (2007).
Connolly, E. C., Freimuth, J. & Akhurst, R. J. Complexities of TGF-beta targeted cancer therapy. Int J. Biol. Sci. 8, 964–978 (2012).
Matsuyama, S. et al. SB-431542 and Gleevec inhibit transforming growth factor-beta-induced proliferation of human osteosarcoma cells. Cancer Res. 63, 7791–7798 (2003).
DaCosta Byfield, S., Major, C., Laping, N. J. & Roberts, A. B. SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol. Pharm. 65, 744–752 (2004).
Hjelmeland, M. D. et al. SB-431542, a small molecule transforming growth factor-beta-receptor antagonist, inhibits human glioma cell line proliferation and motility. Mol. Cancer Ther. 3, 737–745 (2004).
Tanaka, H. et al. Transforming growth factor beta signaling inhibitor, SB-431542, induces maturation of dendritic cells and enhances anti-tumor activity. Oncol. Rep. 24, 1637–1643 (2010).
Ehata, S. et al. Ki26894, a novel transforming growth factor-beta type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci. 98, 127–133 (2007).
Shinto, O. et al. Inhibitory effect of a TGFbeta receptor type-I inhibitor, Ki26894, on invasiveness of scirrhous gastric cancer cells. Br. J. Cancer 102, 844–851 (2010).
Bandyopadhyay, A. et al. Inhibition of pulmonary and skeletal metastasis by a transforming growth factor-beta type I receptor kinase inhibitor. Cancer Res. 66, 6714–6721 (2006).
Zhang, B., Halder, S. K., Zhang, S. & Datta, P. K. Targeting transforming growth factor-beta signaling in liver metastasis of colon cancer. Cancer Lett. 277, 114–120 (2009).
Korpal, M. et al. Imaging transforming growth factor-beta signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat. Med. 15, 960–966 (2009).
Melisi, D. et al. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 7, 829–840 (2008).
Fernandez, T. et al. Disruption of transforming growth factor beta signaling by a novel ligand-dependent mechanism. J. Exp. Med. 195, 1247–1255 (2002).
Capocasale, R. J. et al. Reduced surface expression of transforming growth factor beta receptor type II in mitogen-activated T cells from Sezary patients. Proc. Natl Acad. Sci. USA 92, 5501–5505 (1995).
Sawyer, T. K. Novel oncogenic protein kinase inhibitors for cancer therapy. Curr. Med. Chem. Anticancer Agents 4, 449–455 (2004).
Deshayes, F. & Nahmias, C. Angiotensin receptors: a new role in cancer? Trends Endocrinol. Metab. 16, 293–299 (2005).
Ali, M. S. et al. Dependence on the motif YIPP for the physical association of Jak2 kinase with the intracellular carboxyl tail of the angiotensin II AT1 receptor. J. Biol. Chem. 272, 23382–23388 (1997).
George, A. J. et al. A functional siRNA screen identifies genes modulating angiotensin II-mediated EGFR transactivation. J. Cell Sci. 126, 5377–5390 (2013).
George, A. J., Hannan, R. D. & Thomas, W. G. Unravelling the molecular complexity of GPCR-mediated EGFR transactivation using functional genomics approaches. FEBS J. 280, 5258–5268 (2013).
Lopez-Ilasaca, M., Crespo, P., Pellici, P. G., Gutkind, J. S. & Wetzker, R. Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase gamma. Science 275, 394–397 (1997).
Haendeler, J. et al. GIT1 mediates Src-dependent activation of phospholipase Cgamma by angiotensin II and epidermal growth factor. J. Biol. Chem. 278, 49936–49944 (2003).
Kintscher, U. et al. Angiotensin II induces migration and Pyk2/paxillin phosphorylation of human monocytes. Hypertension 37, 587–593 (2001).
