
Abstract
Neuropeptide Y (NPY) signaling via limbic NPY1 and 2 receptors (NPY1R and NPY2R, respectively) is known to modulate binge-like ethanol consumption in rodents. However, the role of NPY signaling in the medial prefrontal cortex (mPFC), which provides top-down modulation of the limbic system, is unknown. Here, we used “drinking-in-the-dark” (DID) procedures in C57BL/6J mice to address this gap in the literature. First, the impact of DID on NPY immunoreactivity (IR) was assessed in the mPFC. Next, the role of NPY1R and NPY2R signaling in the mPFC on ethanol consumption was evaluated through site-directed pharmacology. Chemogenetic inhibition of NPY1R+ neurons in the mPFC was performed to further evaluate the role of this population. To determine the potential role of NPY1R+ neurons projecting from the mPFC to the basolateral amygdala (BLA) this efferent population was selectively silenced. Three, 4-day cycles of DID reduced NPY IR in the mPFC. Intra-mPFC activation of NPY1R and antagonism of NPY2R resulted in decreased binge-like ethanol intake. Silencing of mPFC NPY1R+ neurons overall, and specifically NPY1R+ neurons projecting to the BLA, significantly reduced binge-like ethanol intake. We provide novel evidence that (1) binge-like ethanol intake reduces NPY levels in the mPFC; (2) activation of NPY1R or blockade of NPY2R reduces binge-like ethanol intake; and (3) chemogenetic inhibition of NPY1R+ neurons in the mPFC and NPY1R+ mPFC neurons projecting to the BLA blunts binge-like drinking. These observations provide the first direct evidence that NPY signaling in the mPFC modulates binge-like ethanol consumption.
Similar content being viewed by others


1.0 Introduction
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) defines binge drinking as a pattern of consumption that produces blood ethanol concentrations (BECs) greater than 0.08% (80 mg/dL) within a 2-h period [1]. Binge consumption is linked to a variety of health consequences and is a risk factor for the eventual development of ethanol dependence [2,3,4,5]. For over a decade now the neurobiological mechanisms underlying binge drinking have been studied in rodents using the “drinking in the dark” (DID) paradigm [6], which involves providing C57BL/6J mice 2- to 4-h of access to 20% ethanol in place of water beginning 3-h into the animal’s 12-h dark cycle [7, 8].
The extended amygdala (EA) is composed of an interconnected set of brain regions that integrate emotional behaviors and which modulate both initial ethanol consumption and the transition to dependence following repeated use [9,10,11]. Regions of the EA, such as the central nucleus of the amygdala (CeA) and the bed nucleus of the stria terminalis (BNST), have served as a focus in investigating the neural substrates underlying binge drinking [for example [12,13,14]. However, brain regions which regulate EA activity have received comparatively less attention. The medial prefrontal cortex (mPFC) is a key region involved in “top-down” regulation of EA activity [13, 15, 16]. Importantly, this region has been implicated in modulating ethanol consumption [17, 18], and dysregulation of mPFC top-down regulation is thought to significantly contribute to the loss of inhibitory control over drinking [13, 19]. A critical pathway in this top-down regulation is thought to be the glutamatergic projection from the mPFC to the basolateral nucleus of the amygdala (BLA), which serves as the primary input nucleus of the amygdala and is poised to regulate CeA and BNST activity [20]. Given the observed role of these regions in binge-like ethanol intake, this projection may serve as a key site of top-down regulation of excessive ethanol intake [21,22,23].
Neuropeptide Y (NPY) is a 36-amino acid molecule that is known to be critically involved in ethanol-directed behaviors from initial consumption to withdrawal from chronic exposure [24]. NPY is widely expressed throughout the brain where it acts primarily on Gi-coupled NPY 1 (NPY1R), NPY 2 (NPY2R), and NPY 5 (NPY5R) receptors [24, 25]. The NPY1R and NPY5R are typically postsynaptically expressed on glutamatergic principal cells and γ-aminobutyric acid (GABA)-ergic interneurons, while NPY2R is located presynaptically and can serve as either an auto- or heteroreceptor on interneurons [25]. Important to this work, NPY is produced by a subpopulation of GABAergic interneurons distributed throughout the cortical layers of the mPFC [26]. These interneurons play an important role in shaping neurobiological responses through modulation of both glutamatergic and GABAergic mPFC signaling [27, 28]. While there is a growing literature implicating NPY signaling in modulating ethanol intake, to our knowledge the role of NPY signaling in the mPFC in the top-down regulation of excessive ethanol intake has yet to be evaluated. This is notable as, similar to the EA, rats selectively bred for high ethanol intake show low levels of NPY in the mPFC [29], suggest that NPY signaling in this region plays a critical role in ethanol consumption. Given this, we predict enhancing NPY signaling in the mPFC will blunt binge-like ethanol intake.
To address this gap in the literature, we use the DID paradigm to model binge-like ethanol intake in male and female mice on a C57BL/6J background in combination with histological, pharmacological, and chemogenetic approaches. We first show that a history of binge-like ethanol intake reduces NPY levels in both the prelimbic (PL) and infralimbic (IL) subregions of the mPFC. Next, we show that enhancing NPY signaling via site-directed infusion of a NPY1R agonist or a NPY2R antagonist (which increases endogenous NPY release via autoreceptor blockade) both blunt binge-like ethanol drinking in mice. Since NPY1R are Gi-coupled, we then chemogenetically silenced (1) the entire population of NPY1R-expressing (NPY1R+) glutamatergic and GABAergic neurons in the mPFC and next (2) specifically NPY1R+ mPFC neurons projecting to the BLA. In both cases we observed reductions of binge-like ethanol drinking. Effects of pharmacological and chemogentic manipulations did not interact with the effect of sex of the animal, and these treatments did not alter consumption of sucrose or locomotor behavior. These studies provide the first evidence that NPY signaling in the mPFC is critical in the modulation of binge-like ethanol drinking.
