
Abstract
The recruitment of vascular endothelial cells from the tumor microenvironment (TME) to promote angiogenesis plays key roles in the progression of renal cell carcinoma (RCC). The potential impact of immune cells in the TME on RCC angiogenesis, however, remains unclear. Here, we found that recruitment of mast cells resulted in increased RCC angiogenesis in both in vitro cell lines and in vivo mouse models. Mechanistic analyses revealed that RCC recruited mast cells by modulating PI3K→AKT→GSK3β→AM signaling. A clinical survey of human RCC samples also showed that higher expression of the PI3K→AKT→GSK3β→AM signaling pathway correlated with increased angiogenesis. Interruption of PI3K→AKT→GSK3β→AM signaling via specific inhibitors led to decreased recruitment of mast cells, and targeting this infiltrating mast cell-related signaling via an AKT-specific inhibitor suppressed RCC angiogenesis in xenograft mouse models. Together, these results identified a novel role of infiltrating mast cells in RCC angiogenesis and metastasis and suggest a new strategy for treating RCC by targeting this newly identified signaling pathway.
Similar content being viewed by others


Introduction
Renal cell carcinoma (RCC) is a common malignant disease of the human genitourinary system.1 In recent decades, the incidence of RCC has been steadily increasing by 2–4% each year and accounts for 2–3% of human cancers.2 Approximately 20–25% of diagnosed RCC patients have already reached the metastatic phase. Due to the resistance to radiotherapy and chemotherapy, anti-angiogenesis drugs have been used as the major therapeutic approach for metastatic RCC in the past two decades and have prolonged the survival of patients. However, resistance to targeted therapy has been frequently reported in recent years.3, 4, 5 The 5-year survival of metastatic RCC is estimated at only 5–15%.6
RCC is a highly vascular tumor arising from epithelial cells within the proximal tubules of nephrons. Angiogenesis plays a crucial role in RCC initiation and progression.7, 8 In RCC cells, frequent inactivation of the Von Hippel–Lindau gene and aberrant activation of the PI3K→AKT→mTOR signaling pathway contribute to hypoxia-inducible factor expression. hypoxia-inducible factors then accelerate angiogenesis through transcription of its target genes, such as vascular endothelial growth factor (VEGF) and platelet derived growth factor,7 and lead to the new vascular vessel formation. In addition, recent studies demonstrated significant infiltration of immune cells, such as mast cells,9 macrophages10 and lymphocytes,11 in RCC, indicating vital roles of the surrounding tumor microenvironment (TME) in RCC progression.
Mast cells are key mediators of angiogenesis,12 and early studies indicated that mast cells in the TME might function through various cytokines/chemokines.13 Our recent reports also found that infiltrating mast cells could promote prostate cancer metastasis and influence the sensitivity to chemotherapy and radiation therapy.14, 15 The detailed mechanism of how mast cells infiltrate RCC and the possible role of mast cells in RCC angiogenesis, however, remain unknown.
Here, we found that infiltrating mast cells could promote RCC angiogenesis via modulation of PI3K→AKT→GSK3β→AM signaling. Targeting mast cell-mediated inflammatory signals with specific inhibitors could suppress RCC progression and may represent a potential therapeutic approach for treatment of RCC.
Results
Infiltrating mast cells are strongly associated with RCC angiogenesis
To investigate the potential association of RCC angiogenesis and infiltrating mast cells, the major immune cells in the kidney TME, we performed immunohistochemistry analyses with anti-CD31 (vascular endothelial cell marker) and tryptase (specific marker of mast cells)16 antibodies of 125 RCC tissues and 52 adjacent normal kidney tissues. The results revealed that mast cell density (MCD) was significantly increased in RCC tissues compared to adjacent normal kidney tissues (2.67±0.22 per high power field (HPF) vs 0.63±0.14 per HPF, P <0.05, Figures 1a and b). CD31 staining in RCC tissues (Figure 1c) indicated that the microvessel density (MVD) increased with MCD in RCC tissues (Figure 1d). Linear regression analysis showed that MVD in RCC was positively correlated with MCD in human RCC tissues (r=0.541, P<0.001) (Figure 1e).

Mast cell infiltration in RCC was correlated with angiogenesis. One hundred twenty-five RCC and 52 adjacent kidney samples were immunohistochemically stained for tryptase and CD31. Each section was examined under a high-power field (× 400) in a double-blinded manner. Microvessel density (MCD) and mast cell density (MVD) were calculated as the average measurement of 10 random fields. (a) Representative photograph of tryptase-positive mast cells in RCC and adjacent normal kidney tissues. (b) MCD in RCC and adjacent normal kidney tissues. (c) Representative photograph of two RCC cases with high MCD/MVD and low MCD/MVD. (d) Comparison of MVD in RCC tissues with different MCD levels. Low: MCD<0.63, n=26 cases; Mid: 0.63⩽MCD<2.67, n=39 cases; High: MCD⩾2.67, n=60 cases. (e) Correlation analysis of MVD and MCD by linear regression. *P<0.05.
Taken together, the results from Figures 1a and e of human clinical RCC samples indicated that infiltrating mast cells are positively correlated with RCC angiogenesis.
Mast cells promote angiogenesis of RCC in vitro and in vivo
Next, to confirm the effects of infiltrating mast cells on angiogenesis in RCC, human umbilical vein endothelial cell (HUVEC) recruitment assays were performed. In brief, conditioned medium (CM) from 2 × 106 RCC OSRC-2 cells or from 1.8 × 105 OSRC-2 plus 2 × 104 mast HMC-1 cells was collected and then added to the lower chamber with the HUVECs in the upper chamber of the transwell system. The results showed that HMC-1 co-cultured CM enhanced the OSRC-2-induced HUVEC recruitment compared to the CM from OSRC-2 cells alone (62.3±7.05 per HPF vs 116.7±10.14 per HPF, P<0.05, Figure 2a). Importantly, CM from co-cultured cells significantly enhanced the OSRC-2-induced HUVEC capacity to form capillary tubes on matrigel compared to RCC medium alone (12.33±2.03 per HPF vs 25.0±2.00 per HPF, P <0.05, Figure 2b).