Natarajan, K., Yin, G. & Berk, B. C. Scaffolds direct Src-specific signaling in response to angiotensin II: new roles for Cas and GIT1. Mol. Pharm. 65, 822–825 (2004).
van Nieuw Amerongen, G. P. et al. GIT1 mediates thrombin signaling in endothelial cells: role in turnover of RhoA-type focal adhesions. Circ. Res. 94, 1041–1049 (2004).
Yin, G., Haendeler, J., Yan, C. & Berk, B. C. GIT1 functions as a scaffold for MEK1-extracellular signal-regulated kinase 1 and 2 activation by angiotensin II and epidermal growth factor. Mol. Cell Biol. 24, 875–885 (2004).
Greco, S. et al. Angiotensin II activates extracellular signal regulated kinases via protein kinase C and epidermal growth factor receptor in breast cancer cells. J. Cell Physiol. 196, 370–377 (2003).
Uemura, H. et al. Angiotensin II receptor blocker shows antiproliferative activity in prostate cancer cells: a possibility of tyrosine kinase inhibitor of growth factor. Mol. Cancer Ther. 2, 1139–1147 (2003).
Imanishi, T., Hano, T. & Nishio, I. Angiotensin II potentiates vascular endothelial growth factor-induced proliferation and network formation of endothelial progenitor cells. Hypertens. Res. 27, 101–108 (2004).
Otani, A., Takagi, H., Oh, H., Koyama, S. & Honda, Y. Angiotensin II induces expression of the Tie2 receptor ligand, angiopoietin-2, in bovine retinal endothelial cells. Diabetes 50, 867–875 (2001).
Suzuki, Y. et al. Inflammation and angiotensin II. Int. J. Biochem. Cell Biol. 35, 881–900 (2003).
De Paepe, B., Verstraeten, V. L., De Potter, C. R., Vakaet, L. A. & Bullock, G. R. Growth stimulatory angiotensin II type-1 receptor is upregulated in breast hyperplasia and in situ carcinoma but not in invasive carcinoma. Histochem. Cell Biol. 116, 247–254 (2001).
De Paepe, B., Verstraeten, V. M., De Potter, C. R. & Bullock, G. R. Increased angiotensin II type-2 receptor density in hyperplasia, DCIS and invasive carcinoma of the breast is paralleled with increased iNOS expression. Histochem. Cell Biol. 117, 13–19 (2002).
Takeda, H. & Kondo, S. Differences between squamous cell carcinoma and keratoacanthoma in angiotensin type-1 receptor expression. Am. J. Pathol. 158, 1633–1637 (2001).
Kikkawa, F. et al. Activation of invasiveness of cervical carcinoma cells by angiotensin II. Am. J. Obstet. Gynecol. 190, 1258–1263 (2004).
Suganuma, T. et al. Functional expression of the angiotensin II type 1 receptor in human ovarian carcinoma cells and its blockade therapy resulting in suppression of tumor invasion, angiogenesis, and peritoneal dissemination. Clin. Cancer Res. 11, 2686–2694 (2005).
Fujita, M., Hayashi, I., Yamashina, S., Itoman, M. & Majima, M. Blockade of angiotensin AT1a receptor signaling reduces tumor growth, angiogenesis, and metastasis. Biochem. Biophys. Res. Commun. 294, 441–447 (2002).
Egami, K. et al. Role of host angiotensin II type 1 receptor in tumor angiogenesis and growth. J. Clin. Investig. 112, 67–75 (2003).
Arrieta, O. et al. Blockage of angiotensin II type I receptor decreases the synthesis of growth factors and induces apoptosis in C6 cultured cells and C6 rat glioma. Br. J. Cancer 92, 1247–1252 (2005).
Lokman, N. A. et al. 4-Methylumbelliferone inhibits cancer stem cell activation and overcomes chemoresistance in ovarian cancer. Cancers 11, 1187 (2019).
Kohli, A. G., Kivimae, S., Tiffany, M. R. & Szoka, F. C. Improving the distribution of Doxil(R) in the tumor matrix by depletion of tumor hyaluronan. J. Control Release 191, 105–114 (2014).
