
Abstract
ATP9A, a lipid flippase of the class II P4-ATPases, is involved in cellular vesicle trafficking. Its homozygous variants are linked to neurodevelopmental disorders in humans. However, its physiological function, the underlying mechanism as well as its pathophysiological relevance in humans and animals are still largely unknown. Here, we report two independent families in which the nonsense mutations c.433C>T/c.658C>T/c.983G>A (p. Arg145*/p. Arg220*/p. Trp328*) in ATP9A (NM_006045.3) cause autosomal recessive hypotonia, intellectual disability (ID) and attention deficit hyperactivity disorder (ADHD). Atp9a null mice show decreased muscle strength, memory deficits and hyperkinetic movement disorder, recapitulating the symptoms observed in patients. Abnormal neurite morphology and impaired synaptic transmission are found in the primary motor cortex and hippocampus of the Atp9a null mice. ATP9A is also required for maintaining neuronal neurite morphology and the viability of neural cells in vitro. It mainly localizes to endosomes and plays a pivotal role in endosomal recycling pathway by modulating small GTPase RAB5 and RAB11 activation. However, ATP9A pathogenic mutants have aberrant subcellular localization and cause abnormal endosomal recycling. These findings provide strong evidence that ATP9A deficiency leads to neurodevelopmental disorders and synaptic dysfunctions in both humans and mice, and establishes novel regulatory roles for ATP9A in RAB5 and RAB11 activity-dependent endosomal recycling pathway and neurological diseases.
Similar content being viewed by others


Introduction
Attention deficit and hyperactivity disorders (ADHD) and Intellectual disability (ID) are common neurodevelopmental disorders in children and adolescents. ID is mainly characterized by an intelligence quotient (IQ) lower than 70, accompanied by intellectual dysfunction and adaptive behavior disorder [1, 2], whereas ADHD is characterized by inattention and/or hyperactivity and impulsivity causing functional impairment [3, 4]. Some patients who have ADHD also display ID, and the diagnosis and treatment of these people can be more challenging due to the complex etiology, unclear pathogenic mechanism and lack of effective therapeutic drugs [4]. The etiology of ID and ADHD is strongly influenced by genetic factors, and both of these conditions are highly heritable neuropsychiatric disorders with heritability estimated at approximately 67% (ID) and 76% (ADHD) [5, 6]. Due to autosomal recessive inheritance in ID and ADHD, pathogenic genes related to these conditions have been poorly studied due to the lack of obvious genetic tendency. Therefore, discovering new pathogenic genes and exploring their pathogenic mechanisms can provide an important basis for clinical diagnosis and treatment.
We identified three individuals suffering from hypotonia, ID, ADHD and other neurological diseases in two independent families during 2017–2021. Whole exon capture sequencing (WES) showed that this disease may be caused by autosomal recessive inheritance of nonsense variants in ATP9A (NM_006045.3). This gene has not been implicated in any human disease according to OMIM [7] until 2017, but three very recent case reports described that biallelic truncating variants in ATP9A cause neurodevelopmental disorders involving ID, postnatal microcephaly (POM) and Multiple Sclerosis (MS) in humans [8,9,10], providing more evidences for the association between ATP9A and neurodevelopment. However, the clinical relevance and physiological role of ATP9A in the neurological system require further investigation.
ATP9A, together with ATP9B, belongs to the second class of P4-ATPase family members. P4-type ATPases are also called phospholipid flippases for their ability to flip phospholipids from the outside of the cell membrane or the luminal surface of the organelles to the cytoplasmic surface. Structural analysis of P4-ATPases indicates that the family members contain a similar structure with 10 transmembrane helices and A (actuator), N (nucleotide binding), P (phosphorylation) domains [11, 12]. These kinds of proteins exist only in eukaryotes and are important components of organelles with critical roles in membrane biogenesis by regulating vesicle transport among the plasma membrane, endosomes and the Golgi [13]. P4-ATPase deficiency is associated with neurological dysfunction [14,15,16], liver disorders [17, 18], diabetes [19], immune deficiency [20], and reduced fertility [21].
Previous studies have shown that ATP9A homologs are essential genes in Saccharomyces cerevisiae (NEO1) and Caenorhabditis elegans (TAT-5) [22, 23] and are critical in cytoplasmic vesicle transport and extracellular vesicle budding in S. cerevisiae [24] and C. elegans [25], respectively. Recently, ATP9A was shown to be required for the recycling pathway from the endosome to the plasma membrane and to mediate Wntless sorting, Wnt secretion and exosome release [26,27,28], indicating that ATP9A may be involved in cellular vesicle transport. However, the regulatory mechanism of ATP9A in mammalian endosomal transport is still unknown.
Endocytic recycling is the major pathway to maintain the abundance of receptors and transporters on the cell surface and is critical for several important cellular processes such as cell migration and cytokinesis as well as the maintenance of polarity in neurons [29]. Endocytic recycling is the process by which cargo molecules are transported from the cell surface into RAB5-positive early endosomes and recycled directly back to the plasma membrane (fast recycling) or subsequently delivered to RAB11-positive recycling endosomes, which release them outside the plasma membrane (slow recycling) [30]. Dysregulation of the expression and activity of the small GTPases RAB5 and RAB11 leads to disruption of endosomal recycling and has been linked to a variety of diseases, including cancer and central or peripheral neurological disorders [31,32,33].
Here, for the first time, we successfully recapitulated human neurological disease phenotypes in an ATP9A knockout mouse model and found that ATP9A deficiency causes synaptic dysfunction in primary motor cortex and hippocampus. Further studies suggest that the underlying pathogenic mechanism involves inactivation of RAB5 and RAB11. Our work highlights the critical role of ATP9A in maintaining synaptic function associated with endosome trafficking and creates a new animal model that simulates human hypotonia, ID, and ADHD disorders.
Materials and methods
Materials and methods used in this study are available in Supplementary Information.
Results
Clinical investigation
In family 1 (Fig. 1A and Supplementary Table 1) from Gansu Province, China, a possible autosomal recessive inheritance pattern for ID was demonstrated. In this family, a healthy nonconsanguineous Chinese couple gave birth to two affected children. The proband (II -1, Fig. 1A, B) is male and the first child with a term birth and showed no abnormalities at birth. After 6 months, the boy showed developmental delays and was diagnosed with “cerebral palsy”. Rehabilitation treatment failed to produce satisfactory results. At 15 years of age, the boy was diagnosed with ID, ADHD, absent speech, severe receptive language delay, lack of social interactions, and sleep disturbances (Supplementary Table 1). Physical examination revealed that the boy had coordination deficits, muscle weakness of the limbs, hyporeflexia, and joint hypermobility, as well as knee and elbow flexion contracture. His parents gave birth to a girl (II-2, Fig. 1A, B) when he was five years old, and the sister had a similar phenotype to her brother. The proband (II-3, Fig. 1A, B) of family 2 was born to a healthy consanguineous Pakistani couple by an unremarkable vaginal delivery. The affected girl showed global developmental delay, ID, ADHD, hypotonia, delayed fine motor skills, microcephaly, speech problems, and sleep disturbances. She has a healthy brother, and both parents are healthy.

