
Abstract
The α2a-adrenergic receptor (α2a-AR) agonist guanfacine has been investigated as a potential treatment for substance use disorders. While decreasing stress-induced reinstatement of cocaine seeking in animal models and stress-induced craving in human studies, guanfacine has not been reported to decrease relapse rates. Although guanfacine engages α2a-AR autoreceptors, it also activates excitatory Gi-coupled heteroreceptors in the bed nucleus of the stria terminalis (BNST), a key brain region in driving stress-induced relapse. Thus, BNST α2a-AR heteroreceptor signaling might decrease the beneficial efficacy of guanfacine. We aimed to determine the role of α2a-AR heteroreceptors and BNST Gi-GPCR signaling in stress-induced reinstatement of cocaine conditioned place preference (CPP) and the effects of low dose guanfacine on BNST activity and stress-induced reinstatement. We used a genetic deletion strategy and the cocaine CPP procedure to first define the contributions of α2a-AR heteroreceptors to stress-induced reinstatement. Next, we mimicked BNST Gi-coupled α2a-AR heteroreceptor signaling using a Gi-coupled designer receptor exclusively activated by designer drug (Gi-DREADD) approach. Finally, we evaluated the effects of low-dose guanfacine on BNST cFOS immunoreactivity and stress-induced reinstatement. We show that α2a-AR heteroreceptor deletion disrupts stress-induced reinstatement and that BNST Gi-DREADD activation is sufficient to induce reinstatement. Importantly, we found that low-dose guanfacine does not increase BNST activity, but prevents stress-induced reinstatement. Our findings demonstrate a role for α2a-AR heteroreceptors and BNST Gi-GPCR signaling in stress-induced reinstatement of cocaine CPP and provide insight into the impact of dose on the efficacy of guanfacine as a treatment for stress-induced relapse of cocaine use.
Similar content being viewed by others

Introduction
Stress is a precipitating factor for craving and relapse in cocaine use disorder (CUD) [1,2,3]; however, there are no FDA-approved medications for the treatment of relapse in CUD. α2a-adrenergic receptor (α2a-AR) agonists inhibit stress-induced reinstatement of operant drug-seeking and conditioned place preference (CPP), animal models useful in the study of stress-induced relapse [4,5,6,7]. In clinical laboratory studies, these compounds have been investigated for stress-induced cocaine craving [8, 9], but the application of full α2a-AR agonists for the treatment of CUD has been limited due to adverse effects such as sedation and hypotension [10, 11]. Pretreatment with the α2a-AR partial agonist guanfacine reduces stress-induced craving in female and stress-induced sympathetic tone in male CUD patients [12]. Guanfacine also decreases stress-induced craving of nicotine and alcohol in male and female patients, suggesting a potentially broad applicability for the treatment of stress-induced drug use [12, 13]. Although guanfacine decreases stress, craving, and withdrawal symptoms in clinical trials, it has not been reported to reduce relapse rates [14, 15].
The mechanisms underlying the anti-drug craving effects of guanfacine are unknown, but it has been suggested that guanfacine blunts stress responses through its actions at presynaptic Gi protein-coupled (Gi-coupled) autoreceptors, which decrease norepinephrine (NE) release [16, 17]. Pre-clinical studies show that at low doses, guanfacine blocks stress-induced activation of the extended amygdala, a group of brain regions that contains the central amygdala and bed nucleus of the stria terminalis (BNST)[18]. NE-signaling within the BNST is critical for stress-induced reinstatement of CPP, and previous research suggests that α2a-AR autoreceptors might suppress stress-induced reinstatement by decreasing NE-mediated activation of BNST beta-adrenergic receptors [19,20,21,22].
Guanfacine also activates postsynaptic α2a-AR heteroreceptors [23]. α2a-AR heteroreceptors are expressed in non-adrenergic cells and regulate many of the pharmacological and physiological effects of α2a-AR agonists, including analgesia, sedation, and improvements in cognition [24]. We previously reported that within the dorsal BNST (dBNST), a high dose of guanfacine increased cFOS, a proxy marker of cellular activity, via activation of α2a-AR heteroreceptors and the subsequent blockade of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels [23, 25, 26]. Furthermore, functionally mimicking intra-dBNST α2a-AR heteroreceptor signaling using virally expressed Gi-coupled designer receptors exclusively activated by designer drug (Gi-DREADDs) also increased dBNST cFOS proportionally to high-dose guanfacine, and produced anxiety-like responses [26]. Given the prominent role of BNST activity in stress-induced reinstatement of cocaine seeking behavior, and the interest in guanfacine as a potential treatment for CUD [9], understanding how α2-AR heteroreceptor signaling may regulate these behaviors is imperative. In this study, we demonstrate that α2-AR heteroreceptors are necessary for stress-induced reinstatement of CPP, and that mimicking their signaling in the dBNST using Gi-coupled DREADDs is sufficient to induce reinstatement. Finally, we show that a low dose of guanfacine does not increase dBNST activity but blocks stress-induced reinstatement.
Methods and materials
Animals
Male and female wild-type (WT), α2a-AR knockout (KO), and KO mice re-expressing α2a-ARs under the DBH promoter (HeteroKO), bred in house and maintained on a C57BL/6 J background were used [26, 27]. For chemogenetic studies, male C57BL/6 J mice (Jackson Laboratories; Bar Harbor, ME) were delivered at 6 or 7 weeks of age and acclimated for at least 1 week before surgical manipulations. For all conditioning experiments, mice were singly-housed at least 2 weeks prior to experiments. Male and female C57BL/6 J mice were used for immunohistochemical experiments and were group-housed with 2–5 mice per cage. All procedures were approved by the Vanderbilt University animal care and use committee.
