
Abstract
SH3TC2, known as the causative gene of autosomal recessive demyelinating Charcot-Marie-Tooth type 4C (CMT4C), was also found linked to a mild mononeuropathy of the median nerve with an autosomal dominant inheritance pattern. Using DNA microarray, Illumina MiSeq, and Ion proton, we carried out gene panel sequencing among 1483 Japanese CMT patients, containing 397 patients with demyelinating CMT. From seven patients with demyelinating CMT, we identified eight recessive variants in the SH3TC2 gene, consisting of five novel (pathogenic/likely pathogenic) and three reported variants. Additionally, from two patients with axonal CMT, we detected a reported recessive variant, p.Arg77Trp, which was herein reclassified as variant with unknown significance. Of the seven CMT4C patients (six females and one male), 2/7 patients developed symptoms at their first decade, and 5/7 patients lost their ambulation around age 50. Scoliosis was observed from more than half (4/7) of these patients, whereas hearing loss is the most common symptom of central nervous system (6/7). No median nerve mononeuropathy was recorded from their family members. We identified recessive variants in SH3TC2 from 1.76% of demyelinating CMT patients. An uncommon gender difference was recognized and the wild spectrum of these variants suggests mutational diversity of SH3TC2 in Japan.
Similar content being viewed by others


Introduction
Charcot-Marie-Tooth (CMT) is the most common type of inherited peripheral neuropathies (IPNs). Electrophysiological and pathological findings have led to CMT being categorized into demyelinating (median nerve motor conduction velocities, MNCV < 38 m/s) and axonal (MNCV >= 38 m/s) forms, respectively. To date, demyelinating autosomal recessive (AR)-CMT, designated as CMT4, have been linked to twelve causative genes and referred to from CMT4A to CMT4K (see http://neuromuscular.wustl.edu/time/hmsn.html; accessed June 2017). Generally, AR-CMT exhibit a more severe disease course than dominant (AD) form, characterized by earlier onset and more rapid clinical progression that results in more marked distal limb deformities and major spinal deformities.
SH3TC2 gene, coding for the SH3 domain and tetratricopeptide repeats 2, has been linked to CMT type 4C (CMT4C) [1]. Mutations in SH3TC2 were found with high frequency of 11.7% (7/60) in demyelinating CMT patients of Czechoslovakia [2], and 15.7% (16/102) of CMT4/intermediate AR-CMT patients in France [3]. In contrast, SH3TC2 mutation was found account for 2.8% (3/109) of demyelinating CMT from southern Italy [4], or 2.7% (21/765) in demyelinating CMT of German [5]. With regard to Japan, we have reported two CMT4C cases with SH3TC2 mutations in 2012 and 2016, respectively [6, 7]. On the basis of another study, CMT4C was found in 1.9% (2/103) Japanese patients with demyelinating CMT [8]. Among these reports, several founder mutations were also described, represented by p.Arg1109* in European Gypsies and p.Arg954* from French–Canadians and other European countries [2, 9,10,11].
In 2010, two heterozygous variants of SH3TC2, p.Arg954* and p.Tyr169His, were linked to a subtle mild mononeuropathy of the median nerve (MNMN) consistent with carpal tunnel syndrome in family carriers of patients with CMT4C [12]. In this study, we retrospect nine patients harboring recessive SH3TC2 variants, who were identified in our laboratory from a Japanese nationwide cohort of CMT patients. We summarize their clinical, eletrophysiological features, and mutational spectrum of SH3TC2.
Materials and methods
Patients
From 2007 to September 2016, 1483 unrelated Japanese CMT cases were referred to our genetic laboratory by their neurologists or physicians from other departments. These cases consisted of 397 demyelinating CMT (MNCV < 38 m/s), 906 axonal CMT (MNCV >= 38 m/s), and 180 cases with unclassified subtype. All demyelinating CMT cases were proved negative for PMP22 duplication/deletion using fluorescence in situ hybridization or multiplex ligation-dependent probe amplification (MLPA).
We extracted genomic DNA using QIAGEN’s Puregene Core Kit C (QIAGEN, Valencia, CA, USA) or Oragene DNA self-collection kit (DNA Genotech, Ottawa, Ontario, Canada). The protocol was reviewed and approved by the Institutional Review Board of Kagoshima University. All patients and family members provided written informed consents to participate in this study.