Pedigrees (A) and photographs/MRI images (B) of patients with inherited ATP9A mutations from families 1 and 2. Informed consent for publication of the images was signed by the patients’ legal guardians, and stripes over the eyes were added in order to anonymize the children’s faces. White arrows represent restricted diffusion within the posterior fossa, and red arrows show expansion of the extra-axial (mainly anterior) CSF spaces. Squares denote male family members, circles represent female family members, solid symbols represent affected family members, and open symbols represent unaffected family members. All individuals were analyzed by whole exome sequencing and Sanger sequencing. C Sanger sequencing traces show three types of mutations in the patients’ ATP9A gene, highlighting the C>T/G>A heterozygous (Y stands for C/T and N stands for G/A) or homozygous point mutations (red nucleotide). D A schematic shows that the three types of nonsense mutations generate premature termination codons and terminate translation of ATP9A. Whole venous blood was collected from 4 independent donors (controls, C1-C4) and family 1 members. The mRNA expression of the indicated amplicons from different exon sites of ATP9A was detected by RT-PCR (E) and qRT-PCR (F, n = 3 replicates) in leukocytes from blood. Representative immunoblots (G) of ATP9A protein expression in leukocytes extracted from blood were detected by western blots, and the immunoblots are representative of four experiments. E1–7/10–18/19–28: amplicons from exon 1–7/10–18/19–28 of ATP9A mRNA. All values are presented as mean ± SEM (**P < 0.01 and ***P < 0.001, one-way ANOVA with Bonferroni post hoc test for F).
The last assessment was conducted at the ages of 11 years 1 month (II-1), 10 years 2 months (II-2), and 11 years (II-3). Anthropometric measurements (Supplementary Table 1) revealed that these subjects had smaller occipitofrontal head circumference (OFC-II-1: 53 cm, <5% ile; OFC-II-2: 50 cm, <2% ile; OFC-II-3: 47 cm, <1% ile) and lower body mass index (BMI-II-1: 15.01 kg/m2, <1% ile; BMI-II-2: 12.54 kg/m2, <1% ile; BMI-II-3: 16.5 kg/m2, <1% ile). Activities of daily living (ADLs) include the basic skills typically required to manage basic physical needs (Supplementary Table 2). The total scores (44, 43 and 42.5) reflected that the patients had severe disorders and were highly dependent on their caregivers [34]. II-1 and II-2 were unable to perform an intelligence measurement because of their severe ID, and the IQ of II-3 was 58, indicating moderately severe ID. The developmental tests (Gesell Development Schedules, GDS) for the three children are shown in Supplementary Table 3. The developmental quotient (DQ) of 5 schedules represents severe to very severe developmental delays [35, 36].
Both II-1 and II-2 (family 1) had mild abnormalities on routine magnetic resonance imaging (MRI), including abnormal signals in the bilateral maxillary sinuses and thickening of soft tissue in the posterior wall of the nasopharynx. II-3 (family 2) had mild abnormalities on MRI, including widening of the extra-axial CSF spaces (mainly anterior) and abnormal signals in the caudate and putamen bilaterally, scattered abnormal signals in the cortices, and restricted diffusion in the posterior fossa (II-3, Fig. 1B and Supplementary Fig. 1A). Congenital metabolic abnormalities were excluded by liquid chromatography and tandem mass spectrometry (LC-MS-MS) of blood and gas chromatography-mass spectrometry (GC-MS) of urine. Electrocardiogram (ECG), ultrasound, and other biochemical tests were unremarkable.
Genetic testing and identification of ATP9A mutations
Chromosome karyotyping (CK) and/or copy number variation (CNV) analysis was performed in all members of families 1 and 2. The results showed no pathogenic or likely pathogenic CNVs. WES and Sanger sequencing were performed in all members of the two families (Fig. 1C and Supplementary Fig. 1B). The compound heterozygous mutation c.433C>T/c.658C>T (p. Arg145*/p. Arg220*) and the homozygous mutation c.983G>A (p. Trp328*) in ATP9A (NM_006045), which encodes probable phospholipid-transporting ATPase IIA (ATP9A), were identified in the three affected children. In addition, the three types of nonsense variants in ATP9A were inherited from the parents, consistent with autosomal recessive inheritance (Fig. 1A, C). A schematic shows that the three types of nonsense mutations generate premature termination codons and cause translational termination of ATP9A (Fig. 1D).
To test whether nonsense mutations in ATP9A occurring in the patients disrupted its translation, we first separated leukocytes from either venous whole blood of 4 healthy subjects or the patients from family 1, and then, tested the expression of ATP9A. Compared with the healthy subjects, the expression of ATP9A in the cell lysates of the patients from family 1 was significantly decreased at both the mRNA (Fig. 1E, F; P < 0.01 for I1 and I2, P < 0.001 for II-1 and II-2) and protein levels (Fig. 1G); in addition, the 2 affected siblings expressed less ATP9A than their parents (P < 0.01). Because of the low specificity of ATP9A antibodies, a weak normal band (~100 kDa) is always seen in the Western blot of patients II-1 and II-2, but no truncated forms of ATP9A were found in either. Venous whole blood samples from family 2 are not available due to pandemic COVID -19.
Atp9a null mice display similar neurological phenotypes as the patients
To test and verify the association between ATP9A deficiency and neurological phenotype, we generated Atp9a gene knockout mice using CRISPR-Cas9 technology. The open reading frame shifts after exon 3 and exon 4 in the mouse Atp9a gene and is cut by Cas9 (Supplementary Fig. 2A). Genotyping of mice was performed 7–10 days after birth and confirmed by RT-PCR and DNA sequencing (Supplementary Fig. 2A, B). A Kaplan–Meier survival curve was used to assess the effect of Atp9a deficit on mouse survival (Supplementary Fig. 2C). The survival curve [72 wild-type mice (WT) and 30 ATP9A−/− mice] demonstrated that the median survival time of the ATP9A−/− mice was 252.5 days, much lower than that of the WT mice, which was 627 days. Comparison of the survival curves with the Breslow test also showed that the survival rate of the mice was greatly reduced by Atp9a deficiency (P = 0.0474). In addition, we found that mating homozygous knockout mice led to complete infertility (P < 0.001; Supplementary Fig. 2D). Therefore, only a small percentage of homozygous knockout mice can be obtained by mating with heterozygotes. The surviving knockout mice had no obvious differences in physical characteristics, and no microcephaly was observed throughout their lifespan.
Homozygous Atp9a knockout mice (ATP9A−/−) and background-matched WT mice were used for subsequent experiments. RT-qPCR confirmed that the brains of ATP9A−/− mice had very low mRNA levels of Atp9a (P < 0.01; Fig. 2A). In addition, we also found significantly low Atp9a mRNA expression in subregions of the mouse brain (cortex, hippocampus, striatum, midbrain, and thalamus) and in the liver, spleen, and kidney (P < 0.001; Supplementary Fig. 2E). In situ hybridization confirmed that Atp9a was more abundant in the six layers of the cortex and in the CA3 region of the hippocampus in the mice from WT than in the Atp9a-null mice (Fig. 2B).

Atp9a gene knockout mice (ATP9A−/−) were engineered using CRISPR/Cas9 technology. A Atp9a mRNA transcription in mouse brain was detected by RT-qPCR (n > 3). B Representative images of Atp9a mRNA transcription (red) in mouse cerebral cortex and hippocampus were detected by in situ hybridization. Scale bar, 200 μm. Two-month-old mice, half male and half female, were used for all behavioral tests. Muscle strength of mice was evaluated using coat hanger tests (C) and wire hang tests (D) (n = 8). E–I MWM tests were conducted including a 5-day navigation test and a spatial probe test on day 6. A schematic of the MWM and representative mouse movement tracks at day 6 (E), escape latency (F) and path length (G) over 5 days are shown. P: platform; N, E, S and W represent the four quadrants. The times of crossing the removed-platform area (H) and the percentage of time mice spent in the target quadrant (I) were assessed in a spatial probe test (n > 8). J A schematic of the Y-maze test and the representative movement tracks of mice are shown. A, B and C represent three different maze arms. Spontaneous alternations (K), total arm entries (L), total distance moved (M) and mean velocity (N) of mice were assessed in the Y-maze test (n > 8). O Schematic representation of the open-field tests and the representative movement tracks of mice. P: peripheral area; C: central area. Number of crossing the central area (P) and time spent in the central area (Q), the total distance moved (R) and mean velocity (S) of the mice were evaluated in the open-field test (n > 8). All values are presented as mean ± SEM (*P < 0.05, **P < 0.01, and ***P < 0.001, unpaired t-test for A, C, D, H, I, K–N, P–S, and two-way ANOVA with Bonferroni post hoc test for F and G).