Mast cells promote RCC angiogenesis in vitro and in vivo. (a). Conditioned medium from 2 × 106 OSRC-2 cells or 1.8 × 105 OSRC-2 cells mixed with 2 × 104 HMC-1 cells was collected. Then, the conditioned medium was added to the lower chamber of a transwell system to evaluate the capacity of HUVEC recruitment (Left: cartoon for HUVEC recruitment assay; Mid: representative pictures; Right: quantitative data). (b) HUVEC tube formation assays upon conditioned medium treatment (Left: representative pictures; Right: quantitative data). (c) 1 × 106 OSRC-2 cells or 1 × 105 HMC-1 cells or a combination of these two cell lines was inoculated subcutaneously into the right dorsal region of nude mice. Animals were killed 2 weeks after injection. Immunohistochemistry staining against CD31 was performed to evaluate MVD in xenografts (Left: representative pictures; Right: quantitative data). *P<0.05.
To determine whether infiltrating mast cells contributed to RCC angiogenesis in vivo, 2 × 107 OSRC-2 cells or 2 × 106 HMC-1 cells or a mixture of these two cells were subcutaneously injected into the right dorsal region of nude mice. Although HMC-1 cells alone did not form tumors (data not shown), co-injection with HMC-1 cells resulted in increased microvessel formation compared to OSRC-2 cells alone (20.8±3.06 per HPF vs 60.2±8.11 per HPF, P<0.05, Figure 2c).
Since VEGF is the major target of RCC anti-angiogenic therapy, we tested HUVEC recruitment and capillary tube formation in the presence of bevacizumab (anti-VEGF neutralizing antibody) and/or cromolyn (mast cell degranulation inhibitor) in the in vitro cell model. The results showed that treatment with either bevacizumab or cromolyn inhibited both HUVEC recruitment and capillary tube formation. However, the combination of bevacizumab and cromolyn was superior to monotherapy with either agent (Supplementary Figures 1a–d). Similarly, when the mice injected with OSRC-2 plus HMC-1 cells were treated with bevacizumab and/or cromolyn, we observed that administration of bevacizumab or cromolyn alone could inhibit the growth and MVD of tumors. However, the combination of bevacizumab and cromolyn had a significant synergistic inhibitory effect (Supplementary Figures 2a–e). These data indicated that VEGF may be involved in mast cell-mediated RCC angiogenesis and tumor growth.
Together, the results from Figures 2a–c, Supplementary Figures 1a-d and 2a-e showed that the recruitment of mast cells to the RCC enhanced the migration of vascular endothelial cells to the RCC to promote RCC angiogenesis.
Mechanistic analysis of RCC recruitment of mast cells
To investigate the mechanisms of RCC recruitment of mast cells to enhance angiogenesis, we first used transwell co-culture migration assays (Figure 3a). The results revealed that RCC OSRC-2 cells could recruit more HMC-1 cells than normal kidney HK5 cells and RCC 769-P cells (Figure 3b). We then performed Q-PCR assays to examine cytokines/chemokines related to the mast cell recruitment (Supplementary Table 2), and found that adrenomedullin (AM) mRNA expression in OSRC-2 cells was much higher (more than five-fold) than that found in HK5 cells and 769-P cells (Supplementary Table 2). Similar results were also obtained when we examined AM protein using enzyme linked immunosorbent assay (ELISA) (Figure 3c), suggesting that AM may play a key role in mediating mast cell recruitment to the RCC.

RCC recruited mast cells into the tumor microenvironment via AM expression. (a) Cartoon of mast cell recruitment by the transwell system. (b) The in vitro mast cell recruitment assay by RCC and normal kidney tubular epithelial cells. (c) AM expression in RCC and tubular epithelial cell lines detected by ELISA. (d) Administration of the AM antagonist AM22-52 in the lower chamber inhibited OSRC-2 cell-mediated mast cell recruitment. Physiological saline was used as a negative control. (e) ELISA showed AM expression in 769-P cells following forced expression of AM. The lentiviral vector LV5 was used as a control. (f) Mast cells were recruited by 769-P cells overexpressing AM. *P<0.05 vs control group; **P<0.05 vs OSRC-2.
Importantly, using an inhibition assay with AM22-52, a specific antagonist of AM, in the Boyden chamber migration system, we found that suppression of AM with 0.5 μM AM22-52 in the lower chamber significantly blocked RCC OSRC-2 cell-induced mast cell recruitment (Figure 3d). Using the opposite approach, we also found that forced expression of AM in 769-P cells could increase mast cell recruitment (Figures 3e and f).
Together, the results from Figures 3a–f showed that RCC cells could recruit mast cells by increasing the expression of AM.
AM mediated mast cell recruitment by activating PI3K→AKT→GSK3β signaling in RCC cells
To further determine how RCC cells alter AM signals to enhance the recruitment of mast cells to RCC, inhibitors of multiple signaling pathways related to RCC progression were used in our study. We found that except for the PI3K→AKT pathway, blocking the JAK→STAT3 pathway, MEK→ERK pathway, transforming growth factor-β pathway or JNK pathway all failed to alter the AM expression (Supplementary Figure 3d), indicating that AM-mediated mast cell recruitment to RCC might be due to activation of the PI3K→AKT pathway. Thus, we focused on the PI3K→AKT pathway, a central player controlling angiogenesis in cancer.7, 17 We found that AKT was constitutively activated in OSRC-2 cells compared to 769-P and HK5 cells (Figure 4a), and inhibition of AKT phosphorylation by the specific PI3K inhibitor LY294002 (Figure 4b) in OSRC-2 cells decreased the expression of AM (Figure 4c), which abolished the recruitment of mast cells (Figure 4d).