Gene panel sequencing
In chronological order, we performed mutation screening using a purpose-built GeneChip CustomSeq Resequencing Array (30 genes; Affymetrix, Inc., Santa Clara, CA) [13], Illumina MiSeq platform (60 genes; Illumina Inc., San Diego, CA, USA) [14], and a custom Ion AmpliSeq gene panel sequencing system (72 genes; Life Technologies, Carlsbad, CA, USA) [15], following the protocols described previously. The SH3TC2 gene was targeted in all these three screening systems.
Variant validation and interpretation
Variants were interpreted according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) guideline published in 2015 [16]. All variants were checked against the public databases, including Exome Sequencing Project, 1000 genomes project, and Exome Aggregation Consortium (ExAC) database (moderate evidence of pathogenicity, PM2). We also checked variants in Japanese population databases, comprising Human Genetic Variation Database (HGVD; whole exome sequencing variants of 1208 Japanese individuals) and Integrative Japanese Genome Variation Database (iJGVD; whole genome sequencing variants of 2049 Japanese individuals) (PS4-moderate). All uncommon variants (minor allele frequency, MAF < 0.05) were further analyzed by multiple pathogenicity prediction programs, PolyPhen2 (0 to 1 scale), SIFT (cutoff = 0.05), PROVEAN (cutoff = −2.5), and MutationTaster. (Supporting evidence of pathogenicity, PP3) Candidate variants were validated by Sanger sequencing. Segregation studies of family members were performed whenever available (PP1/PM3).
Results
Genetic analysis
Among 1483 Japanese patients with CMT, we identified 10 recessive SH3TC2 variants in nine patients using GeneChip (case 1 and 2), Illumina MiSeq (case 3 ~ 6), and Ion Proton (case 7 ~ 9). Of those variants, five had never been reported, consisting of p.Tyr898fs, p.Tyr1182*, p.Ala595Glu, p.Leu571Pro, and p.Glu1164_Phe1169delinsHis (Table 1). Additionally, in case 9, a novel homozygous variant p.Gln2251Lys was detected in SACS gene, which has been linked to early-onset spastic ataxia, Charlevoix-Saguenay type (ARSACS). However, clinical diversity has been recognized in ARSACS, and a patient with prominent sensorimotor neuropathy has been reported [17]. Therefore, although no clinical presentations of spasticity or cerebellar ataxia were identified in our patient until the age of 65, we considered this SACS variant as unknown significance (Data not shown). Besides, no other uncommon variants were identified in any targeted genes by three mutation screening systems. Among these patients, seven were electrophysiologically grouped as demyelinating CMT, whereas the other two cases were found comparable to axonal CMT phenotype (Table 2).
One novel and four reported variants were found existing in the ExAC or HGVD databases, including p.Ala595Glu (HGVD, 1/2204), p.Arg77Trp (ExAC, 29/121392; HGVD, 27/2022), p.Arg904* (ExAC, 7/121302), p.Trp925* (ExAC, 1/121254), and p.Arg1109* (ExAC, 6/121388). However, no homozygote of these variants was recorded in these databases. All of the missense variants were predicted to be deleterious through multiple in silico analysis tools (Table 1). All variants were validated using Sanger sequencing, whereas phenotype–genotype cosegregation was proved in pedigrees of case 5, 7, and 9 (Fig. 1).

Pedigree analysis and sequencing chromatograms of five CMT4C patients with novel SH3TC2 variants. Case 3, 7, and 8 are born to consanguineous parents. Asymptomatic heterozygous carriers are identified in family members of case 5, 7, and 9. “+”: variant, “−”: wild type (Color figure online)
Clinical features
Of the seven patients manifesting with demyelinating CMT, male/female ratio was 1:6, and four patients were born to healthy consanguineous parents. Their onset age varied, occurring in the first/second decade of three patients, or in the adult age of four patients. As primary symptom, scoliosis was observed in case 1, whereas all the other patients initially developed lower limbs motor dysfunctions. These motor dysfunctions manifested as leg weakness or walking/running disabilities. Motor deficit was more pronounced distally and more severe in the lower limbs in all patients. Hypalgesia or hyperpathia was recorded in distal limbs from four cases, whereas vibration sensation was found decreased in five cases. Areflexia was noted in all six patients with examination record. Scoliosis, verified by physical and/or radiographic examination, was ascertained in 4/7 of our patients. Four patients developed foot deformities, manifesting of pes cavus, pes planus, or tiptoe foot. Five patients became wheelchair-dependent after the age of 50 years (Table 1). There has no record of the MNMN phenotype in any family members of these patients.