Whatcott, C. J., Han, H., Posner, R. G., Hostetter, G. & Von Hoff, D. D. Targeting the tumor microenvironment in cancer: why hyaluronidase deserves a second look. Cancer Discov. 1, 291–296 (2011).
Hingorani, S. R. et al. HALO 202: randomized phase II Study of PEGPH20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J. Clin. Oncol. 36, 359–366 (2018).
Doherty, G. J., Tempero, M. & Corrie, P. G. HALO-109-301: a Phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Future Oncol. 14, 13–22 (2018).
Castellani, P. et al. The fibronectin isoform containing the ED-B oncofetal domain: a marker of angiogenesis. Int J. Cancer 59, 612–618 (1994).
Glukhova, M. A., Frid, M. G., Shekhonin, B. V., Balabanov, Y. V. & Koteliansky, V. E. Expression of fibronectin variants in vascular and visceral smooth muscle cells in development. Dev. Biol. 141, 193–202 (1990).
Rybak, J. N., Roesli, C., Kaspar, M., Villa, A. & Neri, D. The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res. 67, 10948–10957 (2007).
Carnemolla, B., Leprini, A., Allemanni, G., Saginati, M. & Zardi, L. The inclusion of the type III repeat ED-B in the fibronectin molecule generates conformational modifications that unmask a cryptic sequence. J. Biol. Chem. 267, 24689–24692 (1992).
Lo, K. M. et al. huBC1-IL12, an immunocytokine which targets EDB-containing oncofetal fibronectin in tumors and tumor vasculature, shows potent anti-tumor activity in human tumor models. Cancer Immunol. Immunother. 56, 447–457 (2007).
Rudman, S. M. et al. A phase 1 study of AS1409, a novel antibody-cytokine fusion protein, in patients with malignant melanoma or renal cell carcinoma. Clin. Cancer Res. 17, 1998–2005 (2011).
Carnemolla, B. et al. Phage antibodies with pan-species recognition of the oncofoetal angiogenesis marker fibronectin ED-B domain. Int J. Cancer 68, 397–405 (1996).
Carnemolla, B. et al. Enhancement of the antitumor properties of interleukin-2 by its targeted delivery to the tumor blood vessel extracellular matrix. Blood 99, 1659–1665 (2002).
Johannsen, M. et al. The tumour-targeting human L19-IL2 immunocytokine: preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma. Eur. J. Cancer 46, 2926–2935 (2010).
Eigentler, T. K. et al. A dose-escalation and signal-generating study of the immunocytokine L19-IL2 in combination with dacarbazine for the therapy of patients with metastatic melanoma. Clin. Cancer Res. 17, 7732–7742 (2011).
Berndorff, D. et al. Radioimmunotherapy of solid tumors by targeting extra domain B fibronectin: identification of the best-suited radioimmunoconjugate. Clin. Cancer Res. 11, 7053s–7063s (2005).
Tijink, B. M. et al. Radioimmunotherapy of head and neck cancer xenografts using 131I-labeled antibody L19-SIP for selective targeting of tumor vasculature. J. Nucl. Med. 47, 1127–1135 (2006).
Sauer, S. et al. Expression of the oncofetal ED-B-containing fibronectin isoform in hematologic tumors enables ED-B-targeted 131I-L19SIP radioimmunotherapy in Hodgkin lymphoma patients. Blood 113, 2265–2274 (2009).
Locher, R. et al. Abundant in vitro expression of the oncofetal ED-B-containing fibronectin translates into selective pharmacodelivery of (131)I-L19SIP in a prostate cancer patient. J. Cancer Res. Clin. Oncol. 140, 35–43 (2014).
Saw, P. E. et al. Aptide-conjugated liposome targeting tumor-associated fibronectin for glioma therapy. J. Mater. Chem. B 1, 4723–4726 (2013).
Gu, G. et al. PEG-PLA nanoparticles modified with APTEDB peptide for enhanced anti-angiogenic and anti-glioma therapy. Biomaterials 35, 8215–8226 (2014).