Behavior
Cocaine CPP training, extinction, and reinstatement testing was conducted as previously described [19]. Testing took place in open-field arenas with two-chamber preference inserts. During conditioning, mice received injections of cocaine (15 mg/kg i.p.) or saline and confined to alternating sides of the CPP apparatus. On the post-conditioning testing day and subsequent days, mice were allowed to move freely between sides. During extinction training, mice were placed in the CPP apparatus daily until they reached the extinction criterion. Mice underwent reinstatement testing 24 h after reaching the extinction criterion. For stress-induced reinstatement of CPP, mice underwent forced swim stress in a beaker of warm water (22–26ο C) for 6 min and were placed in the CPP apparatus. For pharmacological studies, WT mice were injected with guanfacine (0.15 mg/kg, i.p.) 30 min prior to forced swim stress. To control for potential stress-induced biases in side occupancy, mice underwent a modified mock CPP procedure in which they only received injections of saline during the conditioning phase (see Supplement for more details on all procedures).
Stereotaxic surgery
Intracranial virus injections bilaterally targeting the dBNST with AAV5-CaMKIIα-hM4Di-mcherry or AAV5-CaMKIIα-mcherry were performed as previously described [26, 28]. All mice were allowed to recover for at least 3 weeks prior to behavioral testing.
Chemogenetic induction of reinstatement testing
hM4Di- and mcherry-injected mice underwent cocaine CPP conditioning and extinction as described above. Twenty-four hours after reaching the extinction criterion, mice received injections of clozapine-N-oxide (CNO) (3 mg/kg i.p.) [26] 30 min prior to being placed in the CPP apparatus.
Guanfacine-induced cFOS upregulation assay
The cFOS upregulation assay was conducted as previously described [26, 29]. WT mice received i.p. injections of either vehicle (saline) or guanfacine (0.15 mg/kg or 1 mg/kg), and 90 min later, were transcardially perfused. Brain sections containing the dBNST were harvested and processed using immunohistochemistry (see Supplement for details).
Statistics
Data are represented as means, means ± SEM, or medians. All statistics were run using Prism 8 (GraphPad, La Jolla, CA). Differences between groups were assessed using t-tests, one-way, or 2-way repeated measures ANOVAs. Differences in standard deviation between groups were assessed using the Brown-Forsythe test, when significant differences in standard deviation were found using this test, they are reported in the text. Significance was set at α = 0.05. When significant main effects were obtained using ANOVA testing, appropriate post-hoc comparisons between groups were performed (see Supplement for detailed list of statistical results).
Results
α2a-AR full or heteroreceptor deletion does not impact the acquisition or extinction of cocaine CPP
Consistent with previous studies [30, 31], we determined that conditioning increased the amount of time WT, KO, and HeteroKO mice spent on the cocaine-paired side during the post-conditioning session, with a main effect of conditioning (pre-conditioning test session (PRE) vs post-conditioning test session (POST), (F2, 30 = 128.7, p < .0001; two-way RM ANOVA), and a trend towards a main effect of genotype (WT, KO, or HeteroKO), (F2, 30 = 2.95, p = .057) that is likely due to the HeteroKO mice spending less time in the cocaine-paired side when compared to WTs and KOs (in sec: 819 ± 113 for WT, 821 ± 138 for KO, and 723 ± 59 for HeteroKO). The genotype x conditioning interaction was not significant (F2, 30 = .99, p = .38) (Fig. 1c). We found no between-genotype differences in the increase in time on the paired side following conditioning, represented by the preference score (one-way ANOVA), or differences in locomotion during the post-conditioning session (two-way RM ANOVA) (Fig. 1d, e).

a Genetic model used to determine relative α2A-AR auto- and heteroreceptor function. b Timeline and schematic of conditioned place preference (CPP) procedure. c Conditioning increased the amount of time mice from all genotypes spent on the paired side when compared to the pre-conditioning session. Data displayed as means with superimposed individual points. d There was no difference between genotypes in the preference scores (% change in time spent on the paired side from the pre- and post-conditioning sessions). Data displayed as individual points overlaid on top of medians (solid lines) and quartiles (dashed lines). e There were no intra-genotype differences in the distance travelled in the CPP apparatus during the pre- and post-conditioning sessions. f There were no inter-genotype differences in the latency to reach the extinction criterion. Data displayed as means plus individual points, or means and individual data points ± SEM (e) (WT n = 9, KO n = 8, HeteroKO n = 12) (****p < .0001) (PRE: pre-conditioning session, POST: post-conditioning session).
Next, we determined the impact of full or heteroreceptor deletion on the extinction of cocaine CPP (see Materials and Methods, Fig. 1f). We found no differences in the latency to reach the extinction criterion between groups (one-way ANOVA; in days: 3 ± 1 for WT, KO, and HeteroKO) (Fig. 1f). Taken together, these findings suggest that α2a-AR auto- and heteroreceptors do not play a role in the acquisition or extinction of cocaine CPP.
α2a-AR full or heteroreceptor deletion disrupts stress-induced reinstatement of CPP
We next determined the impact of α2a-AR KO or HeteroKO on stress-induced reinstatement of cocaine CPP. Twenty-four hours after reaching the extinction criterion, WT, KO, and HeteroKO mice underwent stress-induced reinstatement testing (Fig. 2a). There was a main effect of stress (extinction session (EXT) vs stress-induced reinstatement session (STRESS)) but no significant genotype × stress interaction (stress F1, 28 = 15.01, p = .0006 interaction F2, 28 = 2.31, p = .11, 2-way RM ANOVA). Sidak’s multiple comparisons post-hoc test revealed that stress only significantly increased time in the paired side in WT mice when compared to the last day of extinction (EXT vs STRESS: WT p < .01, KO p = .78, HeteroKO p = .15) (Fig. 2b). Stress induced a significant difference in the variance of the preference score of WT, KO, and HeteroKO mice (Brown Forsythe test; F2, 27 = 5.67, p < .001) that was driven by a stress-induced increase in standard deviation (SD) in KO and HeteroKO mice (WT SD = 7.21, KO SD = 24.51, HeteroKO SD = 26.31) (Fig. 2c). There was a main effect of stress on locomotion, but no effect of genotype, or the interaction (stress F1, 22 = 76.20, p < .0001, genotype F2, 22 = .03, p = .97, interaction F2,22 = .45, p = .64; 2-way RM ANOVA; Sidak’s multiple comparisons post-hoc test, EXT vs STRESS: WT p < .0001 KO p < .0001, HeteroKO p = .0005) (Fig. 2d). These findings suggest that stress reinstates cocaine CPP in WT mice, and that this reinstatement might be disrupted in KO mice. Furthermore, this disruption is not ameliorated by re-expression of α2a-AR autoreceptors in heteroKO mice.