For the behavioral tests, two-month-old mice were used, half male and half female, to exclude the effects of age and sex on the behavior of the mice. The coat hanger test and the wire hang test are common methods for analyzing neuromuscular strength and coordination in animals [37, 38]. These two tests are based on the ability of mice to climb and grasp objects with their claws. Compared to those of the WT mice, both the grasping scores (Fig. 2C) and the latency to fall (Fig. 2D) of the ATP9A−/− mice were dramatically reduced in the two types of tests (P < 0.001). These data indicate that homozygous deletion of Atp9a leads to impaired muscle strength in mice.
The Morris Water Maze (MWM) test was used to examine hippocampal-dependent spatial learning and memory in mice. Representative movement tracks of the mice on day 6 in the spatial probe test component of the MWM are shown in Fig. 2E. Spatial navigation tests showed that ATP9A−/− mice took more time and longer distances than WT mice to find the invisible platform on day 2 and/or 5 (Fig. 2F, G), with no significant change in mean velocity (Supplementary Fig. 3A). In addition, the ATP9A−/− mice crossed the distant platform area less frequently (P < 0.05; Fig. 2H) and spent less time in the target quadrant (P < 0.05; Fig. 2I) than the WT mice undergoing spatial probe testing on day 6. These results suggest that homozygous deletion of Atp9a has a major impact on disrupting hippocampus-dependent spatial learning and memory in mice, consistent with the phenotypes of patients.
To further verify the prefrontal spatial working memory of mice, we conducted the Y-maze test 48 h after the MWM test. The representative movement tracks of mice in three arms of the Y-maze are shown in Fig. 2J. Unexpectedly, compared with the mice from WT, the ATP9A−/− mice showed no changes in spontaneous alternations (Fig. 2K), but they exhibited increased total number of arm entries (P < 0.001; Fig. 2L), movement distance (P < 0.001; Fig. 2M) and mean velocity (P < 0.001; Fig. 2N).
An open-field (OF) experiment was used to further assess the locomotor, exploratory, and anxiety-like behaviors of the mice (Fig. 2O). The ATP9A−/− mice had higher crossing frequency (P < 0.05; Fig. 2P) and residence time (P < 0.05; Fig. 2Q) in the central area than the WT mice and higher total movement distance (P < 0.001; Fig. 2R) and mean velocity (P < 0.001; Fig. 2S). An elevated plus maze (EPM) test was used to further confirm anxiety-related behavior in the mice (Supplementary Fig. 3B). Consistent with the hyperactivity phenotype in the Y-maze tests and the open-field tests, the ATP9A−/− mice showed a higher residence time (P < 0.05; Supplementary Fig. 3C) in the open arms and a greater willingness to move beyond the edges (P < 0.05; Supplementary Fig. 3D), a greater total distance of movement (P < 0.01; Supplementary Fig. 3E) and higher mean velocity (P < 0.01; Supplementary Fig. 3F). These results indicated that the absence of Atp9a has no effect on the spatial working memory and anxiety-like behaviors of the mice but leads to hyperkinetic movements of the mice.
ATP9A deficiency causes synaptic dysfunction in the primary motor cortex and hippocampus of the mouse brain
To investigate the role of ATP9A in mice, we first attempted to observe the morphological changes in neurons in the motor cortex and hippocampus of ATP9A−/− mice. Golgi staining was performed in 2- month- old mouse brains. Representative images of the dendritic morphology of pyramidal neurons in the mouse motor cortex and hippocampal CA1 region are shown in Fig. 3A and Supplementary Fig. 4A. Sholl analysis was used to quantify neurite complexity. The results revealed that the intersection numbers of dendritic arborization in the motor cortex and hippocampus of the ATP9A−/− mice were robustly reduced compared to the WT mice (Fig. 3A–C). In addition, the total intersection number per neuron in the mouse motor cortex (Supplementary Fig. 4B) showed a pronounced reduction (P < 0.01) compared to that in the hippocampus (Supplementary Fig. 4C). Thus, we observed secondary dendritic spines in the pyramidal neurons of mouse motor cortex. The density of secondary dendritic spines was significantly reduced (P < 0.001; Fig. 3D, E), and both mature (indicated by blue triangles) and immature (indicated by green triangles) dendritic spines disappeared dramatically in the cortical neurons of ATP9A−/− mice (Fig. 3D). These results indicate that the Atp9a-null mice have severely damaged neuronal neurite morphology and synaptic structure in the brain.

A–E Golgi staining was conducted on half of the mouse brains after the behavioral tests. Pyramidal neurons from the fifth- layer of the mouse motor cortex and the CA1 region of the hippocampus were studied for Sholl analysis after Golgi staining. A A schematic of the dendritic morphology of pyramidal neurons in mouse motor cortex and hippocampus. Scale bar, 100 μm. B, C The intersection number per neuron in mouse motor cortex (B, n = 16 neurons from 6 mice/genotype) and hippocampus (C, n = 16 neurons from 6 mice/genotype) was calculated. Representative images of secondary dendritic spines (D) and spine density (E) in mouse pyramidal neurons (n > 14). Blue triangles indicate mature spines and green triangles indicate immature spines. Scale bar, 20 μm. F–J Pyramidal neurons in primary motor cortex (Cor-M1) and the hippocampus (Hip-CA1) were clamped in whole-cell configuration. Miniature excitatory postsynaptic currents (mEPSCs) were recorded. F Representative mEPSC traces. Scale bars, 20 pA and 1 s. Neurons (n = 20) from 5 WT and 5 ATP9A−/− mice in the primary motor cortical region M1 (G, H). Neurons (n = 20) from 5 WT and 5 ATP9A−/− mice in the hippocampal CA1 region (I, J). K–N Atp9a knockdown was performed by siRNA transfection in DIV5 rat primary cortical neurons and N2a cells. (K) Atp9a mRNA expression in rat primary cortical neurons was detected by RT-qPCR after transfection for 48 h (n = 5). L Representative images show that ATP9A knockdown leads to neurite fracture and retraction. The white boxes in the overview images are enlarged on the right, the yellow arrows and box show the fractured neurites and the red arrows and box show the beading change of neurites in ATP9A-depleted primary cortical neurons, and the normal morphology of neurites is shown in the purple box. Scale bar, 50 μm. M Neuron neurites length (more than 50 cells from 8 fields) was measured by ImageJ. N Cell viability of rat primary cortical neurons (n = 5) was determined using a CellTiter-Glo® Luminescent Kit. Atp9a mRNA expression (O, n = 5) and cell viability (P, n = 9) of N2a cells are shown. All values are presented as mean ± SEM (*P < 0.05, **P < 0.01, and ***P < 0.001, two-way ANOVA with Bonferroni post hoc test for B and C; unpaired t-test for E, G–J, K, M and N; one-way ANOVA with Bonferroni post hoc test for O and P).