Parajuli, H. et al. Integrin alpha11 is overexpressed by tumour stroma of head and neck squamous cell carcinoma and correlates positively with alpha smooth muscle actin expression. J. Oral. Pathol. Med. 46, 267–275 (2017).
Franco-Barraza, J. et al. Matrix-regulated integrin alphavbeta5 maintains alpha5beta1-dependent desmoplastic traits prognostic of neoplastic recurrence. Elife https://doi.org/10.7554/eLife.20600 (2017).
Kale, S. et al. Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via alpha9beta1 integrin. Oncogene 34, 5408–5410 (2015).
Zhou, W. et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 17, 170–182 (2015).
Schnittert, J., Bansal, R., Storm, G. & Prakash, J. Integrins in wound healing, fibrosis and tumor stroma: high potential targets for therapeutics and drug delivery. Adv. Drug Deliv. Rev. 129, 37–53 (2018).
Gutheil, J. C. et al. Targeted antiangiogenic therapy for cancer using Vitaxin: a humanized monoclonal antibody to the integrin alphavbeta3. Clin. Cancer Res. 6, 3056–3061 (2000).
Ricart, A. D. et al. Volociximab, a chimeric monoclonal antibody that specifically binds alpha5beta1 integrin: a phase I, pharmacokinetic, and biological correlative study. Clin. Cancer Res. 14, 7924–7929 (2008).
Bell-McGuinn, K. M. et al. A phase II, single-arm study of the anti-alpha5beta1 integrin antibody volociximab as monotherapy in patients with platinum-resistant advanced epithelial ovarian or primary peritoneal cancer. Gynecol. Oncol. 121, 273–279 (2011).
Figlin, R. A., Kondagunta, G. V., Yazji, S., Motzer, R. J. & Bukowski, R. M. Phase II study of volociximab (M200), an α5β1 anti-integrin antibody in refractory metastatic clear cell renal cell cancer (RCC). J. Clin. Oncol. 24, 4535–4535 (2006).
Evans, T. et al. Final results from cohort 1 of a phase II study of volociximab, an anti-α5β1 integrin antibody, in combination with gemcitabine (GEM) in patients (pts) with metastatic pancreatic cancer (MPC). J. Clin. Oncol. 25, 4549–4549 (2007).
Paolillo, M. et al. An RGD small-molecule integrin antagonist induces detachment-mediated anoikis in glioma cancer stem cells. Int. J. Oncol. 53, 2683–2694 (2018).
Raguse, J. D., Gath, H. J., Bier, J., Riess, H. & Oettle, H. Cilengitide (EMD 121974) arrests the growth of a heavily pretreated highly vascularised head and neck tumour. Oral. Oncol. 40, 228–230 (2004).
Mas-Moruno, C., Rechenmacher, F. & Kessler, H. Cilengitide: the first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 10, 753–768 (2010).
Reardon, D. A. et al. Cilengitide: an RGD pentapeptide alphanubeta3 and alphanubeta5 integrin inhibitor in development for glioblastoma and other malignancies. Future Oncol. 7, 339–354 (2011).
Nabors, L. B. et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. 17, 708–717 (2015).
Cox, D., Brennan, M. & Moran, N. Integrins as therapeutic targets: lessons and opportunities. Nat. Rev. Drug Discov. 9, 804–820 (2010).
Gao, H. et al. Multi-organ site metastatic reactivation mediated by non-canonical discoidin domain receptor 1 signaling. Cell 166, 47–62 (2016).
Kim, H. G., Hwang, S. Y., Aaronson, S. A., Mandinova, A. & Lee, S. W. DDR1 receptor tyrosine kinase promotes prosurvival pathway through Notch1 activation. J. Biol. Chem. 286, 17672–17681 (2011).
Yang, J. C. et al. TM4SF1 promotes metastasis of pancreatic cancer via regulating the expression of DDR1. Sci. Rep. 7, 45895 (2017).
Aguilera, K. Y. et al. Inhibition of Discoidin Domain Receptor 1 Reduces Collagen-mediated Tumorigenicity in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 16, 2473–2485 (2017).
