
Abstract
PALB2 loss-of-function variants play an important role in breast, pancreatic and possibly, ovarian and gastric cancer susceptibility. Their frequency can be influenced by founder effects, already described in some populations. Herein, we have assessed the possible founder effect of PALB2 c.2257C>T (p.Arg753*) truncating variant among Greek breast cancer patients, while investigating possible correlations with cancer diagnoses. Following a lead deriving from a background study of highly selected Greek breast cancer patients, a total of 2496 breast and 697 ovarian cancer patients were directly genotyped for the PALB2 c.2257C>T truncating variant. Consequently, haplotype analysis was conducted on identified carriers, using seven microsatellite markers. The prevalence of the PALB2 variant was 0.24% (6/2496) and 0.14% (1/697) among breast and ovarian cases, respectively. Family history seems to be an important factor for the variant identification, although not reaching statistical significance. Microsatellite analysis on 12 carriers revealed a common shared haplotype, spanning a chromosomal region of ~1.2 Mb; the variant was possibly introduced in the Greek population ~1600 years ago. The variant confers high breast cancer risk, as illustrated by comparison with publicly available control groups. Genetic testing for PALB2, especially for the Greek founder c.2257C>T truncating variant, should be seriously considered in Greek breast cancer cases, since such findings could assist appropriate clinical management for the patients and their families.
Similar content being viewed by others


Introduction
Partner and localizer of BRCA2 (PALB2) is a tumor suppressor gene that originally was identified as a Fanconi Anemia gene (FANCN) [1]. PALB2 plays a crucial role at the repair of double-strand DNA breaks by homologous recombination (HR), while acting as a molecular scaffold for BRCA2 and BRCA1 in order to form a, necessary for HR, complex [2, 3]. PALB2 biallelic pathogenic variants were identified in patients with similar phenotype to biallelic BRCA2/FANCD1 patients, displaying growth retardation and tumors, such as Wilms tumors and neuroblastomas [4, 5].
The association of monoallelic, loss-of-function (LoF) PALB2 variants to breast cancer (BrCa) predisposition was first reported in 2007 [6]. Subsequently, in a large polycentric study, BrCa lifetime risk was estimated to be 33% and 58% for PALB2 carriers without and with family history of BrCa, respectively. The latter is comparable to the lifetime BrCa risk of BRCA2 carriers [7]. Recent large sequencing studies reported association of PALB2 pathogenic alleles with high BrCa risk, with respective odds ratios (ORs) ranging from 5.53 to 9.53 [8,9,10,11].
Over the last years, gene panel testing enabled the evaluation of the contribution of PALB2 germline damaging variants in other malignancies, such as ovarian, male BrCa, pancreatic, and gastric cancer. Association of PALB2 variants to ovarian cancer (OvCa) predisposition is controversial. Quite recently, two studies determined ORs of 3.18 and 4.55 and therefore a moderate association, although with no statistical power, was reported [12, 13]. On the contrary, a single study, analyzing ~2000 OvCa patients, where a relatively high frequency of PALB2 LoF variants (0.63%) was observed, reported 10-fold and 4-fold increase, when using NHLBI Exome Sequencing Project (ESP) and Exome Aggregation Consortium (ExAC) data as control groups, respectively [14].
Beyond BrCa and possibly, OvCa predisposition, germline PALB2 damaging alleles are known to be associated with increased pancreatic cancer risk [15, 16], while additional potential associations to gastric [17] and male BrCa (~six-fold increased risk) [18] have been observed. It seems that PALB2 pathogenic variants are rare among male BrCa patients, while not detected in a cohort of 102 Greek male BrCa patients tested [19].
Frequency of PALB2 LoF variants varies among populations and it can possibly be influenced by founder effects. So far, four PALB2 founder pathogenic variants have been described namely, c.1592delT in Finland, with the cumulative BrCa risk being estimated as 40% [20], c.2323C>T in French-Canadians, accounting for ~0.5% of premenopausal BrCa patients [21], and c.1027C>T and c.2167_2168delAT in Italian families, with the latter also being reported in individuals of Hispanic and African ancestry [22]. Additionally, the damaging allele, PALB2 c.3113G>A, detected in Australia, which has a possible British origin, has been shown to be associated with a very high cumulative BrCa risk, reaching up to 90% [23].
