
Abstract
Protein arginine methyltransferase 5 (PRMT5) is an emerging epigenetic enzyme that mainly represses transcription of target genes via symmetric dimethylation of arginine residues on histones H4R3, H3R8 and H2AR3. Accumulating evidence suggests that PRMT5 may function as an oncogene to drive cancer cell growth by epigenetic inactivation of several tumor suppressors. Here, we provide evidence that PRMT5 promotes prostate cancer cell growth by epigenetically activating transcription of the androgen receptor (AR) in prostate cancer cells. Knockdown of PRMT5 or inhibition of PRMT5 by a specific inhibitor reduces the expression of AR and suppresses the growth of multiple AR-positive, but not AR-negative, prostate cancer cells. Significantly, knockdown of PRMT5 in AR-positive LNCaP cells completely suppresses the growth of xenograft tumors in mice. Molecular analysis reveals that PRMT5 binds to the proximal promoter region of the AR gene and contributes mainly to the enriched symmetric dimethylation of H4R3 in the same region. Mechanistically, PRMT5 is recruited to the AR promoter by its interaction with Sp1, the major transcription factor responsible for AR transcription, and forms a complex with Brg1, an ATP-dependent chromatin remodeler, on the proximal promoter region of the AR gene. Furthermore, PRMT5 expression in prostate cancer tissues is significantly higher than that in benign prostatic hyperplasia tissues, and PRMT5 expression correlates positively with AR expression at both the protein and mRNA levels. Taken together, our results identify PRMT5 as a novel epigenetic activator of AR in prostate cancer. Given that inhibiting AR transcriptional activity or androgen synthesis remains the major mechanism of action for most existing anti-androgen agents, our findings also raise an interesting possibility that targeting PRMT5 may represent a novel approach for prostate cancer treatment by eliminating AR expression.
Similar content being viewed by others


Introduction
Protein arginine methyltransferase 5 (PRMT5) is a type II arginine methyltransferase that epigenetically regulates gene transcription by symmetrically dimethylating histone H4 arginine 3 (H4R3me2s), histone H3 arginine 8 (H3R8me2s) or histone H2A arginine 3 (H2AR3me2s).1, 2 PRMT5 also modulates the function of non-histone protein substrates by dimethylating arginine residues on the proteins. By regulating transcription of target genes or post-translational modifications of signaling proteins, PRMT5 is implicated in the regulation of many cellular processes such as cell cycle progression, apoptosis and DNA-damage response. Accumulating evidence shows that PRMT5 is overexpressed in several human cancers, and its expression positively correlates with disease progression and poor outcomes.3, 4, 5, 6, 7, 8 Mechanistic studies have suggested that PRMT5 may function as an oncogene by epigenetic repression of several tumor suppressor genes or by post-translational modification of signaling molecules.9, 10
Prostate cancer remains the most common non-cutaneous cancer among American men.11 Although many molecules and signaling pathways that regulate prostate cancer development and progression have been identified and characterized, androgen receptor (AR) signaling is the most important factor that drives prostate cancer development and progression.12, 13, 14 Thus, targeting AR signaling, such as androgen deprivation therapy (ADT), is a standard treatment for patients with locally advanced and metastatic disease. Despite the initial response to ADT, the majority of prostate cancers progress to a lethal status known as castration resistant prostate cancer (CRPC) owing to AR reactivation, which includes AR gene amplification, AR mutations, AR splice variants, androgen-independent activation of AR by AR modulators and intratumoral de novo androgen synthesis in prostate cancer cells.13, 15, 16 Recent evidence further shows that AR reactivation is also the major mechanism of resistance to the two next-generation anti-androgen agents abiraterone and enzalutamide.17, 18 Therefore, the expression of wild-type or mutant AR is absolutely required in both hormone naive prostate cancer and CRPC. However, compared with extensive studies of AR co-activators and co-repressors including epigenetic regulators,19, 20, 21, 22, 23, 24 how AR expression is regulated, particularly at the epigenetic level, remains largely unknown.