a Timeline and schematic of stress-induced reinstatement of CPP procedure. b during the stress test session (STRESS), 6 min of forced swim stress increased the time WT mice spent on the paired side compared to the last day of extinction (EXT) and this increase was different from heteroKO mice. Data displayed as means plus individual points. c There was a difference between genotypes in the variance of preference scores (Brown-Forsythe test for differences in SD, **$ < .01). Data displayed as individual points overlaid on top of medians (solid lines) and quartiles (dashed lines). d There was an intra-genotype difference in distance travelled in the CPP apparatus during EXT and STRESS. Data displayed as means plus individual points (WT n = 9, KO n = 8, HeteroKO n = 12) (**p < .001, ****p < .0001) (EXT: last extinction session, STRESS: stress-induced reinstatement session).
α2a-AR full or heteroreceptor deletion does not produce stress-dependent biases in side occupancy in cocaine-naïve mice
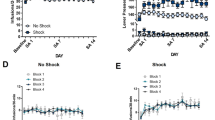
The above findings suggest that, by suppressing locomotion, stress may spuriously increase time on one side of the chamber in a subset of KO and HeteroKO mice, independent of the retrieval of cocaine CPP. We tested this hypothesis by conducting a mock saline CPP experiment followed by stress-induced reinstatement testing. Mice were subjected to a CPP, extinction, and reinstatement procedure as described above, but only received injections of saline during the conditioning stage (mock conditioning) (Fig s1A). Mock conditioning, genotype, or the mock conditioning × genotype interaction, did not increase time in a randomly-paired side (two-way RM ANOVA), or alter the preference score of WT, KO, or HeteroKO mice (one-way ANOVA) (Fig s1B, C). Interestingly, mock conditioning decreased locomotion in WT and HeteroKO mice, but failed to do so in full KO mice (in cm: 3247 ± 937 for WT, 5115 ± 537 for KO, and 3537 ± 1618 for HeteroKO). This decrease in locomotion was statistically significant, with main effects for both the genotype and mock conditioning factors as well as the interaction (mock conditioning F1, 27 = 4.12, p = .017, genotype F2, 27 = 11. 54, p = .029, interaction F2,27 = 4.88p < .01; two-way RM ANOVA, Sidak’s multiple comparisons test, PRE vs POST: WT p < .01, KO p = .92, HeteroKO p < .05) (Fig s1D). The higher level of locomotion in KO mice following repeated exposure to the chamber is consistent with previous findings suggesting that autoreceptors play a role in suppressing spontaneous hyper-locomotion [27].
Twenty-four hours after the last mock extinction training session, mice were subjected to 6 min of forced swim stress and placed in the CPP apparatus (Fig. 3a). Cocaine-naïve mice did not show a significant change in preference for a randomly-paired side of the chamber after stress (two-way RM ANOVA), and we found no inter-genotype differences in preference score of the cocaine-naïve WT, KO, and HeteroKO mice (one-way ANOVA) (Fig. 3b, c). Interestingly, stress increased the SD of the preference score across genotypes when compared to the mock post-conditioning test session (26.65 vs 12.56 for WT, 21.76 vs 14.25 in KO, and 19 vs 11.51 for HeteroKO). This increase in SD demonstrates an enhanced variability in how much time mice spent on each side of the chamber. KO mice showed increased locomotion during the last day of mock extinction, which was significantly different from WT, with main effects of genotype and stress but no interaction (genotype F2,20 = 3.77, p < .05, stress F1,20 = 68, p < .0001, interaction F2,20 = 1.71, p = .205, two-way ANOVA; Sidak’s multiple comparisons test, EXT vs STRESS: WT p < .05, KO p < .0001, HeteroKO p < .01). However, mice across all genotypes showed a stress-induced suppression in locomotion during the mock reinstatement test with a main effect of stress (F1, 20 = 68, p < .0001; two-way RM ANOVA). (Fig. 3d). Taken together, these findings indicate that stress suppresses locomotion and increases variability in side occupancy in cocaine-naïve mice and suggests that variability increases due to decreased movement between sides of the CPP chamber.

a Timeline and schematic of mock conditioning, extinction, and stress-induced reinstatement test. b Stress did not change time in a randomly paired side across genotypes in cocaine-naïve mice. c Stress did not produce differences in the preference score between genotypes. d Stress suppressed locomotion across genotypes and there was a difference in locomotion between WT and KO mice during the last day of mock extinction (EXT) (WT n = 8, KO n = 8, HeteroKO n = 11). Data displayed as means with individual data points (B and C) or medians (solid lines) and quartiles (dashed lines with individual data points (C and G) (*p < .05, **p < .01, ****p < .0001) (EXT: last extinction session, STRESS: stress-induced reinstatement session).