Herein, the possible Greek founder effect of the PALB2 truncating variant c.2257C>T, p.(Arg753*), rs180177110, which was initially detected through screening of a highly selected cohort of BrCa patients, was investigated, while assessment of possible correlations with diagnoses was performed.
Materials and methods
Study group
This study involved 2496 Greek BrCa patients (mean age 50.7 years, range 22–87 years) and 697 Greek OvCa patients (54.9 years, range 17–87 years). A control group of 1114 cancer-free women of Greek descent was included in the analysis. The study was approved by the Bioethics Committee of National Center for Scientific Research ‘Demokritos’ (Reference number: NCSRD-BC report 14/02/2014) in agreement with the 1975 Helsinki statement. All participants provided written informed consent prior to genetic analysis. Among the BrCa cohort, 873 patients reported as having at least one relative diagnosed with BrCa and/or OvCa and/or pancreatic cancer, while 1623 patients reported no family history or did not provide the relevant information. Among OvCa patients, 220 reported as having family history of BrCa and/or OvCa and/or pancreatic cancer, while 477 patients reported no family history.
Background study
As a first step and in a distinct project assessing predisposition in a highly selected cohort of Greek BrCa patients, i.e. very young age at diagnosis (<35 years) and/or strong family history of breast and/or OvCa, were massively parallel sequenced using a multigene cancer predisposition panel [24] (Fostira et al., under review). Through this screening, six distinct PALB2 pathogenic variants were identified, among 11 patients, of which, five carried the c.2257C>T truncating variant (Supplementary Table 1).
Genotyping for the PALB2 c.2257C>T truncating variant
DNA was extracted from peripheral blood lymphocytes using the salt-extraction procedure. In total, 2496 BrCa patients and 697 OvCa patients referred to Molecular Diagnostics Laboratory of NCSR Demokritos and previously tested negative for germline pathogenic alleles in the BRCA1 and BRCA2 genes were analyzed. At this point, screening for the c.2257C>T truncating variant was performed with the SYBR green dye-based quantitative real-time PCR assay using the CFX96 Touch Real-Time PCR detection system (BIO-RAD, Life Science, CA, USA). A 1004-bp DNA fragment was amplified and detection was performed with the KAPA SYBR FAST qPCR Master Mix Universal Kit (KapaBiosystems, Cape Town, South Africa). Negative and positive control DNA samples were included in every assay. All positive samples were confirmed by Sanger sequencing. Primers and protocols are available upon request.
Haplotype analysis
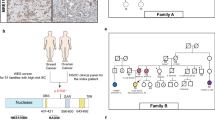
Haplotype analysis was performed using six extragenic (D16S3046, D16S403, 23037GT23, D16S417, D16S420, and D16S401) and one intragenic (23622TCTA14) microsatellite markers covering a genomic area of 3.8 Mb on chromosome 16, surrounding PALB2 gene. A schematic representation of their position, relative to PALB2 is shown in Fig. 1.

Schematic diagram of microsatellite markers positions, along chromosome 16, relative to the PALB2 gene. Microsatellite markers that constitute the shared haplotype are highlighted in bold
The analysis included all 23 c.2257C>T Greek carriers and 55 non-c.2257C>T carriers, of which five were family relatives. Additionally, two c.2257C>T carriers from a single Italian family were included in the analysis. The intragenic marker was specifically analyzed in 45 additional non-c.2257C>T carriers, due to its relatively low variability observed. PCR was performed using the aforementioned markers (the 5′-end of each forward primer was labeled by 6-FAM or HEX), followed by capillary electrophoresis on an ABI 3130XL Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, USA). Primers and protocols are available upon request.
Age estimation of PALB2 c.2257C>T Greek founder truncating variant
The DMLE2.2 software was used to estimate the age of the c.2257C>T truncating variant. The program uses the Markov Chain Monte Carlo algorithm to allow Bayesian estimation of the variant age, based on linkage disequilibrium between disease-causing variants and linked markers on patients and non-affected individuals [25].
Case-control study and statistical analysis
Case-control study involved the genotyping of 1114 Greek cancer-free females (control group), while additional comparisons were implemented, using FLOSSIES European, ExAC non-TCGA non-Finnish European (NFE) and gnomAD non-cancer-NFE population datasets. Odds ratios were calculated using Fisher’s exact test. Association of PALB2 c.2257C>T truncating variant to early BrCa diagnosis and family history were investigated with Wilcoxon rank-sum test. Statistical significance was reached in p values below 0.05.