2.0 Methods
2.1 Animals
We used male and female C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) or NPY1R-Cre mice (positive for Cre-recombinase expression under the NPY1R promoter, determined by standard polymerase chain reaction genotyping) on a C57BL/6J background >10 weeks old at experiment start (bred in house heterozygous X C57BL/6J (Jackson), originally developed by [30]). Mice were housed individually in an AAALAC accredited vivarium (22 °C with a reversed 12:12 h light:dark cycle, lights on at 20:00). Animals had ad libitum access to Prolab® RMH 3000 (Purina LabDiet®; St. Louis, MO) and water, unless otherwise stated. Prior to experiments, animals acclimated to the experimental housing environment for ≥1 week. All procedures were approved by the University of North Carolina Institutional Animal Care and Use Committee and followed the Guidelines for the Care and Use of Laboratory Animals.
2.2 “Drinking in the Dark” procedures
Binge drinking was modeled using a 4-day DID paradigm as previously described [31]. Three hours into the dark cycle (11:00), water bottles were removed and replaced with sipper tubes containing 20% (v/v) ethanol or 3% (w/v) sucrose in tap water for 2 h. On the fourth day of each cycle (test day), animals were treated with drug or appropriate vehicle ~30 min prior to bottles on (except in the immunohistochemistry (IHC) study, where no treatment was given). Each 4-day DID cycle was separated by a 3-day abstinence. In pharmacological/chemogenetic studies tail blood samples (≈60 μL) were taken immediately following DID procedure to determine BECs with a AM1 Alcohol Analyzer (Analox, London, UK).
2.3 Immunohistochemistry
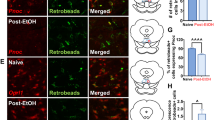
Only male animals were used in IHC analysis. Mice underwent three cycles of ethanol DID or three cycles with water alone [Fig. 1a]. Three cycles was selected based on preliminary mPFC data and previous work in our lab [12]. Immediately following the final DID session (at approximately 13:00 h) each mouse was administered 0.1 mL intraperitoneal (i.p.) injection ketamine/xylazine (6.67 mg/0.1 mL; 0.67 mg/0.1 mL; in 0.9% saline) and perfused transcardially using 0.1 M phosphate buffer saline (PBS; pH = 7.4) and 4% paraformaldehyde in PBS (pH = 7.4). After extraction, brains were postfixed in 4% paraformaldehyde for 24–48 h then sectioned at 40 µm thickness (Leica VT1000S vibratome; Wetzlar, Germany). At this time random identification numbers were assigned to blind experimenter to treatment. Following antigen retrieval (1 h incubation in citrate buffer (10 mM citric acid; 0.05% Tween 20; pH = 6.0) at 66 °C), sections were used for NPY immunoreactivity detection. Sections were blocked in 3% horse serum (0.005% Tween20 in phosphate buffered saline pH = 7.4) for 1 h then incubated in primary anti-NPY (1:1000) (ab6173 sheep polyclonal produced/specificity verified by Abcam, Cambridge, UK) for 72 h at 5 °C. Sections were then incubated 2 h at room temperature in secondary solution (1:5000) (DI-3088 DyLight 488 horse anti-goat, Vector Laboratories, Burlingame, CA) then mounted onto glass slides and coverslipped in Shur Mount (General Data). One tissue set was run without primary antibody to confirm lack of nonspecific florescence. Color images of mPFC subregions were captured through a digital camera (Roper Scientific), mounted on an optical microscope (Leica DM6000), and positive fluorescence was quantified using ImageJ (NIH) (max entropy threshold). Experimenter remained blinded to treatment group until following ImageJ quantification. Unilateral percent area of florescence for 2 sections per animal were averaged together and used for analysis (bregma 1.94–1.54). Representative images in figures were digitally adjusted (brightness/contrast/saturation) for best appearance in publication. All images were enhanced as a group (identical settings).

Three cycles of Binge-Like Ethanol Intake Reduces mPFC NPY Expression. a Timeline of three cycles of DID ethanol or water exposure. b Three cycles of binge-like ethanol intake reduced NPY immunoreactivity relative to animals exposed only to water in the prelimbic (PL) and infralimbic (IL) subregions of the mPFC. c Representative images of NPY IHC in the PL and IL in each treatment group. d NPY immunoreactivity and total binge-like ethanol consumption on the final day of cycle 3 (g/kg) were found to be significantly correlated in the PL, e but not the IL. f NPY immunoreactivity and total water consumption on the final day of cycle 3 (ml/kg) did not correlate in the PL g or IL subregion of the mPFC. (* = main factor: liquid p < 0.05) (scale bar = 100 µm)
2.4 Surgeries
For all surgeries, animals were given intraperitoneal (i.p.) injections (1.5 mL/kg) of an anesthetizing cocktail of xylazine (10 mg/kg) and ketamine (100 mg/kg). For detailed cannula and DREADD injection information see Supplementary methods.