Activation of dBNST Gi-coupled GPCR signaling reinstates cocaine CPP
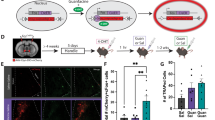
We previously reported that α2a-AR heteroreceptors mediate increases in dBNST activity produced by α2a-AR agonists such as guanfacine, as measured by cFOS upregulation [26]. Due to the fact that α2a-AR receptors are Gi-coupled GPCRs, we previously employed a Gi-coupled DREADD strategy to mimic α2a-AR heteroreceptor signaling and found hM4Di, expressed under the CaMKIIα promoter, activation is both sufficient to increase dBNST cFOS in a similar proportion of cells as guanfacine and occlude guanfacine-induced cFOS upregulation, suggesting the recruitment of an overlapping populations of neurons [26]. Due to the prominent role of the BNST in stress-induced reinstatement [32, 33], we investigated whether hM4Di enhancement of dBNST activity would be sufficient to reinstate cocaine CPP. C57BL/6 J mice were micro-injected in the dBNST with either AAV5-CaMKIIα-mcherry or AAV5-CaMKIIα-hM4Di:mcherry, which produced robust expression in this region (Fig. 4b, c, sF2). Following 4 weeks of recovery, mice underwent CPP training, testing, and extinction as described above (sF3. A). Conditioning increased time in the paired side for both mcherry- and hM4Di-expressing mice with a main effect of conditioning, but no effects of virus (mcherry vs hM4Di) or the interaction (conditioning F1, 12 = 66.28, p < .0001, virus F1,12 = .00, p = .996, interaction F1,12 = 1.26, p = .282; two-way RM ANOVA; Sidak’s multiple comparisons test, PRE vs POST: mcherry p < .01, hM4Di. p < .0001) (Fig s3B). We also found no inter-condition differences in preference score (unpaired t-test, p = .275) (Fig s3C), or latency to reach the extinction criterion (unpaired t-test, P = .593) (Fig s3D). Twenty-four hours after reaching the extinction criterion, mcherry- and hM4Di-expressing mice were injected with CNO (3 mg/kg, i.p.) as previously described [26] and following a 30-min period, mice underwent reinstatement testing (Fig. 4a). CNO treatment significantly increased the time hM4Di-expressing mice spent on the paired side when compared to the last day of extinction training, but failed to do so in mcherry-expressing mice (Fig. 4d). There was a significant main effect for CNO (EXT vs CNO sessions) and a trend towards significance for the interaction, but no effect of virus (CNO F1, 11 = 8, p < .05, interaction F1, 11 = 4.8, p = .0506; virus F1,11 = 1.43, p = .25, two-way RM ANOVA, Sidak’s multiple comparisons test, EXT vs CNO: mcherry, p = .9, hM4Di p < .01). There was a significant difference between the preference score of hM4Di- and mcherry-expressing mice (unpaired t-test, t2,11 = 2.239, p < .05) (Fig. 4e). CNO-treatment did not alter locomotion in hM4Di- or mcherry-expressing mice (two-way RM ANOVA) (Fig. 4f). Further, we found that in an additional group of mice in which the location of mcherry or hM4Di was off target (Fig s4A), CNO did not induce reinstatement, or alter locomotion (Fig s4B-D). Taken together, these findings suggest that engagement of Gi-GPCR signaling in the dBNST is sufficient to reinstate cocaine CPP without impacting locomotion.

a Timeline and schematic showing surgery, recovery and CNO-induced reinstatement of CPP procedure. b Schematic and coordinates of injection site for mcherry and mcherry-tagged hM4Di. c Low magnification (10×) images showing spread of mcherry and mcherry-tagged hM4Di in BNST. Scale bar = 200 μm. d CNO treatment (CNO) increased the time hM4Di-expressing mice spent on the paired side when compared to the last day of extinction (EXT) but did not increase the time in mcherry-expressing controls. Data displayed as means plus individual data points. e CNO treatment increased the CPP score of hM4Di-expressing mice compared to mcherry-expressing controls. Data displayed as individual points overlaid on top of medians (solid lines) and quartiles (dashed lines). f CNO treatment did not alter locomotion in mcherry- or hM4Di-expressing mice when compared to the EXT session. Data displayed as means with individual data points (mcherry n = 5, hM4Di = 8) (p < .05) (EXT: last extinction session, CNO: CNO-induced reinstatement session).
A low dose of guanfacine that does not increase cFOS within the BNST blocks stress-induced reinstatement of cocaine CPP
While non-specific α2-AR agonism blocks stress-induced reinstatement of drug seeking, high doses of these compounds have negative effects, which limit their clinical applications [10]. We previously reported that a 1 mg/kg dose of the α2a-AR partial agonist guanfacine, which produces a strong sedative effect, increased dBNST activity as measured by cFOS upregulation [26]. Given the prominent role of BNST activity in reinstatement of drug seeking and anxiety-like behaviors [34,35,36], we hypothesized that a lower dose of guanfacine that shows antidepressant- and anxiolytic-like effects (0.15 mg/kg), would not increase cFOS in the dBNST but would block stress induced reinstatement [37, 38]. To determine dose-dependent changes in dBNST activity, we assessed cFOS expression within the dBNST following guanfacine administration [26]. Mice were injected with either saline (vehicle, VEH), low (0.15 mg/kg), or high dose (1 mg/kg) guanfacine (Fig. 5a). As we previously reported, high dose guanfacine increased the number of cFOS positive cells within the dBNST, however, this effect that was not present in VEH- or low dose-treated mice (F2,14 = .154, p < .0001; one-way ANOVA, Sidak’s multiple comparisons test: VEH vs 0.15 mg/kg p = .88, VEH vs 1 mg/kg p < .0001) (Fig. 5b, c).