AM-mediated mast cell recruitment was dependent on PI3K→AKT→GSK3β signaling in RCC. (a) The expression of total and p-AKT Ser473 in HK5, 769-P and OSRC-2 cells was assayed by western blotting. (b) The expression of total and p-AKT Ser473 in OSRC-2 cells with or without LY294002 treatment for 24 h. (c) ELISA of AM concentration in the medium of OSRC-2 cells treated with or without (as control) LY294002 for 24 h. (d) OSRC-2 cells were pretreated with or without (as control) LY294002 for 24 h and then subjected to transwell assays for mast cell recruitment. (e) The expression of total and p-Akt Ser473 in 769-P cells treated with or without (as control) the AKT activator SC-79 for 2 h. (f) ELISA of AM concentration in the medium of 769-P cells treated with or without (as control) SC-79. (g) 769-P cells were pretreated with SC-79 for 2 h, and mast cell recruitment was assayed with or without AM22-52. (h) Mast cell recruitment was assayed by adding the inhibitor of AM (AM22-52) with or without AKT activator (SC-79) (i) The expression of p-GSK3β in RCC cells pretreated with LY294002 or SC-79 was assayed by western blotting. (j) The expression of p-GSK3β in RCC cells pretreated with LiCl was assayed by western blotting. (k) AM expression was detected by qRT-PCR assays in 769-P cells with or without 50 μm LiCl treatment. *P<0.05.
Using an opposite approach with a PI3K→AKT activator (SC-79) for the mast cell recruitment assay, we found that addition of a PI3K→AKT activator (Figure 4e) could increase AM expression and enhanced the mast cell recruitment to the 769-P cells (Figures 4f and g), and pre-treatment with the AM antagonist AM22-52 inhibited SC-79-induced recruitment of mast cells (Figure 4h).
To further link PI3K→AKT to AM signaling, GSK3β signaling was examined because early studies have demonstrated that GSK3β is one of the downstream signals for AKT in cancer cells.18 We first confirmed that activation of AKT following addition of SC-79 could increase the phosphorylation of GSK3β at Ser9 in 769-P cells (Figure 4i), and inhibition of AKT activation by LY294002 decreased GSK3β Ser9 phosphorylation in OSRC-2 cells (Figure 4i). We then pre-treated the 769-P cells with LiCl, an inhibitor of GSK3β, and the results showed that LiCl significantly increased p-GSK3 Ser9 and AM expression in 769-P cells (Figures 4j and k).
Taken together, the results from Figures 4a–k indicated that PI3K→AKT→GSK3β signaling may play key roles in modulating AM expression to affect mast cell recruitment in RCC.
PI3K→AKT→AM signaling was correlated with mast cell infiltration and angiogenesis in clinical RCC samples
To confirm the above data in RCC patients, the AM and p-AKT Ser473 expression in 125 RCC cases and 52 adjacent normal kidney tissues was evaluated by immunohistochemical staining (Figure 5a). The results revealed that AM was expressed in 72.8% (91/125) of the RCC tissues and 32.7% (17/52) of the adjacent normal kidney tissues (Supplementary Figure 3a). Mast cell density in AM+ RCC tissues was higher than MCD in AM− RCC tissues (3.51±0.28 per HPF vs 1.89±0.36, Figure 5b). Similarly, MVD in AM+ RCC tissues was significantly higher than MVD in AM-RCC tissues (23.88±1.51 per HPF vs 6.42±1.66 per HPF, Figure 5c). In addition, our results also showed that phosphorylation of AKT at Ser473 in the RCC tissues was increased compared to that in adjacent normal kidney tissues (Supplementary Figure 3b). mast cell density and MVD in p-AKT+ RCC tissues were higher than those in p-AKT− RCC tissues (MCD: 4.02±0.28 per HPF vs 1.43±0.27 per HPF, p<0.05, Figure 5d; MVD: 25.26±1.60 per HPF vs 8.61±1.63 per HPF, Figure 5e). These clinical data also showed that AM expression in RCC samples was positively correlated with p-Akt Ser473+ expression (Supplementary Figure 3c).

The PI3K→AKT→AM signaling pathway is correlated with mast cell infiltration and angiogenesis in RCC samples. (a) Representative immunohistochemistry images of p-Akt Ser473 and AM in RCC tissues. (b) Comparison of MCD in AM+ and AM- RCC tissues. (c) Comparison of MVD in AM+ and AM- RCC tissues. (d) Comparison of MCD in p-Akt Ser473+ and p-Akt Ser473- RCC tissues. (e) Comparison of MVD in p-Akt Ser473+and p-Akt Ser473- RCC tissues.
Together, the results from Figures 5a–e and Supplementary Figures 3a–c suggested that PI3K→AKT→AM signaling in clinical RCC tissues was positively correlated with mast cell infiltration and microvessel formation.
Targeting PI3K→AKT signals suppressed mast cell-induced angiogenesis in an in vivo mouse model
The in vitro and clinical data demonstrated that PI3K→AKT→GSK3β→AM signaling in RCC may promote mast cell-induced angiogenesis in RCC, suggesting that targeting this newly identified PI3K→AKT→GSK3β→AM pathway may be an effective strategy to treat RCC. We then generated a RCC subcutaneous tumor xenograft mouse model to examine the in vivo effects. When the volume of RCC tumor xenografts reached 0.5 cm3, mice were intratumorally injected with 500 μM LY294002 or with 10% dimethylsulphoxide as a control. Mice were killed 48 h after 5 × 106 red fluorescent protein (RFP)-labeled HMC-1 cells were injected via tail vein. The results showed significantly decreased p-Akt Ser473 expression and AM expression in LY294002-treated OSRC-2 tumors compared to dimethylsulphoxide-treated tumors (Figure 6a), and inhibition of AKT in xenograft mice resulted in less RFP+ mast cell recruitment than control mice (Figure 6b).