To further verify whether synaptic transmission is affected in the motor cortex and hippocampus, we recorded miniature excitatory postsynaptic currents (mEPSCs) from pyramidal neurons in M1/M2 layer V (Cor-M1/M2 represent primary/secondary motor cortex, Fig. 3F and Supplementary Fig. 5A) and hippocampal CA1 (Hip-CA1, Fig. 3F) using whole-cell patch-clamp analysis. Reduced mEPSC frequency (P < 0.01) but not amplitude was found in the primary motor cortex of the Atp9a null mouse brain (Fig. 3G, H). Meanwhile, both the frequency (P < 0.01) and amplitude (P < 0.05) of mEPSCs in the hippocampal CA1 pyramidal neurons were decreased dramatically in the Atp9a null mice compared with WT mice (Fig. 3I, J). These results indicate that ATP9A deficit impairs excitatory synaptic transmission in primary motor cortex and hippocampus. However, neither the frequency nor the amplitude of mEPSCs were altered in the M2 pyramidal neurons of Atp9a-null mice (Supplementary Fig. 5B, C). Collectively, these observations indicate a critical role of ATP9A in regulating synaptic function in the brain.
ATP9A is required for the maintenance of neuronal neurite morphology and the viability of neural cells
To further investigate the role of ATP9A in neurodevelopment, we aimed to obtain primary cultured neurons from the ATP9A−/− mice in vitro, but mating of homozygous knockout mice was unsuccessful (Supplementary Fig. 2D). Therefore, we silenced Atp9a by siRNA in rat primary cortical neurons instead. Mean mRNA expression of Atp9a was significantly reduced to 29.04 ± 5.693% in rat primary cortical neurons transfected with siRNA (RNAi) compared to cells transfected with scrambled control siRNA (negative control, NC) (P < 0.001; Fig. 3K). Morphological study suggested that ATP9A knockdown led to severe neurite damage, such as neurite fracture, retraction (indicated by yellow arrows and boxes) and beading change (indicated by red arrows and boxes), in neurons compared to those in the scrambled siRNA treatment group (normal neurite morphology is enlarged in the purple box on the right) (Fig. 3L). We measured the neurite length of primary cortical neurons, which was significantly shortened (P < 0.001) in ATP9A knockdown neurons compared to the NC neurons (Fig. 3M). As a result of the severely disrupted neurite morphology caused by ATP9A knockdown, the cell viability of RNAi neurons was reduced by 46.79% (P < 0.01; Fig. 3N).
To further examine the effect of ATP9A deficit on cell viability, mouse neuroblastoma N2a cells were used. When mRNA expression of Atp9a was silenced by siRNA to 10.02 ± 3.119% (RNAi-1#; P < 0.001 vs. NC) and 6.38 ± 1.052% (RNAi-2#; P < 0.001 vs. NC) in N2a cells (Fig. 3O), the cell viability was reduced to 87.55% and 84.88% (P < 0.01 vs. NC; Fig. 3P), respectively. These results indicate that ATP9A plays a critical role in maintaining neuronal neurite morphology and confirm that ATP9A depletion impairs neurodevelopment and cell viability of both primary neurons and an immortalized neuronal cell line.
ATP9A is mainly localized to endosomes, and pathogenic mutants of ATP9A have altered intracellular distribution
To elucidate the pathogenic mechanism of ATP9A deficit-induced neurological impairment, we first explored its subcellular localization. It has previously been shown that overexpressed HA-ATP9A is mainly localized to early endosomes, recycling endosomes and the trans-Golgi network (TGN) in HeLa cells [39]. To confirm whether endogenous ATP9A is present in endosomes, the crude endosomes were purified from fresh mouse liver based on the protocol developed by Christian J et al. with minor changes [40], as shown in Fig. 4A. We found that ATP9A is enriched in endosomal lysate (EL, RAB5 for early endosomes, RAB7 for late endosomes and RAB11 for recycling endosomes). ATP9A failed to co-fractionate with either lysosomes (LAMP1), cis-Golgi (GM130), plasma membrane (N-cadherin), ER (Calnexin) or nuclei (Lamin B1) (Fig. 4B). This observation further confirms that ATP9A is highly expressed in endocytic organelles.

A, B Crude endosomes were isolated from fresh mouse liver by sucrose gradient centrifugation. A Schematic representation of the extraction protocol for crude endosomes. B ATP9A and organelle markers were analyzed in tissue lysate or endosome components by immunoblotting. C, D Colocalization of ATP9A and organelles in the mCherry-ATP9A-transfected N2a cells was observed by confocal microscopy. Representative immunofluorescence images are shown (C). The white boxes in the overview images are enlarged on the right, and the white arrows indicate colocalization of ATP9A and organelles. D Pearson’s correlation coefficient was applied using Image-Pro Plus to test a correlation between mCherry-tagged ATP9A (red) and organelles (green) (5 randomly selected boxes). Scale bar, 10 μm. (E, F) The mCherry-tagged ATP9A pathogenic mutants were co-transfected with RAB5-GFP, RAB7-GFP or RAB11-GFP in N2a cells. Colocalization of ATP9A pathogenic mutants (C433>T, E; C658>T, F) with RABs in N2a cells was observed by confocal microscopy. Scale bar, 10 μm. Colocalization of ATP9A mutants with RABs on the light blue line in the merged images was analyzed by line profile using ImageJ software and shown on the right. All values are presented as mean ± SEM (*P < 0.05 and **P < 0.01, one-way ANOVA with Bonferroni’s post hoc test for B). EE early endosomes, RE recycling endosomes, LE late endosomes, Trans-G trans-Golgi network, Cis-G cis-Golgi, PM plasma membrane, UC pellet debris and undestroyed cells, P&N plasma membrane and nuclei, E&L endosomes and lysosomes, E endosomes, WCL whole-cell lysate, EL endosomal lysate.
To verify the endosomal distribution of ATP9A in neural cells, we examined the subcellular localization of exogenous ATP9A by immunofluorescence in N2a cells. mCherry-tagged ATP9A forms small puncta distributed in the cytoplasm and colocalizes with RAB7-positive late endosomes, RAB11-positive recycling endosomes, and RAB5-positive early endosomes in N2a cells (Fig. 4C), whereas it does not colocalize with the TGN (TGN38), the cis-Golgi (GM130), the plasma membrane [PHPLCδ-PI (4,5) P2], the mitochondrion (Mito-GFP) or the endoplasmic reticulum (ER, Sec61β) (Fig. 4C). The colocalization of exogenous ATP9A with endosomes (early, recycling and late) was significantly higher than that with other organelles based on Pearson’s correlation coefficient (Fig. 4D).
The above results indicated that exogenous ATP9A is mainly distributed to endosomes in N2a cells. We wondered whether the intracellular localization of ATP9A pathogenic mutants would be altered. The ATP9A pathogenic mutants (C433T and C658T) in family 1 formed aggregated puncta distributed in the cytoplasm of N2a cells (Fig. 4E, F). Unlike the pattern of wild-type ATP9A in Fig. 4C, ATP9A pathogenic mutants rarely colocalized with RAB5, RAB7, or RAB11, indicating that these mutants have altered intracellular distribution. Thus, we proposed that the physiological function of ATP9A may depend on its correct endosomal localization.