a Timeline and schematic of drug injection and immunohistochemistry experiment. Following an hour of acclimation mice were injected with saline (Vehicle, VEH) or guanfacine (GUAN) and sacrificed 90 min post-injection. b High magnification (×20) images showing cFOS staining in dorsal BNST. Scale bar = 50 μm. c 1 mg/kg injection of GUAN increased cFOS within the BNST. (VEH n = 6, 0.15 mg/kg n = 6, 1 mg/kg n = 5). d Timeline and schematic of stress-induced reinstatement blockade experiment. Mice were injected with VEH or GUAN (0.15 mg/kg) 30 min prior to stress. e Stress increased time in the paired side in VEH-treated mice but failed to do so in GUAN-treated mice. Data displayed as means plus individual data points. f There was a difference in the CPP score of VEH-treated and GUAN-treated mice. Data displayed as individual points overlaid on top of medians (solid lines) and quartiles (dashed lines). g Stress suppressed locomotion in VEH- and GUAN-treated mice but there were not inter-treatment differences in locomotion. Data displayed as means plus individual data points (VEH n = 7, GUAN n = 7) (*p < .05, ****p < .0001) (EXT; last extinction session, STRESS; stress-induced reinstatement test session).
We next examined the potential anti-reinstatement effects of low dose guanfacine. WT mice that had undergone CPP training and extinction were injected with either VEH or low dose guanfacine, 30 min prior to stress (Fig. 5d). While we did not find main effects of drug (VEH vs GUAN) or stress, we found a main effect of the treatment × session interaction (Drug F1,12 = .01, p = .9, stress F1,12 = 1.49, p .24, interaction F1,12 = 5.843, p = .032; two-way RM ANOVA), driven by a stress-induced increase in time on the paired side in VEH-treated mice that was not present in guanfacine-treated mice (Fig. 5e). Guanfacine-treated mice also showed a significant decrease in preference score when compared to VEH-treated mice (t = 2.417, df = 12, p < .05; unpaired t-test) (Fig. 5f). While stress significantly suppressed locomotion in both groups, there were no differences in locomotion in VEH- and guanfacine-treated mice following stress, and no main effect of drug, suggesting that low dose guanfacine treatment did not have further sedative effects (interaction F1,12 = 10.34, p < .01, drug F1,12 = .18, p = .67, stress F1,12 = 222.5, p < .0001; two-way RM ANOVA, Sidak’s multiple comparisons test, STRESS: VEH vs guanfacine p = .44, EXT vs STRESS: VEH p < .0001, guanfacine p < .0001) (Fig. 5g). Taken together, these findings suggest low dose guanfacine does not increase the activity of the BNST but blocks stress-induced reinstatement without impacting locomotion.
Discussion
α2a-AR agonists such as guanfacine have risen in popularity for the treatment of several psychiatric conditions [39, 40]. α2a-AR agonists target autoreceptors expressed in adrenergic neurons and heteroreceptors expressed in non-adrenergic cells. Determining the relative contributions of these receptor populations to the pharmacological effects of α2a-AR agonists and behavior has been challenging, due to the inability to distinguish these populations using conventional pharmacological or genetic approaches [27]. In the current study, we used selective genetic deletion models, behavior, histology, and chemogenetics to define the role of α2a-AR heteroreceptors on stress-induced reinstatement of cocaine CPP.
In our unbiased two-sided CPP apparatus, stress reinstated cocaine CPP in all wild-type mice, but only a fraction of α2a-AR full and HeteroKO mice. Following stress, we observed a high degree of variability in the amount of time α2a-AR KO and HeteroKO mice spent on the two sides of the apparatus. This increase in variability was also found in cocaine-naïve mice from all genotypes, but not in cocaine-treated WTs. Notably, stress reduces locomotion in the CPP assay independent of genotype or drug exposure history. Therefore, our data suggest that in α2a-AR full and heteroreceptor knockouts, stress-induced reinstatement of CPP is replaced by increased occupancy in an arbitrary side of the chamber, due to an overall reduction in activity rather than side preference. While this is a parsimonious interpretation of the data, currently we cannot unequivocally rule out the possibility that some of the α2a-AR full and HeteroKO mice show stress-induced reinstatement. Regardless of this limitation, it is still clear that α2a-AR heteroreceptors positively regulate reinstatement behavior.
Our previous findings have demonstrated functional recovery of autoreceptors in the dBNST in this model [26], suggesting the surprising result that α2a-AR heteroreceptor deletion disrupts the ability of stress to drive reinstatement of preference for the cocaine-paired side. It is possible that within the dBNST, α2a-AR auto- and heteroreceptors play opposing roles, where autoreceptors might prevent reinstatement by decreasing NE release, heteroreceptor, and beta-adrenergic signaling [20, 21]. Conditions in which large amounts of NE are released into the BNST, such as chronic stress, might override autoreceptor regulation to lead to reinstatement [41, 42].
We used a chemogenetic approach to mimic dBNST α2a-AR heteroreceptor signaling to determine the effects of Gi-GPCRs on reinstatement of cocaine CPP. We found that acute activation of BNST hM4Di was sufficient to induce reinstatement of CPP. This Gi-DREADD-induced reinstatement is consistent with our findings showing that activation of Gi-DREADDs within the BNST increases activity and anxiety-like responses [26]. Previous work has shown that activation of intra-BNST Gi-DREADDs can inhibit drug consumption or the acquisition of CPP [43,44,45,46]. Notably, these studies focused on alcohol consumption and CPP acquisition without a focus on reinstatement. Thus, our study provides novel insight into the potential role of Gi-GPCR signaling on stress-induced reinstatement of cocaine CPP. Additionally, we previously reported that direct α2a-AR activation or Gi-DREADD mimicking of α2a-AR signaling in excitatory inputs onto BNST CRF cells decreases stress-induced cFOS [29]. Taken together, these findings highlight the heterogeneous nature of α2a-AR regulation of BNST activity. Future studies will aim to determine the role of different BNST cell populations and projections in the pro-reinstatement effects of Gi-coupled GPCR signaling.