Results
Prevalence of PALB2 c.2257C>T truncating variant in Greek breast/ovarian cancer patients
Following the recurrence of the c.2257C>T variant, i.e. identified in five non-related individuals in the background study, direct genotyping for the variant revealed seven additional carriers, of which six and one were diagnosed with BrCa and OvCa, respectively. Therefore, the overall prevalence for the particular PALB2 truncating variant was 0.24% (6/2496) among BrCa patients and 0.14% (1/697) among OvCa patients. No carriers were identified among individuals in the control group (0/1114), which did not reach statistical significance due to the limited number of the controls tested (p = 0.186). Interrogation for the presence of the particular PALB2 variant in public datasets revealed no carriers in the FLOSSIES European dataset (0/7325; p < 0.01), three carriers in the ExAC non-TCGA NFE dataset (3/27171; OR, 21.77; 95%CI, 4.65–135, p < 0.01) and four carriers in the gnomAD non-cancer NFE dataset (4/51,373; OR, 30.88; 95% CI, 7.31–148.7, p < 0.01). Results are summarized in Supplementary Table 2.
Cascade testing for PALB2 c.2257C>T truncating variant in relatives
In total, 16 relatives from 5 families consented to targeted genetic testing (11 females and 5 males). Of these, 11 tested positive for the c.2257C>T truncating variant (nine females and two males), with four having already been diagnosed with BrCa (32, 39, 40, and 54 years), while one was diagnosed with prostate cancer at the age of 80 years.
Association of age at BrCa diagnosis and family history to PALB2 c.2257C>T truncating variant
The mean age at BrCa diagnosis of the patients in our cohort and the six c.2257C>T carriers identified herein was 50.7 years (range 22–87 years) and 43.2 years (range 37–56 years), respectively. The majority of carriers (5/6) were diagnosed with BrCa before the age of 45 years. Despite this observation, no statistically significant association was determined between the c.2257C>T truncating variant and early age at BrCa diagnosis (p = 0.093). Among BrCa patients with reported and no/unknown family history, 0.34% (3/873) and 0.18% (3/1623) carried the c.2257C>T truncating PALB2 variant, respectively, while the only identified OvCa carrier reported no family history. However, association of family history with PALB2 c.2257C>T reached no statistical significance (p > 0.05). It is noteworthy, that among the family relatives of the carriers, malignancies observed included ovarian, gastric, pancreatic, and prostate cancer. Age at diagnosis, along with information on family history of the carriers are summarized in Table 1.
Clinicopathological features of PALB2 c.2257C>T carriers
In an effort to collect all available data on Greek c.2257C>T carriers (five detected in the background study and seven herein), seven pathology reports of BrCa diagnoses were informative. The majority (5/7) of breast tumors were ductal invasive, with most of them (4/7) being high-grade. Four of the patients had positive estrogen and/or progesterone receptors, while there was a single case with a triple negative diagnosis. Histopathology data are summarized in Table 1.
Haplotype analysis and age estimation of PALB2 c.2257C>T truncating variant
Haplotype analysis revealed that all Greek carriers shared a ~1.2 Mb common haplotype, indicating that this variant originated from a single recombination event in a common ancestor 1250–2100 years ago. Specifically, it is estimated that the variant originated 50–84 generations ago with an average generation length of 25 years, since according to demographic data the population growth rate is 0.135. Hence, the variant occurred, on average, 64 generations ago, corresponding to 1600 years. The alleles comprising the haplotype shared among carriers, are summarized on Supplementary Table 3. In total, 46 carrier chromosomes were analyzed, and all were found to share the following core haplotype: “157-228-127-166-263”. The conserved haplotype, defined between markers D16S403 (5′) and D16S420 (3′), possibly derived from recombination events that occurred between markers D16S403-D16S3046 and D16S420-D16S401. On the contrary, the damaging variant detected on the Italian carriers presumably occurred from an independent or a subsequent recombination event, as they share a different, but partially overlapping, haplotype. Haplotype maps for genotyped individuals are depicted in Fig. 2, along with the pedigrees of carrier families.