Ambrogio, C. et al. Combined inhibition of DDR1 and Notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat. Med. 22, 270–277 (2016).
Day, E. et al. Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharm. 599, 44–53 (2008).
Canning, P. et al. Structural mechanisms determining inhibition of the collagen receptor DDR1 by selective and multi-targeted type II kinase inhibitors. J. Mol. Biol. 426, 2457–2470 (2014).
Siddiqui, K. et al. Actinomycin D identified as an inhibitor of discoidin domain receptor 2 interaction with collagen through an insect cell based screening of a drug compound library. Biol. Pharm. Bull. 32, 136–141 (2009).
Rix, U. et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 110, 4055–4063 (2007).
Bantscheff, M. et al. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 25, 1035–1044 (2007).
Jeitany, M. et al. Inhibition of DDR1-BCR signalling by nilotinib as a new therapeutic strategy for metastatic colorectal cancer.EMBO Mol. Med. 10, e7918 (2018).
Hammerman, P. S. et al. Mutations in the DDR2 kinase gene identify a novel therapeutic target in squamous cell lung cancer. Cancer Discov. 1, 78–89 (2011).
Gao, M. et al. Discovery and optimization of 3-(2-(Pyrazolo[1,5-a]pyrimidin-6-yl)ethynyl)benzamides as novel selective and orally bioavailable discoidin domain receptor 1 (DDR1) inhibitors. J. Med. Chem. 56, 3281–3295 (2013).
Tijink, B. M. et al. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 12, 6064–6072 (2006).
Ghatak, S., Misra, S. & Toole, B. P. Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J. Biol. Chem. 280, 8875–8883 (2005).
Ghatak, S., Hascall, V. C., Markwald, R. R. & Misra, S. Stromal hyaluronan interaction with epithelial CD44 variants promotes prostate cancer invasiveness by augmenting expression and function of hepatocyte growth factor and androgen receptor. J. Biol. Chem. 285, 19821–19832 (2010).
Misra, S., Ghatak, S. & Toole, B. P. Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J. Biol. Chem. 280, 20310–20315 (2005).
Misra, S., Toole, B. P. & Ghatak, S. Hyaluronan constitutively regulates activation of multiple receptor tyrosine kinases in epithelial and carcinoma cells. J. Biol. Chem. 281, 34936–34941 (2006).
Zhu, H. et al. The role of the hyaluronan receptor CD44 in mesenchymal stem cell migration in the extracellular matrix. Stem Cells 24, 928–935 (2006).
Misra, S., Hascall, V. C., De Giovanni, C., Markwald, R. R. & Ghatak, S. Delivery of CD44 shRNA/nanoparticles within cancer cells: perturbation of hyaluronan/CD44v6 interactions and reduction in adenoma growth in Apc Min/+ MICE. J. Biol. Chem. 284, 12432–12446 (2009).
Wang, C. et al. A low MW inhibitor of CD44 dimerization for the treatment of glioblastoma. Br. J. Pharm. 177, 3009–3023 (2020).
Kavousipour, S. et al. Effect of CD44 aptamer on snail metastasis factor and aggressiveness of MDA-MB-231. Breast Cancer Cell Line 21, e94641 (2020).
Gao, C., Liu, S., Wang, Y., Chu, G. & Xu, X. Effect of receptor for hyaluronan-mediated motility inhibition on radiosensitivity of lung adenocarcinoma A549 cells. Transl. Cancer Res. 8, 410–421 (2019).
Esguerra, K. V. et al. Identification, design and synthesis of tubulin-derived peptides as novel hyaluronan mimetic ligands for the receptor for hyaluronan-mediated motility (RHAMM/HMMR). Integr. Biol. 7, 1547–1560 (2015).
Tolg, C. et al. A RHAMM mimetic peptide blocks hyaluronan signaling and reduces inflammation and fibrogenesis in excisional skin wounds. Am. J. Pathol. 181, 1250–1270 (2012).