A pro-reinstatement role of heterosynaptic α2a-ARs contrasts with previous reports suggesting that systemic administration of the nonselective α2-AR antagonist yohimbine and the α2a-AR antagonist BRL44408 reinstate CPP [19]. While yohimbine antagonizes α2a-ARs and increases norepinephrine levels, it is also a 5-HT1a receptor agonist [47, 48]. Yohimbine-induced reinstatement of cocaine- and food-seeking, or cocaine CPP is not blocked by application of the α2a-AR agonist clonidine [19, 49]. Indeed, yohimbine’s effects on drug-associated behaviors require orexin and serotonin 5-HT1a receptor signaling [30, 50]. Furthermore, we have previously reported that within the BNST, yohimbine produces α2a-AR independent, orexin receptor-1 dependent decreases in excitatory transmission [30]. Therefore, our current findings add to the growing body of evidence suggesting that the pro-reinstatement effects of yohimbine may not be dependent on modulation of adrenergic signaling. BRL44408 also reinstated CPP, presumably through inhibition of α2a-AR autoreceptors [7]. The location of these receptors mediating the pro-reinstatement effects of this compound remain unknown, but would be predicted to involve presynaptic terminal α2a-ARs [29].
As we previously found that a high dose of guanfacine increases BNST activity, we next sought to determine if a low dose of guanfacine, which has been previously reported to reduce anxiety-like behaviors, would lack excitatory effects in the BNST [37, 38]. We replicated our previous finding showing that a high dose of guanfacine increases cFOS in the BNST, but found that the low dose did not. This suggests that a low dose of guanfacine does not engage BNST α2a-AR heteroreceptors. We also found that low dose guanfacine blocked stress-induced reinstatement of CPP without impacting locomotion. While previous work has shown that higher doses of the α2A-AR agonist clonidine do not block stress-induced reinstatement [19], the strong hypo-locomotive effects of the 1 mg/kg dose of guanfacine prevented us from testing this dose in the current study. Future studies will aim to determine the effects of intra-dBNST manipulation of α2a-AR signaling. These findings may inform the development of dose-targeted guanfacine for the treatment of substance use disorders and are congruent with recent reports suggesting that the efficacy of guanfacine in some clinical applications is dose-dependent [51, 52].
Notably, while α2A-AR agonism shows sex-dependent effects in patients with CUD [12], we did not find any apparent sex differences in the effects of α2a-AR full or heteroreceptor KO, therefore data from male and female mice were combined in our statistical analyses (see Supplemental Figs. 5–8 for values separated by sex). However, one limitation of the current study is that it is not sufficiently powered to analyze more subtle sex differences. While a recent report showed that the low dose of guanfacine used in the current study equally prevented forced swim-induced reinstatement of nicotine CPP in male and female mice [18], future investigations will be aimed at determining potential sex differences in α2a-AR regulation of BNST activity as well as low-dose guanfacine on cocaine CPP reinstatement.
Our findings demonstrate a previously unknown role of α2a-AR heteroreceptors in stress-induced reinstatement of cocaine-associated behaviors. Additionally, we also expand on previous reports suggesting that guanfacine should be further explored as a potential treatment for CUD and other drug use disorders.
Funding and disclosures
This work was supported by National Institutes of Health Grants R01DA042475, R01DA042475S1 (DGW), and T32GM007628 (REP). Behavioral data acquisition and analyses were performed in part through the use of the Vanderbilt Mouse Neurobehavioral Core. Imaging and image data analyses were performed in part through the use of the Vanderbilt University School of Medicine Cell Imaging Shared Resource (supported by National Institutes of Health Grants CA68485, DK20593, DK58404, DK59637, and EY08126). S.P. has received research contract support from H. Lundbeck A/S unrelated to the current work. S.P is also a scientific consultant for H. Lundbeck A/S, Psy therapeutics and Sophran Therapeutics, unrelated to the present work. All other authors report no financial interests or potential conflicts of interests.
References
Karlsgodt KH, Lukas SE, Elman I. Psychosocial stress and the duration of cocaine use in non-treatment seeking individuals with cocaine dependence. Am J Drug Alcohol Abus. 2003;29:539–51.
Levandowski ML, Tractenberg SG, de Azeredo LA, De Nardi T, Rovaris DL, Bau CHD, et al. Crack cocaine addiction, early life stress and accelerated cellular aging among women. Prog Neuro-Psychopharmacol Biol Psychiatry. 2016;71:83–9.
Sinha, R, Catapano, D, O’Malley S. Stress-induced craving and stress response in cocaine dependent individuals. Psychoneuroendocrinology.1999; 142:343–51.
Erb S, Hitchcott PK, Rajabi H, Mueller D, Shaham Y, Stewart J. Alpha-2 adrenergic receptor agonists block stress-induced reinstatement of cocaine seeking. Neuropsychopharmacology. 2000;23:138–50.
Shaham Y, Highfield D, Delfs J, Leung S, Stewart J. Clonidine blocks stress-induced reinstatement of heroin seeking in rats: An effect independent of locus coeruleus noradrenergic neurons. Eur J Neurosci. 2000;12:292–302.
Lê AD, Harding S, Juzytsch W, Funk D, Shaham Y. Role of alpha-2 adrenoceptors in stress-induced reinstatement of alcohol seeking and alcohol self-administration in rats. Psychopharmacol (Berl). 2005;179:366–73.
Mantsch JR, Baker DA, Funk D, Lê AD, Shaham Y. Stress-induced reinstatement of drug seeking: 20 years of progress. Neuropsychopharmacology. 2016;41:335–56.
Fox HC, Seo D, Tuit K, Hansen J, Kimmerling A, Morgan PT, et al. Guanfacine effects on stress, drug craving and prefrontal activation in cocaine dependent individuals: Preliminary findings. J Psychopharmacol. 2012;26:958–72.