Targeting PI3K→AKT signaling inhibited mast cell-mediated angiogenesis in vivo. (a) p-AKT Ser473 and AM expression in OSRC-2 subcutaneous xenograft tumor samples was determined by immunohistochemistry staining. (b) The RFP-labeled HMC-1 cells were identified and analyzed by fluorescence microscopy in sections of frozen OSRC-2 tumor samples (Left: representative pictures; Right: quantitative data). (c) A representative tumor of xenograft mice injected with 1 × 106 OSRC-2 cells or 1 × 105 HMC-1 cells plus 1 × 106 OSRC-2 cells. (d) Tumor volume was measured after injection of 1 × 106 OSRC-2 cells or 1 × 105 HMC-1 cells or a combination of these two cell lines. *P<0.05.
To further determine whether mast cell infiltration promoted RCC angiogenesis in vivo, 2 × 107 OSRC-2 cells or 2 × 106 HMC-1 cells or a mixture of these two cells were injected subcutaneously into the right dorsal region of nude mice, and the results revealed that the tumor volume and tumor weight were significantly higher in mice co-injected with OSRC-2 cells plus HMC-1 cells than mice only injected with OSRC-2 cells (Figure 6c). Additionally, tumors occurred earlier in mice co-injected with OSRC-2 cells plus HMC-1 cells than mice only injected with OSRC-2 cells (Figure 6d), suggesting that infiltrating mast cells enhance RCC progression.
Taken together, the results from Figures 6a–d demonstrated that inhibition of RCC AKT signaling could suppress mast cell-induced RCC progression in the in vivo mouse model, and targeting AKT with specific inhibitors could be a new potential strategy to treat RCC.
Discussion
In addition to releasing low molecular weight substances, including histamine and leukotrienes, mast cells can also release a variety of cytokines, chemokines or proteinases to influence cancer progression.19 While early studies demonstrated that mast cells have anti-tumor effects by mediating host anti-tumor immune response,20 accumulating evidence has also indicated that mast cells promote cancer initiation and tumor progression.
Mast cells directly secrete various angiogenic cytokines and chemokines to the TME, including stem cell factor (SCF), fibroblast growth factor-2 (FGF-2), VEGF, PDGF, CCL2 and CCL5.21 Mast cell-secreted proteases, such as tryptase and chymase, stimulate the proliferation of endothelial cells, promote vascular tube formation in culture and also degrade connective tissue matrix to provide space for neovascular growth. In addition, mast cells also contain abundant matrix metalloproteinases (MMPs), such as MMP-2 and MMP-9, and tissue inhibitors of metalloproteinases (TIMPs), which promote tissue remodeling during neovascularization.21 Adjuvant treatment targeting mast cells or mast cell-secreted factors might be a promising therapeutic approach in the future.22
Mast cells were first reported in kidney tissues in 1960.23 Infiltration of mast cells in the renal interstitium is associated with kidney diseases, such as glomerular diseases.24 Moreover, mast cell counts in human kidney cortex were significantly increased in patients with chronic renal failure, and there was a significant relationship between serum creatinine and the mast cell granulation index.25 Previously, mast cells were observed to highly infiltrate RCC tissues compared with non-neoplastic kidney tissues.9, 26 However, the relationship between mast cells and angiogenesis in RCC remains controversial. Tuna et al. observed a significant correlation between the number of mast cells and MVD in 71 RCC patients.9 However, studies from Mohseni failed to demonstrate a positive correlation between the number of mast cells and MVD in RCC tumors27 using a smaller number of cases in their studies. Using 125 RCC samples and 52 adjacent kidney tissue samples to assess mast cells and endothelial cells, we found that there were more mast cells accumulated in RCC tissues than adjacent kidney tissues. More importantly, there was a significant correlation between MCD and MVD.
Since mast cells originate from bone marrow and invade the tissues from peripheral blood during homeostasis,28 an association between MCD and MVD in RCC tissues may not be sufficient to support the pro-angiogenic role of mast cells. We therefore further determined the role of mast cells in angiogenesis of RCC in various in vitro cell lines and in vivo mouse models and concluded that mast cells accelerated angiogenesis in RCC.
In tumors, mast cells may be recruited and activated via various factors secreted by tumor cells. For example, activation of the c-Kit receptor by SCF may alter mast cell development,29 migration,30 proliferation31 and survival.31, 32 In solid tumors, such as breast cancer, cancer cell-secreted SCF may play major roles in mast cell accumulation and influence tumor angiogenesis and tumor growth.33 Watanab et al.34 reported that the SCF-positive cells were higher in hemodialysis-RCC than non-hemodialysis-RCC (62.9% vs 33.3%), and the MCD in SCF-positive specimens was significantly higher than that in SCF-negative ones. However, whether SCF alters mast cell infiltration to influence the RCC progression remains unclear. Because SCF expression in non-hemodialysis-RCC is low, it is difficult to explain the high level of mast cell infiltration in RCC. More importantly, SCF expression is higher in oncocytomas and papillary RCC than clear cell RCC.35 It is also unclear why the intratumoral MCD in clear cell RCC was significantly higher compared with the non-clear variety.26 Thus, we hypothesized that there were mast cell chemoattractants other than SCF in RCC.