2.5 Drug administration
On DID test days animals underwent micro- or I.P. injection of the experiment-specific drug or appropriate vehicle ~30 min prior to test start. Drugs were as follows: NPY1R agonist Leu, Pro-NPY (99 pmol/0.5 µl/side in saline); NPY2R antagonist BIIE0246 (3 µg/0.5 µl/side in 10% DMSO in saline) (Tocris, Minneapolis, MN); Clozapine-N-Oxide (CNO; I.P. 3.0 mg/kg; microinjection 900 pmol/0.3 µl/side; Sigma-Aldrich, St. Louis, MO or gifted by the NIDA drug depositary; in 0.5% DMSO in Saline). To limit the number of microinjections animals underwent, single drug doses were chosen based on previous dose–response curve experiments performed in our lab [32]. A 2 × 2 Latin-square design was used in each experiment. Animals were randomly assigned to drug or vehicle treatment on test day 1, then received the alternative treatment on test day 2 of a second DID cycle [See Fig. 2a]. Infusions for microinjected drugs occurred as previously published at a rate of 0.5 μL/min using a Hamilton syringe (Reno, NV) attached to a Harvard Apparatus PHD 2000 infusion pump (Holliston, MS) [14, 33]. After infusion, injectors remained in guide cannula for an additional 0.5–1 min for diffusion. As animals underwent a 3-day abstinence from ethanol or sucrose following each test day, potential carry-over effects 24 h following injection were not specifically evaluated. Following brain extraction and section mounting (see IHC methods) cannula and DREADD placements were verified through use of a digital camera (Roper Scientific), mounted on an optical microscope (Leica DM6000).

NPY1R activation in the mPFC specifically reduces ethanol, but not sucrose, binge-like intake. a Timeline of pharmacology studies. b Microinjection of the NPY1R agonist Leu, Pro-NPY into the mPFC significantly reduced binge-like ethanol intake. c BEC concentration was likewise significantly reduced relative to vehicle. d Leu, Pro-NPY mPFC microinjection did not alter 3% sucrose intake relative to vehicle. e Cannula placement checks with each mark representing a single animal (x = misplacement; • = hit). (** = main treatment effect p < 0.01; + = post hoc effect p < 0.05; # = paired t test p < 0.05; ## = paired t test p < 0.01)
2.6 Statistical analysis
GraphPad Prism (GraphPad Software, Inc. La Jolla, Ca) was used to analyze and graph all data, save three-way ANOVAs which were performed in SPSS statistics (IBM Analytics, Armonk, New York). Two-way ANOVAs and Bonferroni post hoc tests were used to determine the effect of binge cycle on NPY immunoreactivity and treatment versus time during individual DID drinking hours. t Tests were used to evaluate effect of treatment on total intake and BEC. All data are reported as the mean ± standard error of the mean and considered significant if p < 0.05. Animals were removed from analysis if: (1) they were found by a Grubbs test (alpha = 0.05) to be a significant outlier (number of outliers/ethanol experiment: 3.1 = 1, 3.2 = 2, 3.3 = 2, 3.5 = 1); (2) due to cannula misplacement (uni or bilateral); or (3) due to inappropriate DREADD expression. In cases where a cannula became clogged between ethanol and sucrose studies animals were immediately perfused and ethanol results alone reported. Cannula placement was determined by locating the end of the guide cannula and adding injector projection length (2 mm in mPFC and 0.5 mm in BLA). Three-way ANOVA analysis found no significant effect of treatment order in any Latin-square design experiment, therefore these data were collapsed and analyzed by two-way ANOVA. Two-way ANOVA found no significant effect of sex on any data set, therefore data from males and females were collapsed.
3.0 Results
3.1 Three cycles of binge-like ethanol decreased mPFC NPY immunoreactivity
NPY immunoreactivity in the PL and IL subregions of the mPFC following 3 cycles of binge-ethanol exposure (N = 12) or water only exposure (N = 10) were assessed [Fig. 1a]. Two-way ANOVA revealed a significant impact of treatment [F(1,20) = 4.42, p < 0.05], but not mPFC subregion [F(1,20) = 3.61, p > 0.05; interaction: F(1,20) = 0.46, p > 0.05] [Fig. 1b, c] on NPY expression. Interestingly, NPY expression was found to be significantly correlated with ethanol intake in the PL [r = 0.62, p < 0.05] [Fig. 1d], but not the IL [r = 0.30, p > 0.05] [Fig. 1e] subregion. NPY expression was not found to correlate with water intake in either the PL [r = 0.16, p > 0.05] [Fig. 1f] or IL [r = −0.52, p > 0.05] [Fig. 1g].
3.2 Pharmacological activation of mPFC NPY1R selectively decreased binge-like ethanol intake
Two-way (treatment × time) ANOVAs were used to analyze the Latin Square data and paired t test to analyze total intake and BEC. Treatment timeline detailed in Fig. 2a. mPFC microinjection of the selective NPY1R agonist Leu, Pro-NPY reduced total binge-like ethanol intake in mice (N = 21) [treatment: F(1,20) = 10.62, p < 0.01; time: F(1,20) = 0.10, p > 0.05; interaction: F(1,20) = 0.053, p > 0.05; total t test: t = 3.26 df = 20, p < 0.01] [Fig. 2b]. A similar significant reduction in BEC was observed (N = 18) (3 blood samples lost due to mechanical error) [t = 2.22 df = 17, p > 0.05] [Fig. 2c]. In contrast, no impact of Leu, Pro-NPY was detected on sucrose consumption (N = 14) [treatment: F(1,13) = 0.01, p > 0.05; time: F(1,13) = 0.29, p > 0.05; interaction: F(1,13) = 2.37, p > 0.05] [Fig. 2d]. Nor did Leu, Pro-NPY alter general locomotor or anxiety-like behavior [see Supplementary methods and Figure S1].