Fox H, Sinha R. The role of guanfacine as a therapeutic agent to address stress-related pathophysiology in cocaine-dependent individuals. vol. 69. 1st edn. Elsevier Inc.; 2014.
Khan ZP, Ferguson CN, Jones RM. Alpha-2 and imidazoline receptor agonists. Anaesthesia. 1999;54:146–65.
Seedat YK. Clonidine and guanfacine—comparison of their effects on haemodynamics in hypertension. South Afr Med J. 1985;67:557–9.
Fox HC, Morgan PT, Sinha R. Sex differences in guanfacine effects on drug craving and stress arousal in cocaine-dependent individuals. Neuropsychopharmacology. 2014;39:1527–37.
McKee SA, Potenza MN, Kober H, Sofuoglu M, Arnsten AFT, Picciotto MR, et al. A translational investigation targeting stress-reactivity and prefrontal cognitive control with guanfacine for smoking cessation. J Psychopharmacol. 2015;29:300–11.
Krupitsky E, Zvartau E, Blokhina E, Verbitskaya E, Tsoy M, Wahlgren V, et al. Naltrexone with or without guanfacine for preventing relapse to opiate addiction in St.-Petersburg, Russia. Drug Alcohol Depend. 2013;132:674–80.
Haney M, Cooper ZD, Bedi G, Herrmann E, Comer SD, Reed SC, et al. Guanfacine decreases symptoms of cannabis withdrawal in daily cannabis smokers. Addict Biol. 2019;24:707–16.
Brown J, Doxey JC, Handley S. Effects of α-adrenoceptor agonists and antagonists and of antidepressant drugs on pre- and postsynaptic a-adrenoceptors. Eur J Pharm. 1980;67:33–40.
Okada M, Fukuyama K, Kawano Y, Shiroyama T, Suzuki D, Ueda Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology. 2019;156:107547.
Lee AM, Calarco CA, McKee SA, Mineur YS, Picciotto MR. Variability in nicotine conditioned place preference, stress‐induced reinstatement, and effects of guanfacine in male and female mice. Genes, Brain Behav. 2019;e12601:1–17.
Mantsch JR, Weyer A, Vranjkovic O, Beyer CE, Baker Da, Caretta H. Involvement of noradrenergic neurotransmission in the stress- but not cocaine-induced reinstatement of extinguished cocaine-induced conditioned place preference in mice: role for β−2 adrenergic receptors. Neuropsychopharmacology. 2010;35:2165–78.
Vranjkovic O, Gasser PJ, Gerndt CH, Baker DA, Mantsch JR. Stress-Induced Cocaine Seeking Requires a Beta-2 Adrenergic Receptor-Regulated Pathway from the Ventral Bed Nucleus of the Stria Terminalis That Regulates CRF Actions in the Ventral Tegmental Area. J Neurosci. 2014;34:12504–14.
Leri F, Flores J, Rodaros D, Stewart J. Blockade of stress-induced but not cocaine-induced reinstatement by infusion of noradrenergic antagonists into the bed nucleus of the stria terminalis or the central nucleus of the amygdala. J Neurosci. 2002;22:5713–8.
McReynolds JR, Vranjkovic O, Thao M, Baker DA, Makky K, Lim Y, et al. Beta-2 adrenergic receptors mediate stress-evoked reinstatement of cocaine-induced conditioned place preference and increases in CRF mRNA in the bed nucleus of the stria terminalis in mice. Psychopharmacol (Berl). 2014;231:3953–63.
Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, et al. α2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410.
Gilsbach R, Albarrán-Juárez J, Hein L. Pre- versus Postsynaptic Signaling by α 2-Adrenoceptors. Curr Top Membr. 2011;67:139–60.
Alamo C, López-Muñoz F, Sánchez-García J. Mechanism of action of guanfacine: a postsynaptic differential approach to the treatment of attention deficit hyperactivity disorder (adhd). Actas Esp Psiquiatr. 2016;44:107–12.
Harris NA, Isaac AT, Günther A, Merkel K, Melchior J, Eguakun E, et al. Dorsal BNST α2A -adrenergic receptors produce HCN-dependent excitatory actions that initiate anxiogenic behaviors. J Neurosci. 2018;38:8922–42.
Gilsbach R, Röser C, Beetz N, Brede M, Hadamek K, Haubold M, et al. Genetic dissection of α2-adrenoceptor functions in adrenergic versus nonadrenergic cells. Mol Pharmacol. 2009;75:1160–70.
Paxinos G, Franklin B. The mouse brain in stereotaxic coordinates. Academic Press; 2004.
Fetterly TL, Basu A, Nabit BP, Awad E, Williford KM, Centanni SW, et al. α 2A-adrenergic receptor activation decreases parabrachial nucleus excitatory drive onto BNST CRF neurons and reduces their activity In vivo. J Neurosci 2018;39:472–84.
Conrad KL, Davis AR, Silber.man Y, Sheffler DJ, Shields AD, Saleh Sa, et al. Yohimbine depresses excitatory transmission in BNST and impairs extinction of cocaine place preference through orexin-dependent, norepinephrine-independent processes. Neuropsychopharmacology. 2012;37:2253–66.
Davis AR, Shields AD, Brigman JL, Norcross M, McElligott ZA, Holmes A, et al. Yohimbine impairs extinction of cocaine-conditioned place preference in an α2-adrenergic receptor independent process. Learn Mem. 2008;15:667–76.
Erb S, Stewart J. A role for the bed nucleus of the stria terminalis, but not the amygdala, in the effects of corticotropin-releasing factor on stress-induced reinstatement of cocaine seeking. J Neurosci. 1999;19:1–6.
Vranjkovic O, Pina M, Kash TL, Winder DG. The bed nucleus of the stria terminalis in drug-associated behavior and affect: a circuit-based perspective. Neuropharmacology. 2017;122:100–6.