Papageorgis, P. et al. Tranilast-induced stress alleviation in solid tumors improves the efficacy of chemo- and nanotherapeutics in a size-independent manner. Sci. Rep. 7, 46140 (2017).
Polydorou, C., Mpekris, F., Papageorgis, P., Voutouri, C. & Stylianopoulos, T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 8, 24506–24517 (2017).
Vennin, C. et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aai8504 (2017).
Incio, J. et al. Metformin reduces desmoplasia in pancreatic cancer by reprogramming stellate cells and tumor-associated macrophages. PLoS ONE 10, e0141392 (2015).
Martin, J. D. et al. Dexamethasone increases cisplatin-loaded nanocarrier delivery and efficacy in metastatic breast cancer by normalizing the tumor microenvironment. ACS Nano 13, 6396–6408 (2019).
Weniger, M., Honselmann, K. C. & Liss, A. S. The extracellular matrix and pancreatic cancer: a complex relationship. Cancers https://doi.org/10.3390/cancers10090316 (2018).
Kaldjian, E. P., Gretz, J. E., Anderson, A. O., Shi, Y. & Shaw, S. Spatial and molecular organization of lymph node T cell cortex: a labyrinthine cavity bounded by an epithelium-like monolayer of fibroblastic reticular cells anchored to basement membrane-like extracellular matrix. Int. Immunol. 13, 1243–1253 (2001).
Ruco, L. P. et al. Expression and cell distribution of the intercellular adhesion molecule, vascular cell adhesion molecule, endothelial leukocyte adhesion molecule, and endothelial cell adhesion molecule (CD31) in reactive human lymph nodes and in Hodgkin’s disease. Am. J. Pathol. 140, 1337–1344 (1992).
Clark, R. A., Alon, R. & Springer, T. A. CD44 and hyaluronan-dependent rolling interactions of lymphocytes on tonsillar stroma. J. Cell Biol. 134, 1075–1087 (1996).
Schmid, C. & Isaacson, P. G. Proliferation centres in B-cell malignant lymphoma, lymphocytic (B-CLL): an immunophenotypic study. Histopathology 24, 445–451 (1994).
de Rooij, M. F. et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 119, 2590–2594 (2012).
Herman, S. E. et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 117, 6287–6296 (2011).
Ponader, S. et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 119, 1182–1189 (2012).
Brown, J. R. Ibrutinib (PCI-32765), the first BTK (Bruton’s tyrosine kinase) inhibitor in clinical trials. Curr. Hematol. Malig. Rep. 8, 1–6 (2013).
de Claro, R. A. et al. FDA approval: ibrutinib for patients with previously treated mantle cell lymphoma and previously treated chronic lymphocytic leukemia. Clin. Cancer Res. 21, 3586–3590 (2015).
Nagel, S., Hirschmann, P., Dirnhofer, S., Gunthert, U. & Tzankov, A. Coexpression of CD44 variant isoforms and receptor for hyaluronic acid-mediated motility (RHAMM, CD168) is an International Prognostic Index and C-MYC gene status-independent predictor of poor outcome in diffuse large B-cell lymphomas. Exp. Hematol. 38, 38–45 (2010).
Turley, E. A., Belch, A. J., Poppema, S. & Pilarski, L. M. Expression and function of a receptor for hyaluronan-mediated motility on normal and malignant B lymphocytes. Blood 81, 446–453 (1993).
Pilarski, L. M., Miszta, H. & Turley, E. A. Regulated expression of a receptor for hyaluronan-mediated motility on human thymocytes and T cells. J. Immunol. 150, 4292–4302 (1993).
Pilarski, L. M. et al. Potential role for hyaluronan and the hyaluronan receptor RHAMM in mobilization and trafficking of hematopoietic progenitor cells. Blood 93, 2918–2927 (1999).
Till, K. J., Zuzel, M. & Cawley, J. C. The role of hyaluronan and interleukin 8 in the migration of chronic lymphocytic leukemia cells within lymphoreticular tissues. Cancer Res. 59, 4419–4426 (1999).