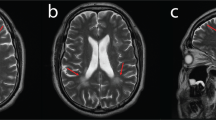
Cranial nerve involvements were documented in 6/7 patients, most commonly manifesting as hearing loss (n = 6), followed by ocular movement symptoms (n = 2), presenting with Adie’s pupil and absence of light reflex (case 3), or bilateral mydriasis and nystagmus (case 7). Besides, facial sensory loss (case 2), reduced pharyngeal reflex (case 8), or tongue deviation (case 7) were also identified. Using brain MRI, leukoencephalopathy and diffused cerebral atrophy were observed in case 2 and case 8, respectively (Data not shown). In nerve conduction studies, MNCV was found slower than 38 m/s in 6/7 cases with an electrophysiological record, thus classified into demyelinating CMT. Additionally, sural nerve biopsy of case 1 and 3 suggested demyelinating neuropathy.
On the other hand, case 4 and 6 developed motor dysfunctions in lower limbs from first decade. No scoliosis or cranial nerve involvement was observed. Muscle weakness was identified in proximal upper and lower limbs from case 4, but no recordable weakness was found in her distal limbs. MNCV of case 4 and 6 were 39.8 m/s and 48.2 m/s, respectively, thus were grouped into axonal CMT (Table 2).
Discussion
Using three gene panel sequencing platforms, we carried out mutation screening in 1483 Japanese CMT patients, who were tested negative for PMP22 duplication/deletion. Therein, among 397 cases comparable with demyelinating CMT, we detected recessive SH3TC2 variants in seven patients. Meanwhile, in another two patients classified as axonal CMT, we also identified recessive SH3TC2 variants.
To date, more than 70 mutations of SH3TC2 have been reported, and scattered along the whole protein (HGMD database, professional release, June 2017). Unlike the studies in European countries or French-Canadians, we could not find any mutation hot spot in the SH3TC2 gene in our cohort patients. It is also not comparable to the patients with CMT4A caused by GDAP1 mutations in Japan, from whom we demonstrated a founder mutation [18]. We identified five novel recessive variants in our patients. Herein, p.Tyr898fs of case 3 was a homozygous null variant, fulfilling her consanguineous family history. The novel p.Tyr1182* variant in case 5, together with another null variant p.Arg904*, were found co-segregated with the disease phenotype in his pedigree. The two novel variants above were considered able to match the pathogenic level according to the ACMG-AMP guidelines. On the other hand, the novel missense (p.Ala595Glu and p.Leu571Pro) and in-frame deletion (p.Glu1164_Phe1169delinsHis) variants were referred to as likely pathogenic owing to their incomplete segregation analysis and unexecuted functional study. Intriguingly, the founder mutation of European Gypsies, p.Arg1109*, was also observed in case 1, who is an ethnically pure Japanese.
We previously reported a compound heterozygous variant, p.Arg77Trp and Try925* of case 6, which was found segregating with the phenotype among her unaffected parents and sibling [7]. During the present study, we identified homozygous p.Arg77Trp variant in case 4. It is of noteworthy that, this p.Arg77Trp variant also present as rare variant in ExAC (MAF = 0.00024), but as an uncommon variant in Japanese exome database HGVD (MAF = 0.013). Furthermore, the clinical phenotype of case 4 is distinguish from the other CMT4C patients, manifesting intact distal muscle strength, even after a long disease duration of approximately 60 years. More importantly, the MNCV of both patients harboring p.Arg77Trp were > 38 m/s, which were distinct from common features of CMT4C. It is unlikely a coincidence in two unrelated patients, we thus reevaluated this variant according to the ACMG-AMP guidelines, and classified it as unknown significance. Further study is required to certify the effect of this variant, and to prove whether the manifestations of these two patients are phenotypic variation.