Crowley NA, Bloodgood DW, Hardaway JA, Kendra AM, McCall JG, Al-Hasani R, et al. Dynorphin Controls the Gain of an Amygdalar Anxiety Circuit. Cell Rep. 2016;14:2774–83.
Avery SN, Clauss JA, Blackford JU. The Human BNST: Functional Role in Anxiety and Addiction. Neuropsychopharmacology. 2016;41:126–41.
Clauss JA, Avery SN, Benningfield MM, Blackford JU. Social anxiety is associated with BNST response to unpredictability. Depress Anxiety. 2019; 36:666–75.
Mineur YS, Bentham MP, Zhou WL, Plantenga ME, McKee SA, Picciotto MR. Antidepressant-like effects of guanfacine and sex-specific differences in effects on c-fos immunoreactivity and paired-pulse ratio in male and female mice. Psychopharmacol (Berl). 2015;232:3539–49.
Mineur YS, Cahuzac EL, Mose TN, Bentham MP, Plantenga ME, Thompson DC, et al. Interaction between noradrenergic and cholinergic signaling in amygdala regulates anxiety- and depression-related behaviors in mice. Neuropsychopharmacology. 2018;43:2118–25.
Fiks AG, Mayne SL, Song L, Steffes J, Liu W, McCarn B, et al. Changing patterns of alpha agonist medication use in children and adolescents 2009–11. J Child Adolesc Psychopharmacol. 2015;25:362–7.
Gowing L, Farrell M, Ali R, Jm W. Alpha2-adrenergic agonists for the management of opioid withdrawal. Cochrane Database of Systematic Reviews 2009. https://doi.org/10.1002/14651858.CD002024.pub3.
Cecchi M, Khoshbouei H, Javors M, Morilak DA. Modulatory effects of norepinephrine in the lateral bed nucleus of the stria terminalis on behavioral and neuroendocrine responses to acute stress. Neuroscience. 2002;112:13–21.
Schmidt KT, Makhijani VH, Boyt KM, Cogan ES, Pati D, Pina MM, et al. Stress-Induced Alterations of Norepinephrine Release in the Bed Nucleus of the Stria Terminalis of Mice. ACS Chem Neurosci. 2019;10:1908–14.
Pleil KE, Rinker Ja, Lowery-Gionta EG, Mazzone CM, McCall NM, Kendra AM, et al. NPY signaling inhibits extended amygdala CRF neurons to suppress binge alcohol drinking. Nat Neurosci 2015;18:545–52.
Rinker JA, Marshall SA, Mazzone CM, Lowery-Gionta EG, Gulati V, Pleil KE, et al. Extended amygdala to ventral tegmental area corticotropin-releasing factor circuit controls binge ethanol intake. Biol Psychiatry. 2017;81:930–40.
Pina MM, Young EA, Ryabinin AE, Cunningham CL. The bed nucleus of the stria terminalis regulates ethanol-seeking behavior in mice. Neuropharmacology. 2015;99:627–38.
Companion MA, Thiele TE. Assessment of ventral tegmental area-projecting GABAergic neurons from the bed nucleus of the stria terminalis in modulating binge-like ethanol intake. Eur J Neurosci. 2018;48:3335–43.
Abercrombie ED, Keller RW, Zigmond MJ. Characterization of hippocampal norepinephrine release as measured by microdialysis perfusion: pharmacological and behavioral studies. Neuroscience. 1988;27:897–904.
Winter JC, Rabin RA. Yohimbine as a serotonergic agent: evidence from receptor binding and drug discrimination. J Pharm Exp Ther. 1992;263:682–9.
Nair SG, Navarre BM, Cifani C, Pickens CL, Bossert JM, Shaham Y. Role of dorsal medial prefrontal cortex dopamine D1-family receptors in relapse to high-fat food seeking induced by the anxiogenic drug Yohimbine. Neuropsychopharmacology. 2011;36:497–510.
Dzung Lê A, Funk D, Harding S, Juzytsch W, Fletcher PJ. The role of noradrenaline and 5-hydroxytryptamine in yohimbine-induced increases in alcohol-seeking in rats. Psychopharmacol (Berl). 2009;204:477–88.
Verplaetse TL, Roberts W, Moore KE, Peltier MR, Oberleitner LM, McKee SA. Pharmacokinetics and pharmacodynamics of immediate-release versus extended-release guanfacine in adult daily smokers. J Clin Psychopharmacol. 2019;39:124–8.
Barcelos NM, Van Ness PH, Wagner AF, MacAvoy MG, Mecca AP, Anderson GM, et al. Guanfacine treatment for prefrontal cognitive dysfunction in older participants: a randomized clinical trial. Neurobiol Aging. 2018;70:117–24.
Acknowledgements
We thank Drs. John D. Allison and Bob Matthews, for technical assistance.
Author information
Authors and Affiliations
Contributions
R.E.P. conceived the study, designed and performed experiments, and co-wrote the paper. A.B. analyzed data, validated viral deposits, and contributed to writing. B.P.N. piloted behavioral studies. N.A.H. performed surgery, assisted in experimental design, and analysis. O.M.F. performed surgery. S.P., R.G., and L.H. provided materials and resources. D.G.W. conceived the study, designed experiments, and co-wrote the paper.
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Perez, R.E., Basu, A., Nabit, B.P. et al. α2A-adrenergic heteroreceptors are required for stress-induced reinstatement of cocaine conditioned place preference. Neuropsychopharmacol. 45, 1473–1481 (2020). https://doi.org/10.1038/s41386-020-0641-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-020-0641-z
This article is cited by
-
An ensemble recruited by α2a-adrenergic receptors is engaged in a stressor-specific manner in mice
Neuropsychopharmacology (2023)
-
Stressing the potential of guanfacine as a treatment for cocaine use disorder
Neuropsychopharmacology (2020)