Girbl, T. et al. CD40-mediated activation of chronic lymphocytic leukemia cells promotes their CD44-dependent adhesion to hyaluronan and restricts CCL21-induced motility. Cancer Res. 73, 561–570 (2013).
Giannopoulos, K. et al. Expression of RHAMM/CD168 and other tumor-associated antigens in patients with B-cell chronic lymphocytic leukemia. Int J. Oncol. 29, 95–103 (2006).
Greiner, J. et al. Identification and characterization of epitopes of the receptor for hyaluronic acid-mediated motility (RHAMM/CD168) recognized by CD8+ T cells of HLA-A2-positive patients with acute myeloid leukemia. Blood 106, 938–945 (2005).
Greiner, J. et al. Expression of tumor-associated antigens in acute myeloid leukemia: Implications for specific immunotherapeutic approaches. Blood 108, 4109–4117 (2006).
Schmitt, M. et al. RHAMM-R3 peptide vaccination in patients with acute myeloid leukemia, myelodysplastic syndrome, and multiple myeloma elicits immunologic and clinical responses. Blood 111, 1357–1365 (2008).
Herishanu, Y. et al. Activation of CD44, a receptor for extracellular matrix components, protects chronic lymphocytic leukemia cells from spontaneous and drug induced apoptosis through MCL-1. Leuk. Lymphoma 52, 1758–1769 (2011).
Eisterer, W. et al. Elevated levels of soluble CD44 are associated with advanced disease and in vitro proliferation of neoplastic lymphocytes in B-cell chronic lymphocytic leukaemia. Leuk. Res. 28, 1043–1051 (2004).
Zarcone, D. et al. Functional and clinical relevance of CD44 variant isoform expression on B-cell chronic lymphocytic leukemia cells. Haematologica 83, 1088–1098 (1998).
Zhang, S. et al. Targeting chronic lymphocytic leukemia cells with a humanized monoclonal antibody specific for CD44. Proc. Natl Acad. Sci. USA 110, 6127–6132 (2013).
Vey, N. et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget 7, 32532–32542 (2016).
Song, G. et al. HI44a, an anti-CD44 monoclonal antibody, induces differentiation and apoptosis of human acute myeloid leukemia cells. Leuk. Res. 28, 1089–1096 (2004).
Gadhoum, Z. et al. CD44: a new means to inhibit acute myeloid leukemia cell proliferation via p27Kip1. Blood 103, 1059–1068 (2004).
da Cruz, L. A. G. et al. Anti-CD44 antibody, ARH460-16-2, binds to human AML CD34+CD38-cancer stem cells and demonstrates anti-tumor activity in an AML xenograft model. Proc. Am. Assoc. Cancer Res. Annu. Meet. 49, 945 (2008).
Lompardia, S. et al. Hyaluronan abrogates imatinib-induced senescence in chronic myeloid leukemia cell lines. Sci. Rep. 9, 10930 (2019).
Uchakina, O. N., Ban, H., Hostetler, B. J. & McKallip, R. J. Inhibition of hyaluronic acid formation sensitizes chronic myelogenous leukemia to treatment with doxorubicin. Glycobiology 26, 1171–1179 (2016).
Goldberg, S. B. et al. Randomized trial of afatinib plus cetuximab versus afatinib alone for first-line treatment of EGFR-mutant non-small-cell lung cancer: final results from SWOG S1403. J. Clin. Oncol. https://doi.org/10.1200/JCO.20.01149 (2020).
Jabbour, E. & Kantarjian, H. Chronic myeloid leukemia: 2012 update on diagnosis, monitoring, and management. Am. J. Hematol. 87, 1037–1045 (2012).
Femel, J. et al. Therapeutic vaccination against fibronectin ED-A attenuates progression of metastatic breast cancer. Oncotarget 5, 12418–12427 (2014).
Lin, E. Y. et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 163, 2113–2126 (2003).
Zhang, W. et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br. J. Cancer 121, 837–845 (2019).