3.3 Pharmacological antagonism of mPFC NPY2R selectively decreased binge-like ethanol intake
Three cohorts of animals were used to evaluate the impact of BIIE0246, a NPY2R antagonist, on binge-like consumption. The first cohort consisted of a subset of animals from the NPY1R agonist experiment which were given 1 week of no drug treatment prior to the NPY2R experiments (N = 9). The second and third cohorts consisted of mice from Jackson Lab (Bar Harbor, ME) with no previous experimental experience which were collapsed together (N = 11). Two-way ANOVA with cohort and treatment as factors revealed no significant effect of cohort [F(1,18) = 0.05, p = 0.82] nor an interaction between cohort and treatment [F(1,18) = 0.02, p = 0.88]. All cohorts were therefore collapsed together for analysis.
Two-way (treatment × time) ANOVAs were used to analyze the Latin Square data and paired t test to analyze total intake and BEC. mPFC microinjection of the selective NPY2R antagonist BIIE0246 reduced total binge-like ethanol intake mice (N = 20) [treatment: F(1,19) = 11.42, p < 0.01; time: F(1,19) = 8.30, p > 0.01; interaction: F(1,19) = 7.17, p > 0.05; total t test: t = 3.45 df = 19, p < 0.01] [Fig. 3a]. This was accompanied by a similar reduction in BEC (N = 16) (4 blood samples lost due to mechanical error) [t = 4.09 df = 15, p < 0.001] [Fig. 3b]. In contrast, no impact of BIIE0246 was observed on sucrose consumption (N = 13) [treatment: F(1,12) = 0.98, p > 0.05; time: F(1,12) = 4.34, p > 0.05; interaction: F(1,12) = 0.13, p > 0.05; total t test: t = 1.0 df = 12, p > 0.05] [Fig. 3c]. Nor did BIIE0246 alter general locomotor or anxiety-like behavior [see Supplementary methods and Figure S2].

NPY2R inhibition in the mPFC specifically reduces ethanol, but not sucrose, binge-like intake. a Microinjection of the NPY2R antagonist BIIE0246 into the mPFC significantly reduced binge-like ethanol intake. b BEC concentration was likewise significantly reduce relative to vehicle. c BIIE0264 mPFC microinjection did not alter 3% sucrose intake relative to vehicle. d Cannula placement checks with each mark representing a single animal (x = misplacement; • = hit). (+++ = post hoc effect p < 0.001; ## = paired t test p < 0.01)
3.4 Chemogenetic inhibition of mPFC NPY1R+ cells selectively decreased binge-like ethanol intake
NPY1R-cre positive animals were used in these experiments. DID timeline was identical to that in the NPY1R pharmacological experiment [Fig. 2a]. Two-way (treatment × time) ANOVAs were used to analyze the Latin Square data and paired t test to analyze total intake and BEC. I.P. CNO injection significantly reduced binge-like ethanol intake compared to vehicle treatment in animals expressing Gi-coupled DREADDs in NPY1R+ mPFC cells (N = 12) [treatment: F(1,11) = 6.68, p < 0.05; time: F(1,11) = 0.004, p > 0.05; interaction: F(1,11) = 2.11, p > 0.05; total t test: t = 2.59 df = 11, p < 0.05] [Fig. 4a]. No such significant decrease in BEC concentration was detected (N = 10) (2 blood samples lost due to mechanical error) [t = 1.55 df = 9, p > 0.05] [Fig. 4b]. CNO treatment in Gi-DREADD animals had no significant impact on amount of consumption of a 3% sucrose solution (N = 10) [treatment: F(1,9) = 0.12, p > 0.05; time: F(1,9) = 6.03, p < 0.05; interaction: F(1,9) = 0.003, p > 0.05] [Fig. 4c].

DREADD inhibition of mPFC NPY1R-expressing neurons reduces binge-like ethanol, but not sucrose intake. a CNO activation of Gi-DREADDs expressed on NPY1R+ mPFC cells reduced binge-like ethanol intake. b CNO Gi-DREADD activation did not significantly reduce BEC levels relative to vehicle treatment. c Nor was sucrose consumption reduced relative to vehicle treatment by CNO treatment in Gi-DREADD animals. d CNO I.P injection in CON DREADD animals did not alter ethanol binge-like intake or e sucrose intake. f Exemplar image of Gi-DREADD expression in the mPFC. (* = main treatment effect p < 0.05; # = paired t test p < 0.05)
In contrast to Gi-DREADD animals, in subjects treated with a control (CON) DREADD virus no significant impact of I.P. CNO was detected on ethanol intake (N = 9) [treatment: F(1,8) = 0.09, p > 0.05; time: F(1,8) = 1.33, p > 0.05; interaction: F(1,8) = 0.22, p > 0.05; total t test t = 0.30 df = 8, p > 0.05] [Fig. 4d], BEC [t test t = 0.32 df = 8, p > 0.05] [data not shown], nor sucrose intake (N = 9) [treatment: F(1,8) = 1.14, p > 0.05; time: F(1,8) = 1.03, p > 0.05; interaction: F(1,8) = 0.50, p > 0.05; total t test t = 1.07 df = 8 p > 0.05] [Fig. 4e]. These results further demonstrate a lack of nonspecific CNO effects.
3.5 Chemogenetic inhibition of NPY1R+ mPFC-BLA projection neurons selectively decreased binge-like ethanol intake
NPY1R-cre positive animals were used in these experiments. DID timeline was identical to that in the NPY1R pharmacological experiment [Fig. 2a]. We next evaluated if chemogenetic inhibition of mPFC NPY1R+ neurons reduced binge-like ethanol intake, in part, by decreasing mPFC signaling to the BLA. Two-way (treatment × time) ANOVAs were used to analyze the Latin Square data and paired t test to analyze total intake and BEC. BLA CNO microinjection significantly reduced ethanol intake compared to vehicle treatment in Gi-coupled DREADD animals (N = 6) [treatment: F(1,5) = 21.73, p < 0.01; time: F(1,5) = 1.18, p > 0.05; interaction: F(1,5) = 0.24, p > 0.05; total t test t = 4.66 df = 5, p < 0.01] [Fig. 5a] and BEC [t = 2.57 df = 5, p < 0.05] [Fig. 5b]. No effect of treatment was found on sucrose intake in Gi-DREADD animals (N = 7) [treatment: F(1,6) = 0.19, p > 0.05; time: F(1,6) = 10.03, p < 0.05; interaction: F(1,6) = 2.63e−005, p > 0.05; total t test: t = 0.44 df = 6, p > 0.05] [Fig. 5c]. In a separate cohort of mice, slice electrophysiology verified mPFC to BLA Gi DREADD activity [S3].