Results of haplotype analysis performed, along with the pedigrees of seven unrelated Greek families and an Italian family, that carry the c.2257C>T truncating variant. Probands are indicated with an arrow. Breast cancer is designated with black color, pancreatic cancer with purple, gastric or stomach with green, prostate cancer with blue, kidney cancer with brown, lung cancer with light blue, leukemia with yellow and thyroid with orange. ca cancer, BrCa breast cancer, PrCa prostate cancer, MT mutant allele, WT wild type allele. Carriers are designated with + and non-carriers with −
Discussion
Testing for PALB2-inherited deleterious variants nowadays occupies a critical part of, at least, BrCa predisposition assessment, based on which specific practice guidelines for risk management can be followed. Herein, a recurrent, PALB2 truncating variant namely, c.2257C>T, has been proven to have a founder effect, illustrated by a shared common 1.2 Mb haplotype, originated from a single recombination event that occurred ~1600 years ago, specifically for individuals of Greek descent. The allele prevalence observed was 0.24% and 0.14% of BrCa and OvCa patients tested, respectively. The truncating variant c.2257C>T, has been registered nine times in ClinVar, but has also been reported in individuals originating from Italy [26], Germany [27], United States [7], and Korea [28]. In a recent meta-analysis, in which results from 48 multigene panel studies were summarized, seven c.2257C>T carriers were reported [29]. It is therefore possible that distinct mutational events occurred, resulting in the presence of c.2257C>T truncating variant in different populations.
Recent large-scale studies on BrCa patients, who met the criteria for genetic testing, revealed that the prevalence of PALB2 LoF variants ranged between 0.8% and 1.2% [9, 30, 31]. Of the founder PALB2 damaging alleles reported in other populations, c.2323C>T [21] and c.1592delT [32] were identified in ~0.9% and ~0.5% of unselected for family history, BrCa patients, respectively, while prevalence of the founder allele in our study is relatively lower.
Interestingly, the calculated cumulative BrCa risks for PALB2 c.3113G>A and c.1592delT were 91% [23] and 40% [20] by the age of 70 years, respectively. It seems, therefore that different PALB2 damaging alleles can be associated with variable cancer risks and although at this point a similar estimation on the Greek founder allele lacks statistical power due to the small number of carriers, it would be rather interesting to pursue it in the near future.
Noteworthy, we have observed that the Greek founder PALB2 damaging variant is associated with high BrCa risk, when compared to ExAc and gnomAD control datasets, respectively. The wide OR range observed can be mainly attributed to the small number of carriers. This finding is important for Greek BrCa patients, as well as to their family relatives in terms of their clinical management.
Family history plays an important role when counseling cancer patients; especially for a PALB2 carrier, strong family history for BrCa, i.e. two first-degree relatives diagnosed before the age of 50 years, increased the cumulative BrCa risk from 33% to 58% [7]. Although, it did not reach statistical significance, ascertainment of our cohort for family history, i.e. at least one relative diagnosed with breast, ovarian, and/or pancreatic cancer, almost doubled the prevalence of the founder allele (0.34%; 3/873) among BrCa cases, indicating that family history is a significant aspect and should be considered when testing for PALB2 LoF variants.
Several studies support that the majority of PALB2 tumors are hormone receptor positive [7, 9]. Quite recently this association was specifically reported in >1000 BrCa patients, where a four-fold association of PALB2 damaging alleles to estrogen receptor-positive diagnosis was illustrated [33]. On the contrary, PALB2 pathogenic variants have been reported as enriched in triple-negative breast cancer (TNBC) diagnoses [11, 34, 35]. Therefore, it can be assumed that there is no clear association of PALB2 tumors with a specific histological BrCa subtype or it could be dependent on the type of the damaging allele [36]. It seems that the association of PALB2-pathogenic variants to TNBC predisposition is comparable to that conferred by BRCA2-pathogenic variants, which for many years was not established [35, 37]. Herein, the majority of our c.2257C>T carriers (4/7) developed hormone positive breast tumors, while there was a single triple negative diagnosis.
Moreover, multiple case-control studies have assessed the association of PALB2 damaging variants to OvCa predisposition, with a single study, up to now, showing statistical significant association [14], while another [13] reporting a non-statistically significant, four-fold increased association. In our cohort of 697 OvCa cases, we have identified a single PALB2 c.2257C>T carrier, indicating a weak to no association of the particular allele to OvCa susceptibility.