Santimaria, M. et al. Immunoscintigraphic detection of the ED-B domain of fibronectin, a marker of angiogenesis, in patients with cancer. Clin. Cancer Res. 9, 571–579 (2003).
Cutler, J. I., Auyeung, E. & Mirkin, C. A. Spherical nucleic acids. J. Am. Chem. Soc. 134, 1376–1391 (2012).
Li, N., Chang, C., Pan, W. & Tang, B. A multicolor nanoprobe for detection and imaging of tumor-related mRNAs in living cells. Angew. Chem. Int. Ed. Engl. 51, 7426–7430 (2012).
Zoumi, A., Yeh, A. & Tromberg, B. J. Imaging cells and extracellular matrix in vivo by using second-harmonic generation and two-photon excited fluorescence. Proc. Natl Acad. Sci. USA 99, 11014–11019 (2002).
Moon, P. G. et al. Fibronectin on circulating extracellular vesicles as a liquid biopsy to detect breast cancer. Oncotarget 7, 40189–40199 (2016).
Hou, B. et al. Engineering stimuli-activatable boolean logic prodrug nanoparticles for combination cancer immunotherapy. Adv. Mater. 32, e1907210 (2020).
Untch, M. et al. NAB-paclitaxel improves disease-free survival in early breast cancer: GBG 69-GeparSepto. J. Clin. Oncol. 37, 2226–2234 (2019).
Schmid, P. et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med 379, 2108–2121 (2018).
Shroff, R. T. et al. Gemcitabine, cisplatin, and nab-paclitaxel for the treatment of advanced biliary tract cancers: a phase 2 clinical trial. JAMA Oncol. 5, 824–830 (2019).
Sahai, V. et al. Nab-paclitaxel and gemcitabine as first-line treatment of advanced or metastatic cholangiocarcinoma: a phase 2 clinical trial. JAMA Oncol. 4, 1707–1712 (2018).
Macarulla, T. et al. Phase I/II trial to evaluate the efficacy and safety of nanoparticle albumin-bound paclitaxel in combination with gemcitabine in patients with pancreatic cancer and an ECOG performance status of 2. J. Clin. Oncol. 37, 230–238 (2019).
Dong, Y. et al. Higher matrix stiffness as an independent initiator triggers epithelial-mesenchymal transition and facilitates HCC metastasis. J. Hematol. Oncol. 12, 112 (2019).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81903143), Innovative Research Groups of National Natural Science Foundation of China (No. 81721091), National S&T Major Project (No. 2017ZX10203205), Zhejiang International Science and Technology Cooperation Project (No. 2016C04003), Research Unit Project of Chinese Academy of Medical Sciences (2019-I2M-5-030), and Major Program of National Natural Science Foundation of China (No. 91542205).
Author information
Authors and Affiliations
Contributions
Y.Q. conceived the idea, designed the study, and supervised our work. S.L. conceived the idea and supervised our work. J.H. retrieved the literature and drafted the paper. L.Z., D.W., L.Z., and S.Z. revised our paper. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, J., Zhang, L., Wan, D. et al. Extracellular matrix and its therapeutic potential for cancer treatment. Sig Transduct Target Ther 6, 153 (2021). https://doi.org/10.1038/s41392-021-00544-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-021-00544-0
This article is cited by
-
Brillouin microscopy monitors rapid responses in subcellular compartments
PhotoniX (2024)
-
Modelling the complex nature of the tumor microenvironment: 3D tumor spheroids as an evolving tool
Journal of Biomedical Science (2024)
-
Transcriptional regulation of cancer stem cell: regulatory factors elucidation and cancer treatment strategies
Journal of Experimental & Clinical Cancer Research (2024)
-
Comprehensive bioinformatics analysis unveils THEMIS2 as a carcinogenic indicator related to immune infiltration and prognosis of thyroid cancer
Scientific Reports (2024)
-
Loss of the extracellular matrix glycoprotein EMILIN1 accelerates Δ16HER2-driven breast cancer initiation in mice
npj Breast Cancer (2024)