To our best knowledge, our study is the original report describing gender difference of CMT4C (male/female = 1:6), in spite of an unknown reason. Clinically, progression of CMT4C was generally slow, and particularly for the lower limbs, muscle atrophy and weakness initially involve distal limbs then progress to proximal limbs, always resulting in loss of ability to walk. Five patients became wheelchair-dependent after 50 years old as a result of involvement of the proximal lower limbs or recurrent fractures. Pronounced sensory deficit was revealed in four CMT4C patients, and sensory nerve action potential (SNAP) could not be evoked in three of them. Severely decreased vibratory sensation was frequent. Scoliosis, which was reported to be the presenting sign in the majority of CMT4C patients, was observed in more than half (4/7) of our patients. For cranial nerves, auditory, ocular movement, trigeminal sensory, hypoglossal, glossopharyngeal, and/or vagus nerves were found involved in our patients.
Regardless of harboring same or different mutation in SH3TC2, CMT4C patients could present with remarkably broad spectrum of clinical phenotypes. Researchers are still dedicate to demonstrate hidden genetic modifiers of SH3TC2, which might be responsible for the CMT4C clinical variability, such as common SNPs in SH3TC2 and transcription regulation factors CREB/SOX10 [19]. MNMN was linked to heterozygous variants of SH3TC2, and haploinsufficiency or toxic gain of function were considered contributing to this phenotype. Unfortunately, we could not find any record of our patients and their family members manifesting an MNMN phenotype, either clinically or electrophysiologically.
SH3TC2 encodes a Rab11 effector molecule, SH3TC2/KIAA1985, which is exclusively found in myelinating Schwann cells, and expresses late during myelination, thus is considered to be required for maintaining the structural integrity of peripheral nerve myelin sheaths [20]. SH3TC2 is targeted to cellular membranes, including plasma membrane and cytoplasmic vesicles, and its SH3 and tetratricopeptide domains are important for protein–protein interactions. Integrin-α6 is an SH3TC2-interacted protein, which regulates the Rab11-dependent endocytic recycling. Whereas mutations in SH3TC2 would prevent Rab11 binding and lead to intracellular mistargeting away from the recycling endosome as the fundamental molecular defect that leads to CMT4C [20]. SH3TC2 was also found interact with neuregulin-1 (Nrg1)/ERBB, which is critical for the proliferation and migration of Schwann cells and the subsequent myelination of peripheral nerve axons. Loss of SH3TC2 function in both mice and CMT4C patients affect ERBB internalization, potentially altering its downstream intracellular signaling pathways [21].
In conclusion, we identified pathogenic or likely pathogenic variants in SH3TC2, from 1.76% demyelinating CMT patients in our cohort, but with no mutation hot spot. This proportion is slightly lower than previous studies in Italy and German, but comparable to the other Japanese study. Although age of onset varied markedly in our patients, they developed comparable clinical process, and the majority lost their ambulance around the age of 50. The gender difference and p.Arg77Trp variant of unknown significance require further research.
References
Senderek J, Bergmann C, Stendel C, Kirfel J, Verpoorten N, De Jonghe P, et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am J Hum Genet. 2003;73:1106–19.
Lassuthova P, Mazanec R, Vondracek P, Siskova D, Haberlova J, Sabova J, et al. High frequency of SH3TC2 mutations in Czech HMSN I patients. Clin Genet. 2011;80:334–45.
Yger M, Stojkovic T, Tardieu S, Maisonobe T, Brice A, Echaniz-Laguna A, et al. Characteristics of clinical and electrophysiological pattern of Charcot-Marie-Tooth 4C. J Peripher Nerv Syst. 2012;17:112–22.
Manganelli F, Tozza S, Pisciotta C, Bellone E, Iodice R, Nolano M, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes in a Southern Italy population. J Peripher Nerv Syst. 2014;19:292–8.
Rudnik-Schoneborn S, Tolle D, Senderek J, Eggermann K, Elbracht M, Kornak U, et al. Diagnostic algorithms in Charcot-Marie-Tooth neuropathies: experiences from a German genetic laboratory on the basis of 1206 index patients. Clin Genet. 2016;89:34–43.
Iguchi M, Hashiguchi A, Ito E, Toda K, Urano M, Shimizu Y, et al. Charcot-Marie-Tooth disease type 4C in Japan: report of a case. Muscle Nerve. 2013;47:283–6.
Ichikawa K, Numasawa K, Takeshita S, Hashiguchi A, Takashima H. Novel mutations in SH3TC2 in a young Japanese girl with Charcot-Marie-Tooth disease type 4C. Pediatr Int. 2016;58:1252–4.
Hayashi M, Abe A, Murakami T, Yamao S, Arai H, Hattori H, et al. Molecular analysis of the genes causing recessive demyelinating Charcot-Marie-Tooth disease in Japan. J Hum Genet. 2013;58:273–8.