DREADD inhibition of mPFC to BLA NPY1R-expressing neurons reduces binge-like ethanol, but not sucrose, intake. a CNO activation of Gi-DREADDs expressed on NPY1R+ mPFC cells projecting to the BLA reduced binge-like ethanol intake. b CNO Gi-DREADD activation significantly reduced BEC levels relative to vehicle treatment. c Sucrose consumption relative to vehicle treatment was not reduced by CNO treatment in Gi-DREADD animals. d CNO microinjection into CON DREADD animals did not alter binge-like ethanol intake, e BEC, or f sucrose intake. g TOP: Cannula placement checks with each mark representing a single cannula (2 per animal) (x = misplacement; • = hit); BOTTOM: exemplar image of mPFC NPY1R+ terminal fluorescence in the BLA of a NPY1R-cre mouse (scale bar = 250 µm). (** = main treatment effect p < 0.01; # = paired t test p < 0.05; ## = paired t test p < 0.01)
In contrast to Gi-DREADD activation, in CON-virus animals BLA CNO microinjection had no effect on ethanol (N = 7) [treatment: F(1,6) = 0.96, p > 0.05; time: F(1,6) = 0.19, p > 0.05; interaction: F(1,6) = 0.09, p > 0.05; total t test: t = 0.98 df = 6, p > 0.05] [Fig. 5d], BEC [t = 0.30 df = 6, p > 0.05] [Fig. 5e], or sucrose intake (N = 7) [treatment: F(1,6) = 0.33, p > 0.05; time: F(1,6) = 0.56, p > 0.05; interaction: F(1,6) = 0.98, p > 0.05; total intake t test: t = 0.58 df = 6, p > 0.05] [Fig. 5f].
4.0 Discussion
Results from the present study fill a critical gap in the literature by providing the first evidence that NPY signaling in the mPFC is critical in the modulating of binge-like ethanol intake. These findings parallel observed reductions of NPY levels in limbic regions critical to regulating ethanol-directed behaviors [11, 12]. Our IHC results suggest that binge-like ethanol consumption reduces mPFC NPY levels. Notably, despite an overall reduction in mPFC NPY immunoreactivity, NPY levels within the PL were found to positively correlate with total ethanol intake. This positive correlation is potentially due to NPY acting as a type of “breaking mechanism” engaged following high levels of ethanol intake [34]. That NPY levels are blunted relative to the water treated group suggest repeated binge cycles compromise PL ability to utilize NPY signaling to off-set the effects of high levels of ethanol intake. In line with this potential counteractive role of the mPFC NPY system, we hypothesized that increasing NPY signaling in the mPFC during the drinking period would blunt ethanol intake. mPFC infusion of a NPY1R agonist or a NPY2R antagonist (which increases endogenous NPY release via autoreceptor blockade) both blunt binge-like ethanol drinking in mice with no impact on sucrose intake or anxiety-like/general locomotor behavior. Since NPY1R are Gi-coupled [25] we used DREADD technology in NPY1R-cre mice to chemogenetically silence NPY1R+ neurons in the mPFC and in mPFC→BLA-projecting NPY1R+ neurons. CNO activation of the inhibitory Gi-coupled DREADD significantly reduced binge-like ethanol intake in both cases, with no effect in sucrose consumption. Importantly, no such change in binge-like ethanol intake was observed following CNO injection in animals expressing a control virus in the mPFC. The lack of CNO effect in the control group is of particular importance given recent reports of potential off-target CNO effects [35]. Of interest, an apparently more robust effect was observed following administration of our NPY1R agonist or the specific inhibition the of mPFC→BLA projection over total mPFC NPY1R+ DREADD inhibition. This may be due to route of CNO administration, as direct microinjection of CNO to the BLA or NPY1R agonist to the mPFC more rapidly interacts with receptors relative to an I.P. CNO injection. Alternatively, I.P. CNO injection may have inhibited distal NPY1R+ mPFC projections throughout the brain in a sequential order which would not be observed following direct microinjection to the mPFC. As the role of other important mPFC projections, such as the nucleus accumbens or BNST, were not evaluated in this work it is possible that inhibition of these projections prior or subsequent to inhibition of mPFC→BLA may impact observed results.
Potential differential roles of the two primary subregions of the mPFC, the PL and IL, in consumption were not directly addressed in this work. However, past work suggests these subregions play distinct roles in ethanol-seeking behavior. For instance, in an operant task, lesions or inactivation of the PL disrupts goal-directed responding and facilitates the development of habitual responding (responding that is insensitive to reward devaluation). In contrast, inactivation of the IL suppresses habitual responding stemming from overtraining and supports a return to goal-directed operant behavior [36,37,38]. While we did not systematically investigate these regions separately in our pharmacological or chemogenetic studies, the observation that NPY levels were blunted in both the PL and IL after a history of binge-like ethanol drinking suggests that NPY signaling in both regions may be involved. Interestingly, however, NPY levels in the PL region of the mPFC alone were found to correlate with ethanol intake, suggesting potentially distinct relationships between NPY signaling and mPFC subregion in binge-like intake. In further support of such a distinct relationship, cannula placement analyses revealed that the NPY1R agonist blunted binge-like ethanol drinking when injectors terminated in the PL, but not the IL subregion [Fig. S4]. Future studies will build on these preliminary observations to systematically examine the contributions of each individual region.