Adrenomedullin is a 52 amino acid peptide36 that belongs to the calcitonin gene peptide superfamily and has high angiogenic activities in solid tumors.37 AM mediates its activities via heterodimeric receptors composed of a G-protein-coupled receptor named calcitonin-receptor-like receptor and a receptor activity modifying protein.38 Clinical studies indicated that AM was highly expressed in RCC, and its expression was associated with VEGF expression and MVD.39, 40 Under hypoxic conditions, both VEGF and AM expression in RCC cell lines was increased. However, the increase in AM was markedly greater than that of VEGF,40 suggesting that AM might be a pro-angiogenic factor in RCC. In our study, we found that AM expression was higher in RCC compared to that in adjacent normal kidney tissues, and RCC tissues with AM-positive expression had higher MVD and MCD.
The PI3K→AKT signaling pathway is activated in many tumors. In RCC, PI3K→AKT may closely interact with mTOR, VEGF and platelet derived growth factor to influence angiogenesis7 and may represent a potential therapeutic target. However, due to side effects and high toxicity, very few PI3K or Akt inhibitors are used for clinical treatment of RCC.41 Recently, tyrosine kinase inhibitors, such as sunitinib and sorafenib, have been used in metastatic RCC patients. However, the effects are limited due to the occurrence of drug resistance.5 Interestingly, early experimental evidence showed an increase of AKT activity in sunitinib-resistant RCC cells.42 Our study showed that PI3K→AKT promotes AM expression through GSK3β and the consequent mast cell recruitment and angiogenesis in RCC. This represents a pro-angiogenic signal pathway that is different from the classic PI3K→AKT→mTOR→VEGF pathway. Whether novel therapeutics can be developed based on this newly identified signaling pathway requires further study.
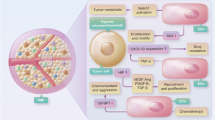
In summary, our studies indicated that in RCC patients, the aberrant activation of PI3K→AKT signaling may upregulate AM expression through GSK3β. Then, increased AM recruits more mast cells from the TME to the RCC. The consequence of the increased mast cells in RCC is enhanced RCC angiogenesis, which promotes RCC progression (Figure 7). Targeting this newly identified pathway may help us to develop novel therapeutics to better treat RCC.

Diagrammatic sketch. PI3K→AKT→GSK3β signal-mediated AM expression could increase mast cells recruitment to the tumor microenvironment, and then, mast cells could recruit vascular endothelial cells by secreting pro-angiogenic factors, such as VEGF and fibroblast growth factor-2,12 or promote tissue remodeling by secreting proteinase, such as tryptase and MMPs,12 and finally accelerate angiogenesis in RCC.
Materials and methods
Chemicals and reagents
Primary antibodies against T-AKT (#2920), p-AKT Ser473 (#3787for immunohistochemistry staining, #4051 for western blot), GSK3β (#9315) and p-GSK3β Ser9 (#5558) were purchased from Cell Signaling Technology (Beverly, MA, USA). A primary antibody against tryptase was purchased from DAKO A/S (Nr. M 7052). Primary antibodies against AM (sc-53153) and IkBα (sc-371) were purchased from Santa Cruz Biotechnology (Paso Robles, CA, USA). LY294002 (L9908) and cromolyn (#1150502) were purchased from Sigma-Aldrich Chemical Co. (St Louis, MO, USA). Bevacizumab was purchased from Roche Pharmaceutical (Basel, Switzerland).
Cell culture
The human renal carcinoma 769-P cell line was obtained from American Type Culture Collection (USA). The human renal carcinoma OSRC-2 cell line was obtained from National Platform of Experimental Cell Resources for Sci-Tech (Beijing, China). The human renal tubular epithelial HK5 cell line was kindly gifted by Prof. Chawnshang Chang (University of Rochester, USA). Cells had been determined to be mycoplasma-free and were propagated in Dulbecco′s modified Eagle’s medium/F12 containing 10% fetal bovine serum (FBS). The human mast cell HMC-1 cell line was propagated in Iscove's Modified Dulbecco's Medium (IMDM) medium containing 10% fetal bovine serum. Cells were maintained at 37 °C in a humidified incubator with 5% CO2 in air. All cell lines were used for fewer than 6 months and were routinely screened for mycoplasma every 4 weeks. No genotypic authentication was conducted. Each cell line was used in the early passages.
Conditioned medium collection
Tumor cells (2 × 106) were seeded into a 10-cm dish with 10% fetal bovine serum and cultured for 6 h. Then, the cells were washed three times with 0.01 M phosphate-buffered saline and cultured with 10 ml serum-free medium for 24 h. The supernatant fluid was collected, and cell debris was eliminated using a 0.44-μm polycarbonate filter.
In vitro mast cell recruitment assay
To evaluate RCC cell-mediated mast cell recruitment, a 24-well transwell assay was used in our study. Briefly, 2.5 × 104 HK5, OSRC-2 or 769- P cells were placed in the lower chambers of the 24-well transwells. The, 1 × 105 mast cells were then seeded in the upper chambers. The upper and lower chambers were separated by an 8 μm polycarbonate filter coated with fibronectin (10 μg/ml, sc-29011 Santa Cruz Biotechnology). The chambers were incubated for 4 h at 37 °C. Filters were then scraped, washed, fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Cell migration was measured by counting the number of cells attached to the lower surface of the filter. Each experiment was assayed in triplicate. The results are expressed as the mean±s.e.m. of the number of migrating cells.