Our study has limitations, the major of which being bias on patient selection. Since our laboratory specializes in hereditary cancer, BrCa patients referred and thus included in the study have been somewhat selected, mainly on young age and family history.
Detection of the Greek founder allele, PALB2 c.2257C>T, in symptomatic and asymptomatic individuals is consequential in terms of clinical management, while the strong association of the allele to BrCa diagnosis, should not be overlooked. To this end, testing at least for the PALB2 c.2257C>T truncating variant in BRCA1/2 negative BrCa patients of Greek descent, should be seriously considered.
References
Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–4.
Buisson R, Masson JY. PALB2 self-interaction controls homologous recombination. Nucleic Acids Res. 2012;40:10312–23.
Fernandes PH, Saam J, Peterson J, Hughes E, Kaldate R, Cummings S, et al. Comprehensive sequencing of PALB2 in patients with breast cancer suggests PALB2 mutations explain a subset of hereditary breast cancer. Cancer. 2014;120:963–7.
Tischkowitz M, Xia B, Sabbaghian N, Reis-Filho JS, Hamel N, Li G, et al. Analysis of PALB2/FANCN-associated breast cancer families. Proc Natl Acad Sci USA. 2007;104:6788–93.
Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia. Cancer Res. 2010;70:7353–9.
Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–7.
Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkas K, Roberts J, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506.
Lu HM, Li S, Black MH, Lee S, Hoiness R, Wu S, et al. Association of breast and ovarian cancers with predisposition genes identified by large-scale sequencing. JAMA Oncol. 2018. https://doi.org/10.1001/jamaoncol.2018.2956.
Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;3:1190–6.
Hauke J, Horvath J, Gross E, Gehrig A, Honisch E, Hackmann K, et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: results of the german consortium for hereditary breast and ovarian cancer. Cancer Med. 2018;7:1349–58.
Castera L, Harter V, Muller E, Krieger S, Goardon N, Ricou A, et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet Med. 2018;20:1677–86.
Ramus SJ, Song H, Dicks E, Tyrer JP, Rosenthal AN, Intermaggio MP, et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J Natl Cancer Inst. 2015. https://doi.org/10.1093/jnci/djv214.
Kotsopoulos J, Sopik V, Rosen B, Fan I, McLaughlin JR, Risch H, et al. Frequency of germline PALB2 mutations among women with epithelial ovarian cancer. Fam Cancer. 2017;16:29–34.
Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016;2:482–90.
Zhen DB, Rabe KG, Gallinger S, Syngal S, Schwartz AG, Goggins MG, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2015;17:569–77.
Borecka M, Zemankova P, Vocka M, Soucek P, Soukupova J, Kleiblova P, et al. Mutation analysis of the PALB2 gene in unselected pancreatic cancer patients in the Czech Republic. Cancer Genet. 2016;209:199–204.
Fewings E, Larionov A, Redman J, Goldgraben MA, Scarth J, Richardson S, et al. Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CDH1 mutation: a whole-exome sequencing study. Lancet Gastroenterol Hepatol. 2018;3:489–98.
Pritzlaff M, Summerour P, McFarland R, Li S, Reineke P, Dolinsky JS, et al. Male breast cancer in a multi-gene panel testing cohort: insights and unexpected results. Breast Cancer Res Treat. 2017;161:575–86.
Fostira F, Saloustros E, Apostolou P, Vagena A, Kalfakakou D, Mauri D, et al. Germline deleterious mutations in genes other than BRCA2 are infrequent in male breast cancer. Breast Cancer Res Treat. 2018;169:105–13.
Erkko H, Dowty JG, Nikkila J, Syrjakoski K, Mannermaa A, Pylkas K, et al. Penetrance analysis of the PALB2 c.1592delT founder mutation. Clin Cancer Res. 2008;14:4667–71.
Foulkes WD, Ghadirian P, Akbari MR, Hamel N, Giroux S, Sabbaghian N, et al. Identification of a novel truncating PALB2 mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. 2007;9:R83.
Catucci I, Casadei S, Ding YC, Volorio S, Ficarazzi F, Falanga A, et al. Haplotype analyses of the c.1027C>T and c.2167_2168delAT recurrent truncating mutations in the breast cancer-predisposing gene PALB2. Breast Cancer Res Treat. 2016;160:121–9.