Though the specific role of NPY in regulating mPFC subregion activity remains an area of important future investigation, we can speculate based on NPY system expression patterns how this neuropeptide contributes to the top-down modulation of limbic activity. NPY is locally produced by a population of GABAergic interneurons within the mPFC [26], which primarily synapses both on other local interneurons and on the dendrites of glutamatergic pyramidal neurons. NPY1R receptors are expressed both on glutamatergic and GABAergic cells within the mPFC [39]. This NPY+ interneuron population is therefore positioned to alter GABAergic and glutamatergic activity through both GABA and NPY signaling mechanisms, and thus potently shape mPFC output and synchronize regional neural activity [40,41,42]. As a Gi-coupled homoreceptor, NPY2R activity serves as an important regulator of mPFC NPY levels, with decreased receptor function facilitating NPY1R activity. As the mPFC is well known to be altered during chronic ethanol exposure/withdrawal [13, 19, 43], NPY within the mPFC may be poised to modulate both the initial binge-like patterns of ethanol intake, as demonstrated in this study, and in later stages of alcohol use disorders through dramatically shaping mPFC output to a variety of downstream targets critically involved in systems that modulate emotions and reward.
Within this work, we cannot specifically determine if there was a total overlap in the population of NPY1R+ glutamatergic and GABAergic cells mediated which actions of our pharmacological study and that which mediated the reduced binge-like ethanol intake resulting from general silencing of NPY1R+ neurons in the mPFC. However, we can confidently conclude that NPY1R+ mPFC→BLA projection neurons are critically involved in the modulation of binge-like ethanol intake. While the mPFC→BLA circuit has been implicated in other neurobiological responses, such as fear and anxiety [16, 44,45,46], the present results are the first direct evidence that a top-down mPFC→BLA circuit modulates binge-like ethanol intake. Future studies will be necessary to further determine the role of NPY1R in microcircuits within the mPFC itself in the modulation of binge-like ethanol intake.
As a Gi-coupled receptor, NPY1R activation serves to decrease mPFC→BLA projection activity. A loss of NPY modulation of this projection, through the reduced NPY expression observed in this work or a potential accompanying decrease in NPY1R function, may therefore decrease mPFC ability to effectively regulate BLA activity. Such disrupted regulation may eventually contribute to the increase in glutamatergic signaling observed at cortical inputs to the BLA during withdrawal from chronic ethanol exposure [47]. Importantly, alterations in mPFC→BLA communication are involved in numerous neuropsychiatric disorders associated with alcohol abuse behaviors, including depression [27], chronic stress [28], and anxiety disorders [48]. Indeed, across a whole brain analysis in humans, anxiety levels were found to be exclusively predicted by mPFC-amygdala functional connectivity, suggesting this pathway plays a critical role in regulating amygdala-driven emotionality [48]. Alterations in mPFC regulation of BLA activity are further critical as the BLA acts as the primary input nucleus of the amygdala and thus serves as an important gateway though which the mPFC regulates activity of downstream limbic targets [20, 49]. Within this present work we evaluated only NPY modulation of the first leg of this circuit, the connection from the mPFC to the BLA. Future studies will be required to determine the precise secondary downstream targets which are functionally altered by changes in mPFC NPY signaling, with an emphasis on alterations in activity within the EA during binge-like consumption of ethanol.
In conclusion, we provide novel evidence for a role of the mPFC NPY system in modulating binge-like ethanol consumption regardless of sex. These findings strengthen evidence that compounds targeting the NPY system may be a viable treatment for alcohol use disorders, and may be effective in correcting abnormal NPY signaling stemming from repeated alcohol use in a variety of brain circuits.
References
NIAAA. National Institute on Alcohol Abuse and Alcoholism Council approves definition of binge drinking. NIAAA Newsletter. 2004.
Fan AZ, Russell M, Stranges S, Dorn J, Trevisan M. Association of lifetime alcohol drinking trajectories with cardiometabolic risk. J Clin Endocrinol Metab. 2008;93:154–61. https://doi.org/10.1210/jc.2007-1395
Hingson R, Heeren T, Winter M, Wechsler H. Magnitude of alcohol-related mortality and morbidity among U.S. college students ages 18-24: changes from 1998 to 2001. Annu Rev Public Health. 2005;26:259–79. https://doi.org/10.1146/annurev.publhealth.26.021304.144652
Miller JW, Naimi TS, Brewer RD, Jones SE. Binge drinking and associated health risk behaviors among high school students. Pediatrics. 2007;119:76–85. https://doi.org/10.1542/peds.2006-1517
Miller P, Plant M. Spreading out or concentrating weekly consumption: alcohol problems and other consequences within a UK population sample. Alcohol Alcohol. 2005;40:461–8. https://doi.org/10.1093/alcalc/agh169
Sprow GM, Thiele TE. The neurobiology of binge-like ethanol drinking: evidence from rodent models. Physiol Behav. 2012;106:325–31. https://doi.org/10.1016/j.physbeh.2011.12.026
Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63.