RNA extraction and quantitative real-time PCR analysis
Total RNA was extracted from cells using a Total RNA Extraction Kit obtained from Fastagen Biotech (Shanghai, China). Total RNA (1 μg) was subjected to reverse transcription using Superscript III transcriptase (Invitrogen, Eugene, OR, USA). Quantitative real-time PCR (qRT-PCR) was conducted using a Bio-Rad CFX96 system with SYBR Green to determine the mRNA expression level of a gene of interest. Human glyceraldehyde-3-phosphate dehydrogenase cDNA was amplified as an internal control. Primer sequences are listed in Supplementary Table 1.
Western blot assay
Total protein was extracted from cells with RIPA lysis buffer containing protease inhibitors. The protein concentration of the lysates was analyzed by the Bradford method. Western blot analysis was performed using a modified version of a previously described method.9 Briefly, 30 μg of protein was separated on a 12% SDS-polyacrylamide gel and blotted onto nitrocellulose filter membranes. The membranes were blocked with 5% nonfat-milk in Tris-buffered saline and Tween 20 for 1 h at room temperature. The blots were then incubated with primary antibodies overnight at 4 °C and horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Immunoreactive signals were detected using the ECL detection system, followed by exposure to X-ray film. Immunoblotting of glyceraldehyde-3-phosphate dehydrogenase was performed as an internal control.
In vivo mast cell recruitment assay
Mast cell recruitment assays in vivo were performed as previously described.15 Briefly, 1 × 106 OSRC-2 cells suspended in matrigel 1 (1:8; BD Biosciences, San Jose, CA, USA) were subcutaneously injected into the right dorsal region of 4-week-old male BALB/c nude mice. When the volume of the xenografts reached 0.5 cm3, animals were averagely divided into two groups (six mice for each group) with random digit. One group was intratumorally injected with 500 μM LY294002, and the other group was intratumorally injected with 10% dimethylsulphoxide as a control. Then, 5 × 106 RFP-labeled HMC-1 cells were injected into tumor-bearing mice through the tail vein. Tumor tissues were surgically excised from the mice 48 h after the injection, and frozen sections were prepared and analyzed by fluorescence microscopy. The average number of RFP-positive cells in each slide was counted from five random 10 × fields. The animal experiments were approved by the institutional review board of the First Affiliated Hospital of Xi’an Jiaotong University.
Mast cell-mediated RCC angiogenesis in vitro
Conditioned medium from 2 × 106 RCC OSRC-2 cells or from 1.8 × 105 OSRC-2 plus 2 × 104 mast HMC-1 cells was collected and was then was added to the lower chamber with 2.5 × 104 HUVECs in the upper chamber of the transwell system or was added to a 24-well plate with 7.5 × 104 HUVECs on the matrigel for papillary tube formation. To determine the synergistic effect of cromolyn and bevacizumab on inhibition of mast cell-mediated angiogenesis, 1.8 × 105 OSRC-2 plus 2 × 104 HMC-1 cells were seeded in 10 cm dishes. After incubation with 25 μM cromolyn, 0.1 mg/mL bevacizumab, or a combination of both drugs for 24 h, the CM was harvested. Then, the CM was added to the lower chamber with 5.0 × 104 HUVEC cells in the upper chamber of the transwell system or added to 24-well plates with 7.5 × 104 HUVEC cells on the matrigel for papillary tube formation.
Mast cell-mediated RCC angiogenesis in vivo
Eighteen BALB/c nude mice (4-week-old, male) were averagely allocated to three groups (six mice for each group) with random digit. Then, these three groups were subcutaneously injected with 2 × 107 OSRC-2 cells, 2 × 106 HMC-1 cells or 2 × 107 OSRC-2 cells mixed with 2 × 106 HMC-1 cells. The body weight and tumor diameter were measured every 3 days. Tumor volume was calculated using the following equation: Volume= (A × B2)/2 (A refers to the long diameter and B refers to the short diameter). Animals were killed 2 weeks later, and tumor tissues were surgically excised from the mice. Tumor weight was evaluated. The samples were fixed in 4% paraformaldehyde and embedded in paraffin. Five-micrometer-thick sections were prepared for immunohistochemical staining. Each section was examined under a high-power field (× 400) in a double-blinded manner by one pathologist.
To determine the synergistic effect of cromolyn and bevacizumab on inhibition of mast cell-mediated angiogenesis, 1 × 105 HMC-1 cells plus 1 × 106 OSRC-2 cells were inoculated subcutaneously into the right dorsal region of 24 BALB/c nude mice (4-week-old, male). Animals were averagely divided into four groups (six mice for each group) with random digit and were treated with saline (100 μl, daily), cromolyn (10 mg/kg daily), bevacizumab (8 mg/kg, once every two weeks) and a combination of cromolyn and bevacizumab. Animals were killed after 4 weeks. Tumors were measured and analyzed as above.
The animal experiments were approved by the institutional review board of the First Affiliated Hospital of Xi’an Jiaotong University.
Clinical specimens and immunohistochemistry
To investigate mast cell infiltration in RCC tissues and its correlation with angiogenesis, 125 clear cell RCC samples and 52 adjacent kidney tissue samples were collected from patients who underwent partial or radical nephrectomy for RCC at the Department of Urology, the First Affiliated Hospital of Xi’an Jiaotong University, between August 2008 and July 2010. The approval of the institutional review board of the First Affiliated Hospital of Xi’an Jiaotong University was obtained before the samples were collected, which was performed with the permission of the patients. Tissues were fixed in 4% paraformaldehyde and embedded in paraffin. Five-micrometer-thick sections were prepared, and immunohistochemical staining of tryptase (dilution 1:200), CD31 (dilution 1:150), p-Akt Ser473 (dilution 1:150) and AM (dilution 1:200) was performed using a DAKO Autostainer Plus system. Each section was examined under a high-power field (× 400) in a double-blinded manner by one pathologist. Mast cell density was defined as the average count of tryptase-positive mast cells from 10 random fields. Any stained CD31+ endothelial cell or CD31-positive cell cluster separated from other microvessel structures was defined as a countable microvessel.43 Microvessel density was defined as the average count of microvessels from 10 random fields. The results of anti-p-Akt Ser473 and anti-AM staining were evaluated using a scoring system that combines intensity and percentage. In each field, intensity was scored as follows: 0 for no staining, 1 for weakly positive staining, 2 for moderately positive staining and 3 for strongly positive staining. The staining percentage of the relative number of cells stained was graded as follows: 0 for 0%, 1 for ⩽25%, 2 for 25–50%, 3 for 50–75% and 4 for ⩾75%. The total score of each section was calculated by multiplying the intensity and percentage scores. In the current study, sections with total scores ⩾4 were defined as positive staining.