WT, but not pathogenic forms of ATP9A, play a pivotal role in efficient endosomal recycling in neural cells
Previous reports have shown that ATP9A is crucial for endocytic recycling in HeLa cells [27], although the exact mechanism is not fully understood. Since aberrant endosomal recycling has been implicated in a variety of the most common neurological disorders [30], we hypothesized that the pathogenesis of ATP9A in neurological diseases might be related to its function in regulating endosomal recycling. To assess the influence of ATP9A deficiency or mutation on endosomal trafficking in neural cells, we generated a stable heterozygous ATP9A knockout N2a cell line (named ATP9A-KD) by CRISPR/Cas9 technology. Low expression of ATP9A was detected at both mRNA (P < 0.001, Fig. 5A) and protein level (Fig. 5B) compared to the cells infected with vector lentiviruses (negative control, NC). Rescue experiments were conducted by transfecting WT or pathogenic mutants of ATP9A into the ATP9A-KD N2a cells. Efficient transfection of WT (KD + ATP9A WT) or pathogenic mutants (KD + ATP9A C433T and KD + ATP9A C658T) of ATP9A was confirmed by western blotting (Fig. 5C).

A, B N2a cells with ATP9A stable knockdown were generated using CRISPR/Cas9 technology. A Atp9a mRNA expression in stable knockdown N2a cells was detected by RT-qPCR (n = 3). B ATP9A protein expression in control and ATP9A stable knockdown N2a cells was detected by western blots. C Control or ATP9A knockdown N2a cells were transfected with the HA-tagged vector, WT (117.9 kDa) or the pathogenic ATP9A mutant (C433 > T, 18.7 kDa and C658 > T, 26.9 kDa), and immunoblots of ATP9A proteins were performed with a HA antibody. D–M Cells in five groups (NC + Vector; KD + Vector; KD + ATP9A WT; KD + ATP9A C433 > T; KD + ATP9A C658 > T) were placed on ice for 10 min and then incubated with Alexa Fluor 647–conjugated TFN at 37 °C for 20 min. Cells were washed and incubated at 37 °C for the indicated times. They were then fixed and stained for RAB11, EEA1, and DAPI. Representative immunofluorescence images of colocalization of TFN with EEA1 (D, E, H, I, J) or RAB11 (F, G, K, L, M) at three time points (10 min, 30 min, 60 min) are shown. The white boxes within the overview images are shown. Scale bar, 10 μm. Pixel intensities of TFN (N) and colocalization of TFN with EEA1(O) and RAB11 (P) were estimated at the indicated time points using Image-Pro Plus software. Data are shown as mean TFN intensities and Pearson’s correlation coefficient in different groups at each time point. More than 200 cells from 5–8 randomly selected fields of each sample were analyzed. All values are presented as mean ± SEM (*P < 0.05, **P < 0.01, and ***P < 0.001, unpaired t-test for A, and two-way ANOVA with Bonferroni’s post hoc test for N–P).
Alexa Fluor 647–conjugated transferrin (TFN) was used to study the effects of ATP9A depletion on the recycling pathway of endocytosis. Transferrin is internalized into cells and encapsulated by early endosomes, transported to recycling endosomes and recycled back to the plasma membrane [41]. N2a cells were stained with EEA1 (an early endosome marker) or RAB11 after incubation with TFN for 20 min and chasing for an additional 10 min, 30 min, or 60 min respectively (Fig. 5D–M). As shown in Fig. 5D–G, TFN signal in ATP9A-KD cells is distributed throughout the cytoplasm and remains at a relatively high level at all time points compared with control cells, indicating that ATP9A knockdown leads to the intracellular retention of TFN in N2a cells. In addition, the distribution of endosomes, especially recycling endosomes, is more dispersed in the cytoplasm of ATP9A-KD cells, whereas it is concentrated on one side of the nucleus in control cells. Restoration of wild-type ATP9A, but not pathogenic ATP9A mutants (C433T and C658T), largely cures the transport delay of TFN and the abnormal distribution of recycling endosomes caused by ATP9A depletion in N2a cells (Fig. 5H–M). The internalized TFN signals in Fig. 5D–M were quantified and further demonstrate that TFN transport delay occurs in the cytoplasm when cells lack full-length ATP9A (Fig. 5N).
To confirm which parts of the intracellular trafficking process of TFN are disrupted by ATP9A depletion, we analyzed the colocalization of TFN with EEA1 and RAB11 using Pearson’s correlation coefficient. The results are shown in Fig. 5O, P. We found that in ATP9A-KD cells, more TFN was localized to early endosomes but less to recycling endosomes at each time point. These data indicate that ATP9A-KD cells fail to deliver TFN from early endosomes to recycling endosomes, and that most TFN remain trapped in early endosomes over time. In addition, the defects are partially restored by re-expression of full-length ATP9A but not by the pathological mutants. These observations suggest that ATP9A is required for maintenance of recycling endosome distribution and recycling of TFN. The intracellular retention of endocytosed TFN caused by ATP9A defects is due to inhibition of its transport from early endosomes to recycling endosomes. Taken together, these observations demonstrate that ATP9A is required for proper distribution of recycling endosome and efficient recycling of TFN in neural cells.
ATP9A is required for RAB5 and RAB11 activation in vitro and in vivo
Small GTPase RABs serve as master regulators in modulating intracellular vesicle transport. The conversion between an inactive GDP-bound and an active GTP-bound form of RAB5 and RAB11 mediates the early endosomal recycling pathway, as well as the fusion or localization of endosomes [42,43,44]. As mentioned above, ATP9A localizes mainly to recycling endosomes and regulates their trafficking. We investigated whether ATP9A modulates the activity of RAB5 and RAB11. By using GTP-agarose pull-down assay, we found that depletion of ATP9A (KD) significantly reduced the GTP form of RAB5 (P < 0.05) and RAB11 (P < 0.01) in N2a cells (Fig. 6A–C), without affecting the overall protein levels of RAB5 and RAB11 (Fig. 6A). These results were also confirmed in both the M1 (P < 0.01) and hippocampal CA1 (P < 0.05) regions of ATP9A−/− mouse brains (Fig. 6D–I).

A–C The active form of RAB5 and RAB11 (GTP-RAB5 and GTP-RAB11) in control (NC) and ATP9A knockdown (KD) N2a cells was determined by GTP-agarose pull-down assay. A Representative immunoblots of total or active RAB5 and RAB11 in cells were detected by western blots. “Empty agarose” indicates that samples were incubated with blank agarose beads, and “Pull-down” indicates that samples were incubated with GTP-agarose beads. B, C Quantification of RAB5 and RAB11 activity in control and ATP9A knockdown N2a cells (n = 3). GTP-RAB5 and GTP-RAB11 in M1 (D–F) and CA1 regions in the hippocampus (G-I) of WT and ATP9A−/− mice were determined by GTP-agarose pull-down assay. Representative immunoblots of total or active RAB5 and RAB11 in M1 (D) and hippocampal CA1 (G) regions of WT and ATP9A−/− mouse brains were detected by Western blots. (E, F, H, I) Quantification of RAB5 and RAB11 activity in M1 (n = 5) and hippocampal CA1 (n = 4) regions of WT and ATP9A−/− mouse brains. J–L GTP-RAB5 and GTP-RAB11 were determined by GTP-agarose pull-down assay in control (Ctrl) and ATP9A overexpressed (OE) N2a cells (n = 3). M HA-ATP9A were co-transfected with GFP-vector, GFP-RAB5 (Q79L) or GFP-RAB5 (S34N) in N2a cells. GFP-tagged RAB5 was immunoprecipitated from cell lysates and immunoblotted against exogenous ATP9A. N HA-ATP9A was co-transfected with GFP-vector, GFP-RAB11 (Q70L), or GFP-RAB11 (S25N) in N2a cells. GFP-tagged RAB11 was immunoprecipitated from cell lysates and immunoblotted against exogenous ATP9A. All values are presented as mean ± SEM (*P < 0.05, **P < 0.01, and ***P < 0.001, unpaired t-test for B, C, E, F, H, I, K and L). Filled triangles indicate protein markers.