Gooding R, Colomer J, King R, Angelicheva D, Marns L, Parman Y, et al. A novel Gypsy founder mutation, p.Arg1109X in the CMT4C gene, causes variable peripheral neuropathy phenotypes. J Med Genet. 2005;42:e69.
Claramunt R, Sevilla T, Lupo V, Cuesta A, Millan JM, Vilchez JJ, et al. The p.R1109X mutation in SH3TC2 gene is predominant in Spanish Gypsies with Charcot-Marie-Tooth disease type 4. Clin Genet. 2007;71:343–9.
Gosselin I, Thiffault I, Tetreault M, Chau V, Dicaire MJ, Loisel L, et al. Founder SH3TC2 mutations are responsible for a CMT4C French-Canadians cluster. Neuromuscul Disord. 2008;18:483–92.
Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362:1181–91.
Zhao Z, Hashiguchi A, Hu J, Sakiyama Y, Okamoto Y, Tokunaga S, et al. Alanyl-tRNA synthetase mutation in a family with dominant distal hereditary motor neuropathy. Neurology. 2012;78:1644–9.
Maeda K, Idehara R, Hashiguchi A, Takashima H. A family with distal hereditary motor neuropathy and a K141Q mutation of small heat shock protein HSPB1. Intern Med. 2014;53:1655–8.
Higuchi Y, Hashiguchi A, Yuan J, Yoshimura A, Mitsui J, Ishiura H, et al. Mutations in MME cause an autosomal-recessive Charcot-Marie-Tooth disease type 2. Ann Neurol. 2016;79:659–72.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Pyle A, Griffin H, Yu-Wai-Man P, Duff J, Eglon G, Pickering-Brown S, et al. Prominent sensorimotor neuropathy due to SACS mutations revealed by whole-exome sequencing. Arch Neurol. 2012;69:1351–4.
Yoshimura A, Yuan JH, Hashiguchi A, Hiramatsu Y, Ando M, Higuchi Y, et al. Clinical and mutational spectrum of Japanese patients with Charcot-Marie-Tooth disease caused by GDAP1 variants. Clin Genet. 2017;92:274–80.
Brewer MH, Ma KH, Beecham GW, Gopinath C, Baas F, Choi BO, et al. Haplotype-specific modulation of a SOX10/CREB response element at the Charcot-Marie-Tooth disease type 4C locus SH3TC2. Hum Mol Genet. 2014;23:5171–87.
Vijay S, Chiu M, Dacks JB, Roberts RC. Exclusive expression of the Rab11 effector SH3TC2 in Schwann cells links integrin-alpha6 and myelin maintenance to Charcot-Marie-Tooth disease type 4C. Biochim Biophys Acta. 2016;1862:1279–90.
Gouttenoire EA, Lupo V, Calpena E, Bartesaghi L, Schupfer F, Medard JJ, et al. Sh3tc2 deficiency affects neuregulin-1/ErbB signaling. Glia. 2013;61:1041–51.
Acknowledgements
The authors thank the patients and their families for participating in this study and their physicians for submitting the clinical samples. Also the authors thank Mrs. Aya Ebina and Tomoko Onishi for their excellent technical assistant. This study was supported in part by grants from the research on the Nervous and Mental Disorders and Research Committee for Charcot–Marie–Tooth Disease, Neuropathy, Ataxic Disease and Applying Health and Technology of Ministry of Health, Welfare and Labour, Japan. This research is also supported by the Research program for conquering intractable disease from Japan Agency for Medical Research and development (AMED), and JSPS KAKENHI (Grant Number 26461275).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Yuan, JH., Hashiguchi, A., Okamoto, Y. et al. Clinical and mutational spectrum of Japanese patients with recessive variants in SH3TC2 . J Hum Genet 63, 281–287 (2018). https://doi.org/10.1038/s10038-017-0388-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0388-5
This article is cited by
-
Clinical genetics of Charcot–Marie–Tooth disease
Journal of Human Genetics (2023)
-
Screening for SH3TC2 variants in Charcot–Marie–Tooth disease in a cohort of Chinese patients
Acta Neurologica Belgica (2022)
-
Compound heterozygous mutations of SH3TC2 in Charcot–Marie–Tooth disease type 4C patients
Journal of Human Genetics (2019)