Statistical analysis
Results were analyzed using the statistical software SPSS for Windows 10.0.Quantitative data were presented as mean±s.d. Differences in mean values between two groups were analyzed by two-tailed Student’s t-test and the mean values of more than two groups were compared with one-way analysis of variance. Correlation analysis was carried out using χ2 tests. P<0.05 was considered significant.
References
Siegel RL, Miller KD, Jemal A . Cancer statistics, 2015. CA Cancer J Clin 2015; 65: 5–29.
Cairns P . Renal cell carcinoma. Cancer Biomark 2010; 9: 461–473.
Kornakiewicz A, Solarek W, Bielecka ZF, Lian F, Szczylik C, Czarnecka AM . Mammalian target of rapamycin inhibitors resistance mechanisms in clear cell renal cell carcinoma. Curr Signal Transduct Ther 2014; 8: 210–218.
Liu XD, Hoang A, Zhou L, Kalra S, Yetil A, Sun M et al. Resistance to antiangiogenic therapy is associated with an immunosuppressive tumor microenvironment in metastatic renal cell carcinoma. Cancer Immunol Res 2015; 3: 1017–1029.
Zhang L, Bhasin M, Schor-Bardach R, Wang X, Collins MP, Panka D et al. Resistance of renal cell carcinoma to sorafenib is mediated by potentially reversible gene expression. PLoS One 2011; 6: e19144.
Patel PH, Chaganti RS, Motzer RJ . Targeted therapy for metastatic renal cell carcinoma. Br J Cancer 2006; 94: 614–619.
Rini BI, Campbell SC, Escudier B . Renal cell carcinoma. Lancet 2009; 373: 1119–1132.
Shen C, Kaelin WG Jr . The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol 2013; 23: 18–25.
Tuna B, Yorukoglu K, Unlu M, Mungan MU, Kirkali Z . Association of mast cells with microvessel density in renal cell carcinomas. Eur Urol 2006; 50: 530–534.
Toge H, Inagaki T, Kojimoto Y, Shinka T, Hara I . Angiogenesis in renal cell carcinoma: the role of tumor-associated macrophages. Int J Urol 2009; 16: 801–807.
Ning H, Shao QQ, Ding KJ, Gao DX, Lu QL, Cao QW et al. Tumor-infiltrating regulatory T cells are positively correlated with angiogenic status in renal cell carcinoma. Chin Med J (Engl) 2012; 125: 2120–2125.
Maltby S, Khazaie K, McNagny KM . Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochim Biophys Acta 2009; 1796: 19–26.
Crivellato E, Nico B, Ribatti D . Mast cells and tumour angiogenesis: new insight from experimental carcinogenesis. Cancer Lett 2008; 269: 1–6.
Dang Q, Li L, Xie H, He D, Chen J, Song W et al. Anti-androgen enzalutamide enhances prostate cancer neuroendocrine (NE) differentiation via altering the infiltrated mast cells —> androgen receptor (AR) —> miRNA32 signals. Mol oncol 2015; 9: 1241–1251.
Li L, Dang Q, Xie H, Yang Z, He D, Liang L et al. Infiltrating mast cells enhance prostate cancer invasion via altering LncRNA-HOTAIR/PRC2-androgen receptor (AR)-MMP9 signals and increased stem/progenitor cell population. Oncotarget 2015; 6: 14179–14190.
Shi GP, Lindholt JS . Mast cells in abdominal aortic aneurysms. Curr Vasc Pharmacol 2013; 11: 314–326.
Safdari Y, Khalili M, Ebrahimzadeh MA, Yazdani Y, Farajnia S . Natural inhibitors of PI3K/AKT signaling in breast cancer: emphasis on newly-discovered molecular mechanisms of action. Pharmacol res 2015; 93: 1–10.
Wickenden JA, Watson CJ . Key signalling nodes in mammary gland development and cancer. Signalling downstream of PI3 kinase in mammary epithelium: a play in 3 Akts. Breast Cancer Res 2010; 12: 202.
Galinsky DS, Nechushtan H . Mast cells and cancer—no longer just basic science. Crit Rev Oncol Hematol 2008; 68: 115–130.
Siebenhaar F, Metz M, Maurer M . Mast cells protect from skin tumor development and limit tumor growth during cutaneous de novo carcinogenesis in a Kit-dependent mouse model. Exp Dermatol 2014; 23: 159–164.
Ribatti D, Crivellato E . Mast cells, angiogenesis, and tumour growth. Biochim Biophys Acta 2012; 1822: 2–8.
Ribatti D . Mast cells as therapeutic target in cancer. Eur j pharmacol 2016; 778: 152–157.
Pavone-Macaluso M . Tissue mast cells in renal diseases. Acta pathol microbiol Scand 1960; 50: 337–346.
Eddy AA . Mast cells find their way to the kidney. Kidney int 2001; 60: 375–377.
Saruta T, Kondo K, Ohguro T, Ozawa Y, Kato E . Mast cells in human kidney cortex. Keio j med 1977; 26: 163–169.