Thiele TE, Crabbe JC, Boehm SL. “Drinking in the Dark” (DID): a simple mouse model of binge-like alcohol intake. Curr Protoc Neurosci. 2014;68:1–12. https://doi.org/10.1002/0471142301.ns0949s68. 9.49
Koob GF. Alcoholism: allostasis and beyond. Alcohol Clin Exp Res. 2003;27:232–43. https://doi.org/10.1097/01.ALC.0000057122.36127.C2
Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. https://doi.org/10.1016/S0893-133X(00)00195-0
Gilpin NW. Neuropeptide Y (NPY) in the extended amygdala is recruited during the transition to alcohol dependence. Neuropeptides. 2012;46:253–9. https://doi.org/10.1016/j.npep.2012.08.001
Sparrow AM, Lowery-Gionta EG, Pleil KE, Li C, Sprow GM, Cox BR, et al. Central neuropeptide Y modulates binge-like ethanol drinking in C57BL/6J mice via Y1 and Y2 receptors. Neuropsychopharmacology. 2012;37:1409–21. https://doi.org/10.1038/npp.2011.327
Pleil KE, Lowery-Gionta EG, Crowley NA, Li C, Marcinkiewcz CA, Rose JH, et al. Effects of chronic ethanol exposure on neuronal function in the prefrontal cortex and extended amygdala. Neuropharmacology. 2015;99:735–49. https://doi.org/10.1016/j.neuropharm.2015.06.017
Lowery-Gionta EG, Navarro M, Li C, Pleil KE, Rinker JA, Cox BR, et al. Corticotropin releasing factor signaling in the central amygdala is recruited during binge-like ethanol consumption in C57BL/6J mice. J Neurosci. 2012;32:3405–13. https://doi.org/10.1523/JNEUROSCI.6256-11.2012
Likhtik E, Paz R. Amygdala-prefrontal interactions in (mal)adaptive learning. Trends Neurosci. 2015;38:158–66. https://doi.org/10.1016/j.tins.2014.12.007
Likhtik E, Pelletier JG, Paz R, Pare D. Prefrontal control of the amygdala. J Neurosci. 2005;25:7429–37. https://doi.org/10.1523/JNEUROSCI.2314-05.2005
Lei K, Wegner SA, Yu JH, Mototake A, Hu B, Hopf FW. Nucleus accumbens shell and mPFC but not insula orexin-1 receptors promote excessive alcohol drinking. Front Neurosci. 2016;10:400 https://doi.org/10.3389/fnins.2016.00400
Haun HL, Griffin WC, Lopez MF, Solomon MG, Mulholland PJ, Woodward JJ, et al. Increasing brain-derived neurotrophic factor (BDNF) in medial prefrontal cortex selectively reduces excessive drinking in ethanol dependent mice. Neuropharmacology. 2018;140:35–42. https://doi.org/10.1016/j.neuropharm.2018.07.031
Holmes A, Fitzgerald PJ, MacPherson KP, DeBrouse L, Colacicco G, Flynn SM, et al. Chronic alcohol remodels prefrontal neurons and disrupts NMDAR-mediated fear extinction encoding. Nat Neurosci. 2012;15:1359–61. https://doi.org/10.1038/nn.3204
Lalumiere RT. Optogenetic dissection of amygdala functioning. Front Behav Neurosci. 2014;8:107 https://doi.org/10.3389/fnbeh.2014.00107
Little JP, Carter AG. Synaptic mechanisms underlying strong reciprocal connectivity between the medial prefrontal cortex and basolateral amygdala. J Neurosci. 2013;33:15333–42. https://doi.org/10.1523/JNEUROSCI.2385-13.2013
Mcdonald AJ, Mascagni F, Guo L. Projections of the medial and lateral prefrontal cortices to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience. 1996;71:55–75.
Pinard CR, Mascagni F, McDonald AJ. Medial prefrontal cortical innervation of the intercalated nuclear region of the amygdala. Neuroscience. 2012;205:112–24. https://doi.org/10.1016/j.neuroscience.2011.12.036
Robinson SL, Thiele TE. The role of neuropeptide Y (NPY) in alcohol and drug abuse disorders. Int Rev Neurobiol. 2017;136:177–97. https://doi.org/10.1016/bs.irn.2017.06.005
Palmiter RD, Erickson JC, Hollopeter G, Baraban SC, Schwartz MW. Life without neuropeptide Y. Recent Prog Horm Res. 1998;53:163–99.
Kubota Y, Shigematsu N, Karube F, Sekigawa A, Kato S, Yamaguchi N, et al. Selective coexpression of multiple chemical markers defines discrete populations of neocortical GABAergic neurons. Cereb Cortex. 2011;21:1803–17. https://doi.org/10.1093/cercor/bhq252
Carballedo A, Scheuerecker J, Meisenzahl E, Schoepf V, Bokde A, Möller HJ, et al. Functional connectivity of emotional processing in depression. J Affect Disord. 2011;134:272–9. https://doi.org/10.1016/j.jad.2011.06.021
Gilabert-Juan J, Castillo-Gomez E, Guirado R, Moltó MD, Nacher J. Chronic stress alters inhibitory networks in the medial prefrontal cortex of adult mice. Brain Struct Funct. 2013;218:1591–605. https://doi.org/10.1007/s00429-012-0479-1
Ehlers CL, Li TK, Lumeng L, Hwang BH, Somes C, Jimenez P, et al. Neuropeptide Y levels in ethanol-naive alcohol-preferring and nonpreferring rats and in Wistar rats after ethanol exposure. Alcohol Clin Exp Res. 1998;22:1778–82.