An ATP9A-overexpressed stable N2a cell line was used to verify whether the activity of RAB5 and RAB11 is enhanced by a high expression level of ATP9A. Efficient overexpression of ATP9A was confirmed by immunoblot with HA antibody (Supplementary Fig. 6A). The GTP form of RAB5 (P < 0.05) and RAB11 (P < 0.001) increased markedly in HA-ATP9A-overexpressed (OE) N2a cells compared with cells overexpressing HA-vector (Ctrl) (Fig. 6J to L).
Exogenous RAB5 and RAB11 activity was further determined using recombinant RAB5-binding domain of Rabenosyn-5 (GST-R5BD; residues 1–40) and Rabin8 (GST-Rabin8), which specifically interact with active GTP-bound RAB5 and RAB11, respectively [45, 46]. The recombinant proteins of GST, GST-R5BD, and GST-Rabin8 were purified and detected by Coomassie blue staining (Supplementary Fig. 6B). Specific interaction of the purified proteins with the GTP form of RABs was detected by GST pull-down assay in N2a cells (Supplementary Fig. 6C, D). GST-R5BD or GST-Rabin8 interacts with both wild type and the constitutively active form of RAB5 / RAB11 [RAB5 (Q79L) / RAB11 (Q70L)], but not with the dominant-negative inactive form of RAB5 / RAB11 [RAB5 (S34N) / RAB11 (S25N)] (Supplementary Fig. 6C). Next, we found that more GTP-RAB5 or GTP-RAB11 was pulled down by GST-R5BD (P < 0.05 vs. Ctrl; Supplementary Fig. 6E, F) or GST-Rabin8 (P < 0.001 vs. Ctrl; Supplementary Fig. 6G, H), respectively, in OE cells, suggesting that RAB5 or RAB11 has high activity in ATP9A-overexpressed N2a cells.
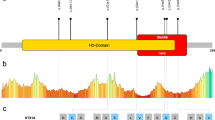
The binding specificity of ATP9A with the inactive GDP-bound and the active GTP-bound forms of RAB5 and RAB11 was verified by co-IP assay (Fig. 6M, N). ATP9A binds more inactive GDP-bound forms of RAB5 or RAB11 than its active GTP-bound and wild-type forms, suggesting that ATP9A mainly plays a key role in the activation of RAB5 and RAB11. In addition, the binding regions of ATP9A to RAB5 and RAB11 were identified by co-IP assay with a panel of HA-tagged ATP9A truncates (Supplementary Fig. 7A). We found that ATP9A truncates lacking the P and N domains, which mediate ATP binding and ADP release [11], showed only a very weak interaction with both RAB5 and RAB11. This suggests that the P domain and the N domain may be the key regions for binding and activation of these RABs (Supplementary Fig. 7B, C).
Discussion
In this study, we describe for the first time a novel human neurological disease caused by ATP9A nonsense mutations. This disorder is characterized by autosomal recessive hypotonia, ID, ADHD, absent speech, severe receptive language delay, lack of social interactions, and severe sleep disturbance. In addition to the 3 cases we have reported here, the correlation of ATP9A mutations with human neurodevelopmental disorders has recently been reported by 3 groups, although the phenotypes of the patients are partially different [8,9,10], and more individuals with homozygous or heterozygous mutations of ATP9A have mild to severe intellectual disability according to GeneMatcher [47]. Moreover, deficiency and overexpression of ATP8A1-, ATP8A2-, ATP8B4-, and other P4-ATPases are closely related to human neurological diseases, such as ID, hypotonia, hyperkinetic movement disorders, cerebellar ataxia, optic atrophy, hearing loss, autism and Alzheimer’s disease [15, 48,49,50,51]. These findings are strong evidence that there is an inseparable link between ATP9A deficiency and neurological diseases. The pathological phenotypes of ATP9A deficiency are partially consistent with those caused by depletion of ATP8A1 or ATP11B (both are P4-ATPases). Disruption of either ATP8A1 or ATP11B leads to altered synaptic strength, electrical activity, autistic-like behavior, delayed hippocampus-dependent learning, and impaired synaptic plasticity in mice [14, 16, 52].
Endosome-mediated vesicle transport is essential for neuronal survival and synaptic plasticity [53]. Mutations in several endosome-associated genes, such as SLC9A6, TBC1D23, ARF1, and ATP13A2, can affect the central or peripheral nervous system [54,55,56,57]. Despite the evidence that ATP9A may be involved in cellular endosomal transport in HeLa cells [27, 28], knowledge of the physiological function of endogenous ATP9A in neural cells and mouse brain is still limited. Our data have shown that the endosomal localization of ATP9A and the altered intracellular localization of its pathogenic mutants in N2a cells, indicating the physiological function of ATP9A are possibly associated with the modulation of endosomal transport.
Transferrin and its receptor TfnR, are widely used to monitor the recycling pathway of endocytosis [41]. In this study, retention of TFN and altered intracellular localization of recycling endosomes occurred in N2a cells with ATP9A knockdown. These results are reported in partial agreement by Yoshiki Tanaka [27]. Supplementing wild-type ATP9A largely restored normal transport of TFN and altered distribution of recycling endosomes, but the pathogenic mutants failed to rescue these processes. In addition, we discovered that the delivery of TFN in ATP9A knockdown cells was blocked especially during transport from early endosomes to recycling endosomes. These observations encourage us to focus on detecting the expression and activation of the small GTPases RAB5 and RAB11, which are critical for early endosome fusion [43], recycling endosome location [44], and transferrin recycling [58].
Sufficient activity of RAB5 and RAB11 is required for BDNF-induced dendritic branching [59] and recruitment of AMPA receptors to the synaptic membrane during long-term potentiation (LTD) [60, 61] in hippocampal neurons to mediate synaptic plasticity and memory formation. Moreover, dysregulation of RAB5 and RAB11 has been implicated in some neurodegenerative diseases, such as Alzheimer’s disease (AD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) [31, 62]. We demonstrated that ATP9A only regulates the activity of RAB5 and RAB11 without affecting their expression in N2a cells and transgenic mouse brains. This suggests that the physiological function of ATP9A in the nervous system may be related to the activation of RAB5 and RAB11. The inactive form of RAB5 and RAB11 binds tightly to ATP9A, confirming that ATP9A mainly plays a key role in activating RAB5/RAB11 but not in down-regulating their hydrolysis. In conjunction with previous studies, it has been shown that activation of RAB11 is required for the correct location of pericentriolar recycling endosomes [44] and the exit of transferrin from early endosomes to the recycling compartment [42], whereas activation of RAB5 is required prior to membrane fusion of early endosomes [43]. It is easy to explain that why the internalized TFNs are trapped in early endosomes but not the ectopic recycling endosomes. However, the question of how ATP9A dynamically regulates this process by activating different components in the endosomal recycling pathway needs further exploration. Although ATP9A also partially colocalizes with RAB7, we did not pursue this further. This is because RAB7 is the key RAB that mainly modulates the endo-lysosomal degradation pathway, which was not affected after knockdown of ATP9A [27]. However, whether ATP9A regulates the activity of other RABs requires further investigation.