Guldur ME, Kocarslan S, Ozardali HI, Ciftci H, Dincoglu D, Gumus K . The relationship of mast cells and angiogenesis with prognosis in renal cell carcinoma. J Pak Med Assoc 2014; 64: 300–303.
Mohseni MG, Mohammadi A, Heshmat AS, Kosari F, Meysamie AP . The lack of correlation between mast cells and microvessel density with pathologic feature of renal cell carcinoma. Int Urol Nephrol 2010; 42: 109–112.
Kitamura Y, Oboki K, Ito A . Development of mast cells. Proc Jpn Acad Ser B Phys Biol Sci 2007; 83: 164–174.
Kinoshita T, Sawai N, Hidaka E, Yamashita T, Koike K . Interleukin-6 directly modulates stem cell factor-dependent development of human mast cells derived from CD34(+) cord blood cells. Blood 1999; 94: 496–508.
Samayawardhena LA, Pallen CJ . Protein-tyrosine phosphatase alpha regulates stem cell factor-dependent c-Kit activation and migration of mast cells. J Biol Chem 2008; 283: 29175–29185.
Hollins F, Kaur D, Yang W, Cruse G, Saunders R, Sutcliffe A et al. Human airway smooth muscle promotes human lung mast cell survival, proliferation, and constitutive activation: cooperative roles for CADM1, stem cell factor, and IL-6. J Immunol 2008; 181: 2772–2780.
Forster A, Grotha SP, Seeger JM, Rabenhorst A, Gehring M, Raap U et al. Activation of KIT modulates the function of tumor necrosis factor-related apoptosis-inducing ligand receptor (TRAIL-R) in mast cells. Allergy 2015; 70: 764–774.
Zhang W, Stoica G, Tasca SI, Kelly KA, Meininger CJ . Modulation of tumor angiogenesis by stem cell factor. Cancer Res 2000; 60: 6757–6762.
Watanabe S, Miyata Y, Matsuo T, Mochizuki Y, Nishikido M, Hayashi T et al. High density of tryptase-positive mast cells in patients with renal cell carcinoma on hemodialysis: correlation with expression of stem cell factor and protease activated receptor-2. Hum Pathol 2012; 43: 888–897.
Horstmann M, Hennenlotter J, Geiger LM, Vogel U, Schmid H, Kuehs U et al. Evaluation of the KIT/stem cell factor axis in renal tumours. Anticancer Res 2012; 32: 4339–4345.
Kitamura K, Kangawa K, Kawamoto M, Ichiki Y, Nakamura S, Matsuo H et al. Adrenomedullin: a novel hypotensive peptide isolated from human pheochromocytoma. Biochem Biophys Res Commun 1993; 192: 553–560.
Deville JL, Salas S, Figarella-Branger D, Ouafik L, Daniel L . Adrenomedullin as a therapeutic target in angiogenesis. Expert Opin Ther Targets 2010; 14: 1059–1072.
Nikitenko LL, Fox SB, Kehoe S, Rees MC, Bicknell R . Adrenomedullin and tumour angiogenesis. Br J Cancer 2006; 94: 1–7.
Deville JL, Bartoli C, Berenguer C, Fernandez-Sauze S, Kaafarani I, Delfino C et al. Expression and role of adrenomedullin in renal tumors and value of its mRNA levels as prognostic factor in clear-cell renal carcinoma. Int J Cancer 2009; 125: 2307–2315.
Fujita Y, Mimata H, Nasu N, Nomura T, Nomura Y, Nakagawa M . Involvement of adrenomedullin induced by hypoxia in angiogenesis in human renal cell carcinoma. Int J Urol 2002; 9: 285–295.
Park JY, Lin PY, Weiss RH . Targeting the PI3K-Akt pathway in kidney cancer. Expert Rev Anticancer Ther 2007; 7: 863–870.
Juengel E, Kim D, Makarevic J, Reiter M, Tsaur I, Bartsch G et al. Molecular analysis of sunitinib resistant renal cell carcinoma cells after sequential treatment with RAD001 (everolimus) or sorafenib. J Cell Mol Med 2015; 19: 430–441.
Trojan L, Thomas D, Friedrich D, Grobholz R, Knoll T, Alken P et al. Expression of different vascular endothelial markers in prostate cancer and BPH tissue: an immunohistochemical and clinical evaluation. Anticancer Res 2004; 24: 1651–1656.
Acknowledgements
This work was supported by The National High Technology Research and Development Program of China (863 Program, NO SS2014AA020607) and National Natural Science Foundation of China grant (No. 81072107, 81472679, 81302227, 81130041), and the National Key Research and Development Program (2016YFC0902603).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Chen, Y., Li, C., Xie, H. et al. Infiltrating mast cells promote renal cell carcinoma angiogenesis by modulating PI3K→AKT→GSK3β→AM signaling. Oncogene 36, 2879–2888 (2017). https://doi.org/10.1038/onc.2016.442
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.442
This article is cited by
-
Mast cells inhibit colorectal cancer development by inducing ER stress through secreting Cystatin C
Oncogene (2023)
-
Identification and development of a novel risk model based on cuproptosis-associated RNA methylation regulators for predicting prognosis and characterizing immune status in hepatocellular carcinoma
Hepatology International (2023)
-
KCNN4 may weaken anti-tumor immune response via raising Tregs and diminishing resting mast cells in clear cell renal cell carcinoma
Cancer Cell International (2022)
-
A novel ferroptosis-related 12-gene signature predicts clinical prognosis and reveals immune relevancy in clear cell renal cell carcinoma
BMC Cancer (2021)
-
PD-1 immunobiology in glomerulonephritis and renal cell carcinoma
BMC Nephrology (2021)

{kind=link}
{kind=link}
{kind=link}