Southey MC, Teo ZL, Dowty JG, Odefrey FA, Park DJ, Tischkowitz M, et al. A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res. 2010;12:R109.
Fostira F, Walsh T, Casadei S, Lee MK, Vratimos A, Fountzilas G, et al., Combination of founder mutation screening and genomic capture using BROCA yield high rate of Loss-of-Function mutation in early onset and familial breast and ovarian cancer in Greece. ASHG meeting abstracts; 2013.
Reeve JP, Rannala B. DMLE+: Bayesian linkage disequilibrium gene mapping. Bioinformatics. 2002;18:894–5.
Papi L, Putignano AL, Congregati C, Piaceri I, Zanna I, Sera F, et al. A PALB2 germline mutation associated with hereditary breast cancer in Italy. Fam Cancer. 2010;9:181–5.
Hellebrand H, Sutter C, Honisch E, Gross E, Wappenschmidt B, Schem C, et al. Germline mutations in the PALB2 gene are population specific and occur with low frequencies in familial breast cancer. Hum Mutat. 2011;32:E2176–88.
Kim H, Cho DY, Choi DH, Oh M, Shin I, Park W, et al. Frequency of pathogenic germline mutation in CHEK2, PALB2, MRE11, and RAD50 in patients at high risk for hereditary breast cancer. Breast Cancer Res Treat. 2017;161:95–102.
Suszynska M, Klonowska K, Jasinska AJ, Kozlowski P. Large-scale meta-analysis of mutations identified in panels of breast/ovarian cancer-related genes - Providing evidence of cancer predisposition genes. Gynecol Oncol. 2019;153:452–62.
Hauke J, Horvath J, Gross E, Gehrig A, Honisch E, Hackmann K, et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018;7:1349–58.
Buys SS, Sandbach JF, Gammon A, Patel G, Kidd J, Brown KL, et al. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer. 2017;123:1721–30.
Erkko H, Xia B, Nikkila J, Schleutker J, Syrjakoski K, Mannermaa A, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–9.
Girard E, Eon-Marchais S, Olaso R, Renault AL, Damiola F, Dondon MG, et al. Familial breast cancer and DNA repair genes: Insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing. Int J Cancer. 2018. https://doi.org/10.1002/ijc.31921.
Slavin TP, Maxwell KN, Lilyquist J, Vijai J, Neuhausen SL, Hart SN, et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer. 2017;3:22.
Shimelis H, LaDuca H, Hu C, Hart SN, Na J, Thomas A, et al. Triple-negative breast cancer risk genes identified by multigene hereditary cancer panel testing. J Natl Cancer Inst. 2018. https://doi.org/10.1093/jnci/djy106.
Nguyen-Dumont T, Hammet F, Mahmoodi M, Tsimiklis H, Teo ZL, Li R, et al. Mutation screening of PALB2 in clinically ascertained families from the breast cancer family registry. Breast Cancer Res Treat. 2015;149:547–54.
Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol. 2015;33:304–11.
Acknowledgements
We thank the patients and their families who participated in this study, as well as the clinicians for their valuable contribution on this project. We would also like to thank Dr. Laura Papi (Department of Experimental and Clinical Biomedical Sciences “Mario Serio”, Medical Genetics Unit, University of Florence, Florence, Italy) and her colleagues for providing DNA samples. Finally, we would like to thank Prof. Roberto Colombo (Faculty of Medicine “Agostino Gemelli”, Catholic University of the Sacred Heart, Rome, Italy) for his useful suggestions regarding haplotyping and microsatellite marker selection.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Vagena, A., Papamentzelopoulou, M., Kalfakakou, D. et al. PALB2 c.2257C>T truncating variant is a Greek founder and is associated with high breast cancer risk. J Hum Genet 64, 767–773 (2019). https://doi.org/10.1038/s10038-019-0612-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0612-6
This article is cited by
-
Frequency of heterozygous germline pathogenic variants in genes for Fanconi anemia in patients with non-BRCA1/BRCA2 breast cancer: a meta-analysis
Breast Cancer Research and Treatment (2020)
-
Germline pathogenic variants in BRCA1, BRCA2, PALB2 and RAD51C in breast cancer women from Argentina
Breast Cancer Research and Treatment (2019)