Overall, these data have revealed a previously unknown role for ATP9A in activating RAB5 and RAB11 to promote the endosomal recycling pathway in N2a cells and mouse brains. Deficiency or mutation of ATP9A leads to inactivation of RAB5 and RAB11, resulting in abnormal endosomal recycling in neural cells. This leads to synaptic dysfunction in the primary motor cortex and hippocampus of the brain, causing neurological disorders with hypotonia, ID, and ADHD as major phenotypes (Supplementary Fig. 8). Our study extends the current knowledge of ATP9A in regulating endosomal recycling and neurodevelopment. Given that variants of ATP9A have been identified in several neurological diseases with different phenotypes, this study will provide important information for evaluating the enhancement of RAB5 and RAB11 activity as a therapeutic strategy for the treatment of neurological disorders caused by ATP9A deficiency.
Data availability
All data are available in the main text or the supplementary information at MP’s website.
References
Mehregan H, Najmabadi H, Kahrizi K. Genetic studies in intellectual disability and behavioral impairment. Arch Iran Med. 2016;19:363–75.
Stevenson RE, Procopio-Allen AM, Schroer RJ, Collins JS. Genetic syndromes among individuals with mental retardation. Am J Med Genet A. 2003;123A:29–32.
Klein M, Onnink M, van Donkelaar M, Wolfers T, Harich B, Shi Y, et al. Brain imaging genetics in ADHD and beyond–mapping pathways from gene to disorder at different levels of complexity. Neurosci Biobehav Rev. 2017;80:115–55.
Association AP. Diagnostic and statistical manual of mental disorders: DSM-5. Washington, DC: American Psychiatric Association; 2013.
van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet. 2011;45:81–104.
Franke B, Neale BM, Faraone SV. Genome-wide association studies in ADHD. Hum Genet. 2009;126:13–50.
Online Mendelian Inheritance in Man. OMIM. Maryland: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; 2020.
Vogt G, Verheyen S, Schwartzmann S, Ehmke N, Potratz C, Schwerin-Nagel A, et al. Biallelic truncating variants in ATP9A cause a novel neurodevelopmental disorder involving postnatal microcephaly and failure to thrive. J Med Genet. 2022;59:662–8.
Mattioli F, Darvish H, Paracha SA, Tafakhori A, Firouzabadi SG, Chapi M, et al. Biallelic truncation variants in ATP9A are associated with a novel autosomal recessive neurodevelopmental disorder. NPJ Genom Med. 2021;6:94.
Fazia T, Marzanati D, Carotenuto AL, Beecham A, Hadjixenofontos A, McCauley JL, et al. Homozygosity haplotype and whole-exome sequencing analysis to identify potentially functional rare variants involved in multiple sclerosis among sardinian families. Curr Issues Mol Biol. 2021;43:1778–93.
Hiraizumi M, Yamashita K, Nishizawa T, Nureki O. Cryo-EM structures capture the transport cycle of the P4-ATPase flippase. Science. 2019;365:1149–55.
Andersen JP, Vestergaard AL, Mikkelsen SA, Mogensen LS, Chalat M, Molday RS. P4-ATPases as phospholipid flippases-structure, function, and enigmas. Front Physiol. 2016;7:275.
Sebastian TT, Baldridge RD, Xu P, Graham TR. Phospholipid flippases: building asymmetric membranes and transport vesicles. Biochim Biophys Acta. 2012;1821:1068–77.
Levano K, Punia V, Raghunath M, Debata PR, Curcio GM, Mogha A, et al. Atp8a1 deficiency is associated with phosphatidylserine externalization in hippocampus and delayed hippocampus-dependent learning. J Neurochem. 2012;120:302–13.
Guissart C, Harrison AN, Benkirane M, Oncel I, Arslan EA, Chassevent AK, et al. ATP8A2-related disorders as recessive cerebellar ataxia. J Neurol. 2019;267:203–13.
Wang J, Li W, Zhou F, Feng R, Wang F, Zhang S, et al. ATP11B deficiency leads to impairment of hippocampal synaptic plasticity. J Mol Cell Biol. 2019;11:688–702.
Cai SY, Gautam S, Nguyen T, Soroka CJ, Rahner C, Boyer JL. ATP8B1 deficiency disrupts the bile canalicular membrane bilayer structure in hepatocytes, but FXR expression and activity are maintained. Gastroenterology. 2009;136:1060–9.
de Waart DR, Naik J, Utsunomiya KS, Duijst S, Ho-Mok K, Bolier AR, et al. ATP11C targets basolateral bile salt transporter proteins in mouse central hepatocytes. Hepatology. 2016;64:161–74.
Dhar MS, Sommardahl CS, Kirkland T, Nelson S, Donnell R, Johnson DK, et al. Mice heterozygous for Atp10c, a putative amphipath, represent a novel model of obesity and type 2 diabetes. J Nutr. 2004;134:799–805.
Siggs OM, Arnold CN, Huber C, Pirie E, Xia Y, Lin P, et al. The P4-type ATPase ATP11C is essential for B lymphopoiesis in adult bone marrow. Nat Immunol. 2011;12:434–40.
van der Mark VA, Elferink RP, Paulusma CC. P4 ATPases: flippases in health and disease. Int J Mol Sci. 2013;14:7897–922.
Hua Z, Fatheddin P, Graham TR. An essential subfamily of Drs2p-related P-type ATPases is required for protein trafficking between Golgi complex and endosomal/vacuolar system. Mol Biol Cell. 2002;13:3162–77.
Lyssenko NN, Miteva Y, Gilroy S, Hanna-Rose W, Schlegel RA. An unexpectedly high degree of specialization and a widespread involvement in sterol metabolism among the C. elegans putative aminophospholipid translocases. BMC Dev Biol. 2008;8:96.
Efe JA, Plattner F, Hulo N, Kressler D, Emr SD, Deloche O. Yeast Mon2p is a highly conserved protein that functions in the cytoplasm-to-vacuole transport pathway and is required for Golgi homeostasis. J Cell Sci. 2005;118:4751–64.
Beer KB, Rivas-Castillo J, Kuhn K, Fazeli G, Karmann B, Nance JF, et al. Extracellular vesicle budding is inhibited by redundant regulators of TAT-5 flippase localization and phospholipid asymmetry. Proc Natl Acad Sci USA. 2018;115:E1127–E1136.
Naik J, Hau CM, Ten Bloemendaal L, Mok KS, Hajji N, Wehman AM, et al. The P4-ATPase ATP9A is a novel determinant of exosome release. PLoS ONE. 2019;14:e0213069.
Tanaka Y, Ono N, Shima T, Tanaka G, Katoh Y, Nakayama K, et al. The phospholipid flippase ATP9A is required for the recycling pathway from the endosomes to the plasma membrane. Mol Biol Cell. 2016;27:3883–93.
McGough IJ, de Groot REA, Jellett AP, Betist MC, Varandas KC, Danson CM, et al. SNX3-retromer requires an evolutionary conserved MON2:DOPEY2:ATP9A complex to mediate Wntless sorting and Wnt secretion. Nat Commun. 2018;9:3737.
Li X, DiFiglia M. The recycling endosome and its role in neurological disorders. Prog Neurobiol. 2012;97:127–41.
O’Sullivan MJ, Lindsay AJ. The endosomal recycling pathway-at the crossroads of the cell. Int J Mol Sci. 2020;21:6074.
Veleri S, Punnakkal P, Dunbar GL, Maiti P. Molecular insights into the roles of rab proteins in intracellular dynamics and neurodegenerative diseases. Neuromol Med. 2018;20:18–36.
Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–25.
Mitra J, Hegde PM, Hegde ML. Loss of endosomal recycling factor RAB11 coupled with complex regulation of MAPK/ERK/AKT signaling in postmortem spinal cord specimens of sporadic amyotrophic lateral sclerosis patients. Mol Brain. 2019;12:55.
Mlinac ME, Feng MC. Assessment of activities of daily living, self-care, and independence. Arch Clin Neuropsychol. 2016;31:506–16.
Yang J, Hu L, Zhang Y, Shi Y, Jiang W, Song C. Gesell Developmental Schedules scores and the relevant factors in children with Down syndrome. J Pediatr Endocrinol Metab. 2020;33:539–46.
Group BMDC. Gesell developmental diagnosis scale. Beijing: Beijing Mental Development Cooperative Group; 1985.
Võikar V, Rauvala H, Ikonen E. Cognitive deficit and development of motor impairment in a mouse model of Niemann-Pick type C disease. Behav Brain Res. 2002;132:1–10.
Matsuo N, Tanda K, Nakanishi K, Yamasaki N, Toyama K, Takao K, et al. Comprehensive behavioral phenotyping of ryanodine receptor type 3 (RyR3) knockout mice: decreased social contact duration in two social interaction tests. Front Behav Neurosci. 2009;3:3.
Takatsu H, Baba K, Shima T, Umino H, Kato U, Umeda M, et al. ATP9B, a P4-ATPase (a putative aminophospholipid translocase), localizes to the trans-Golgi network in a CDC50 protein-independent manner. J Biol Chem. 2011;286:38159–67.
Schröter CJ, Braun M, Englert J, Beck H, Schmid H, Kalbacher H. A rapid method to separate endosomes from lysosomal contents using differential centrifugation and hypotonic lysis of lysosomes. J Immunol Methods. 1999;227:161–8.
Mayle KM, Le AM, Kamei DT. The intracellular trafficking pathway of transferrin. Biochim Biophys Acta. 2012;1820:264–81.
Ren M, Xu G, Zeng J, De Lemos-Chiarandini C, Adesnik M, Sabatini DD. Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc Natl Acad Sci USA. 1998;95:6187–92.
Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–96.
Ullrich O, Reinsch S, Urbe S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135:913–24.
Knodler A, Feng S, Zhang J, Zhang X, Das A, Peranen J, et al. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci USA. 2010;107:6346–51.
Balaji K, Mooser C, Janson CM, Bliss JM, Hojjat H, Colicelli J. RIN1 orchestrates the activation of RAB5 GTPases and ABL tyrosine kinases to determine the fate of EGFR. J Cell Sci. 2012;125:5887–96.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–30.
McMillan HJ, Telegrafi A, Singleton A, Cho MT, Lelli D, Lynn FC, et al. Recessive mutations in ATP8A2 cause severe hypotonia, cognitive impairment, hyperkinetic movement disorders and progressive optic atrophy. Orphanet J Rare Dis. 2018;13:86.
Li H, Wetten S, Li L, St Jean PL, Upmanyu R, Surh L, et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol. 2008;65:45–53.
Zhu X, Libby RT, de Vries WN, Smith RS, Wright DL, Bronson RT, et al. Mutations in a P-type ATPase gene cause axonal degeneration. PLoS Genet. 2012;8:e1002853.
Martin-Hernandez E, Rodriguez-Garcia ME, Camacho A, Matilla-Duenas A, Garcia-Silva MT, Quijada-Fraile P, et al. New ATP8A2 gene mutations associated with a novel syndrome: encephalopathy, intellectual disability, severe hypotonia, chorea and optic atrophy. Neurogenetics. 2016;17:259–63.
Kerr DJ, Marsillo A, Guariglia SR, Budylin T, Sadek R, Menkes S, et al. Aberrant hippocampal Atp8a1 levels are associated with altered synaptic strength, electrical activity, and autistic-like behavior. Biochim Biophys Acta. 2016;1862:1755–65.
Padamsey Z, McGuinness L, Bardo SJ, Reinhart M, Tong R, Hedegaard A, et al. Activity-dependent exocytosis of lysosomes regulates the structural plasticity of dendritic spines. Neuron. 2017;93:132–46.
Ilie A, Gao AY, Reid J, Boucher A, McEwan C, Barriere H, et al. A Christianson syndrome-linked deletion mutation ((287)ES(288)) in SLC9A6 disrupts recycling endosomal function and elicits neurodegeneration and cell death. Mol Neurodegener. 2016;11:63.
Huang W, Liu Z, Yang F, Zhou H, Yong X, Yang X, et al. Structural and functional studies of TBC1D23 C-terminal domain provide a link between endosomal trafficking and PCH. Proc Natl Acad Sci USA. 2019;116:22598–608.
Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–91.
Rocca DL, Amici M, Antoniou A, Blanco Suarez E, Halemani N, Murk K, et al. The small GTPase Arf1 modulates Arp2/3-mediated actin polymerization via PICK1 to regulate synaptic plasticity. Neuron. 2013;79:293–307.
Trischler M, Stoorvogel W, Ullrich O. Biochemical analysis of distinct Rab5- and Rab11-positive endosomes along the transferrin pathway. J Cell Sci. 1999;112:4773–83.
Moya-Alvarado G, Gonzalez A, Stuardo N, Bronfman FC. Brain-derived neurotrophic factor (BDNF) regulates Rab5-positive early endosomes in hippocampal neurons to induce dendritic branching. Front Cell Neurosci. 2018;12:493.
Brown TC, Correia SS, Petrok CN, Esteban JA. Functional compartmentalization of endosomal trafficking for the synaptic delivery of AMPA receptors during long-term potentiation. J Neurosci. 2007;27:13311–5.
Brown TC, Tran IC, Backos DS, Esteban JA. NMDA receptor-dependent activation of the small GTPase Rab5 drives the removal of synaptic AMPA receptors during hippocampal LTD. Neuron. 2005;45:81–94.
Chandran J, Ding J, Cai H. Alsin and the molecular pathways of amyotrophic lateral sclerosis. Mol Neurobiol. 2007;36:224–31.
Acknowledgements
We thank the affected individuals and their parents for their participation in this study. We thank Dr. Qi-Ming Sun’s lab (Zhejiang University) for their assistance in the project.
Funding
National Natural Science Foundation of China 92254307, 91754115, and 32170758 (DF), National Natural Science Foundation of China 82001205 (TM), Natural Science Foundation of Guangdong 2020A1515010022 (TM), Fundamental and Applied Fundamental Research Project of Guangzhou 202102020016 (TM), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (GDUPS, DF), Guangzhou Ling Nan Ying Cai Project 2018, Q847004 (DF).
Author information
Authors and Affiliations
Contributions
TM, X-TC and DF conceived the study and prepared the figures and manuscript. Y-SY, MU, AW, QL and CZ contributed to the recruitment and characterization of the patients. TM, Z-JH, H-FH and S-YL performed the in vivo and in vitro experiments for phenotypic characterization. M-DX performed the electrophysiological recording of brain slices. K-RL, GB and H-XZ performed the biochemical experiments and bioinformatics analysis. X-DS, Y-LZ, H-SH and HL discussed and revised the manuscript. DF supervised the experiments and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Meng, T., Chen, X., He, Z. et al. ATP9A deficiency causes ADHD and aberrant endosomal recycling via modulating RAB5 and RAB11 activity. Mol Psychiatry 28, 1219–1231 (2023). https://doi.org/10.1038/s41380-022-01940-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-022-01940-w