Padilla SL, Qiu J, Soden ME, Sanz E, Nestor CC, Barker FD, et al. Agouti-related peptide neural circuits mediate adaptive behaviors in the starved state. Nat Neurosci. 2016;19:734–41. https://doi.org/10.1038/nn.4274
Marshall SA, McKnight KH, Blose AK, Lysle DT, Thiele TE. Modulation of binge-like ethanol consumption by IL-10 signaling in the basolateral amygdala. J Neuroimmune Pharmacol. 2016. https://doi.org/10.1007/s11481-016-9709-2
Pleil KE, Rinker JA, Lowery-Gionta EG, Mazzone CM, McCall NM, Kendra AM, et al. NPY signaling inhibits extended amygdala CRF neurons to suppress binge alcohol drinking. Nat Neurosci. 2015;18:545–52. https://doi.org/10.1038/nn.3972
Finn DA, Snelling C, Fretwell AM, Tanchuck MA, Underwood L, Cole M, et al. Increased drinking during withdrawal from intermittent ethanol exposure is blocked by the CRF receptor antagonist D-Phe-CRF(12-41). Alcohol Clin Exp Res. 2007;31:939–49. https://doi.org/10.1111/j.1530-0277.2007.00379.x
Brothers SP, Wahlestedt C. Therapeutic potential of neuropeptide Y (NPY) receptor ligands. EMBO Mol Med. 2010;2:429–39. https://doi.org/10.1002/emmm.201000100
MacLaren DA, Browne RW, Shaw JK, Krishnan Radhakrishnan S, Khare P, España RA, et al. Clozapine N-oxide administration produces behavioral effects in long-evans rats: implications for designing DREADD experiments. eNeuro. 2016;3. https://doi.org/10.1523/ENEURO.0219-16.2016
Balleine BW, Dickinson A. Goal-directed instrumental action: contingency and incentive learning and their cortical substrates. Neuropharmacology. 1998;37:407–19.
Balleine BW, O’Doherty JP. Human and rodent homologies in action control: corticostriatal determinants of goal-directed and habitual action. Neuropsychopharmacology. 2010;35:48–69. https://doi.org/10.1038/npp.2009.131
Mendoza J, Sanio C, Chaudhri N. Inactivating the infralimbic but not prelimbic medial prefrontal cortex facilitates the extinction of appetitive pavlovian conditioning in long-evans rats. Neurobiol Learn Mem. 2015;118:198–208. https://doi.org/10.1016/j.nlm.2014.12.006
Vollmer LL, Schmeltzer S, Schurdak J, Ahlbrand R, Rush J, Dolgas CM, et al. Neuropeptide Y impairs retrieval of extinguished fear and modulates excitability of neurons in the infralimbic prefrontal cortex. J Neurosci. 2016;36:1306–15. https://doi.org/10.1523/JNEUROSCI.4955-13.2016
Constantinidis C, Williams GV, Goldman-Rakic PS. A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat Neurosci. 2002;5:175–80. https://doi.org/10.1038/nn799
Ghosal S, Hare B, Duman RS. Prefrontal cortex GABAergic deficits and circuit dysfunction in the pathophysiology and treatment of chronic stress and depression. Curr Opin Behav Sci. 2017;14:1–8. https://doi.org/10.1016/j.cobeha.2016.09.012
Delli Pizzi S, Chiacchiaretta P, Mantini D, Bubbico G, Edden RA, Onofrj M, et al. GABA content within medial prefrontal cortex predicts the variability of fronto-limbic effective connectivity. Brain Struct Funct. 2017. https://doi.org/10.1007/s00429-017-1399-x
Abernathy K, Chandler LJ, Woodward JJ. Alcohol and the prefrontal cortex. Int Rev Neurobiol. 2010;91:289–320. https://doi.org/10.1016/S0074-7742(10)91009-X
Cho JH, Deisseroth K, Bolshakov VY. Synaptic encoding of fear extinction in mPFC-amygdala circuits. Neuron. 2013;80:1491–507. https://doi.org/10.1016/j.neuron.2013.09.025
Arruda-Carvalho M, Clem RL. Pathway-selective adjustment of prefrontal-amygdala transmission during fear encoding. J Neurosci. 2014;34:15601–9. https://doi.org/10.1523/JNEUROSCI.2664-14.2014
Arruda-Carvalho M, Clem RL. Prefrontal-amygdala fear networks come into focus. Front Syst Neurosci. 2015;9:145 https://doi.org/10.3389/fnsys.2015.00145
Christian DT, Alexander NJ, Diaz MR, McCool BA. Thalamic glutamatergic afferents into the rat basolateral amygdala exhibit increased presynaptic glutamate function following withdrawal from chronic intermittent ethanol. Neuropharmacology. 2013;65:134–42. https://doi.org/10.1016/j.neuropharm.2012.09.004
Kim MJ, Gee DG, Loucks RA, Davis FC, Whalen PJ. Anxiety dissociates dorsal and ventral medial prefrontal cortex functional connectivity with the amygdala at rest. Cereb Cortex. 2011;21:1667–73. https://doi.org/10.1093/cercor/bhq237
Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse. 2004;51:32–58. https://doi.org/10.1002/syn.10279
Funding
The authors thank Rhiannon D. Thomas for her technical assistance and Richard Palmiter, PhD for supplying the NPY1R-cre mice. This work was supported by National Institutes of Health grants AA022048, AA013573, AA015148, and F32AA025811. The authors declare no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Robinson, S.L., Marrero, I.M., Perez-Heydrich, C.A. et al. Medial prefrontal cortex neuropeptide Y modulates binge-like ethanol consumption in C57BL/6J mice. Neuropsychopharmacol. 44, 1132–1140 (2019). https://doi.org/10.1038/s41386-018-0310-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-018-0310-7
This article is cited by
-
Basolateral amygdala neuropeptide Y system modulates binge ethanol consumption
Neuropsychopharmacology (2024)
-
Patterns of functional connectivity alterations induced by alcohol reflect somatostatin interneuron expression in the human cerebral cortex
Scientific Reports (2022)
-
Somatostatin neurons control an alcohol binge drinking prelimbic microcircuit in mice
Neuropsychopharmacology (2021)