Here, we report that PRMT5 is highly expressed in prostate cancer tissues and that its expression positively correlates with the expression of AR. Molecular analysis reveals that PRMT5 epigenetically activates the transcription of AR via symmetric dimethylation of H4R3 and promotes prostate cancer cell growth in vitro and xenograft tumor growth in mice. Given that current AR-targeting strategies, which are largely based on the inhibition of AR transcriptional activity or inhibition of androgen synthesis, are ultimately ineffective, our findings raise an interesting possibility that targeting PRMT5 may be explored as a novel therapeutic approach to inhibit or eliminate AR expression for prostate cancer treatment.
Results
PRMT5 expression is required for prostate cancer cell growth in an AR-dependent manner
We and others previously reported that knockdown of PRMT5 inhibited cell growth in LNCaP cells.25, 26 To further investigate this, we examined the role of PRMT5 in DU145 and PC-3 cells by transiently knocking down PRMT5, and did not observe any significant effect on cell growth when compared with scrambled control (SC; Supplementary Figure S1a–d). Knockdown of PRMT5 in LNCaP cells also exhibited a pronounced inhibitory effect on colony formation in soft agar (Supplementary Figure S1e). Next, we established stable cell lines using LNCaP and DU145 that can be induced by doxycycline (Dox) to express short-hairpin RNA (shRNA), and confirmed that inducible knockdown of PRMT5 indeed showed significant growth inhibition in LNCaP cells (Figure 1a), but not in DU145 cells (Figure 1b). Because DU145 and PC-3 cells do not express detectable level of AR,27 these results suggest that PRMT5 may regulate prostate cancer cell growth in an AR-dependent manner. To confirm this, we established Dox-inducible stable cell lines using LNCaP-derived CRPC cell line C4-2 cells that express a higher level of PRMT5 and AR (Supplementary Figure S2), and normal prostate epithelial RWPE-1 cells that do not express detectable AR in the absence of androgen stimulation.28, 29 Again, knockdown of PRMT5 significantly inhibited cell growth in C4-2 cells, but had no effect on cell growth in RWPE-1 cells (Figures 1c and d). Consistent with the growth inhibition in LNCaP and C4-2 cells, PRMT5 knockdown also downregulated AR expression (Figure 1e). As a result, the mRNA level of AR target genes PSA, KLK2 and TMPRSS2 was decreased by PRMT5 knockdown30 (Figure 1f). To further confirm that AR mediates the effect of PRMT5 on the regulation of cell growth, we performed a rescue experiment by expressing FLAG-AR under the control of a CMV promoter, and observed that overexpressed FLAG-AR completely abolished the growth inhibition induced by PRMT5 knockdown (Figures 1g and h). Similar results were obtained when the LNCaP stable cell line was used and the target gene expression was partially rescued (Supplementary Figure S3). Thus, AR downregulation is likely responsible for the growth inhibition induced by PRMT5 knockdown.

PRMT5 regulates prostate cancer cell growth in an AR-dependent manner. (a–d) Induction of PRMT5 knockdown by doxycycline (Dox+) inhibited cell proliferation in AR-expressing LNCaP and C4-2 cells but not in DU145 and RWPE-1 cells that do not express AR. (e) PRMT5 knockdown induced by Dox decreased AR expression in LNCaP and C4-2 stable cell lines. (f) Knockdown of PRMT5 in LNCaP-shPRMT5 cells reduced the mRNA level of the indicated AR target genes measured by qRT-PCR. (g) Restored cell growth by exogenous expression of FLAG-AR in LNCaP cells transiently co-transfected with SC, or pLKO-Tet-On-shPRMT5 (KD) in combination with pFLAG-CMV (Vec) or pFLAG-CMV-AR (AR). (h) Representative Western blots from g to verify the expression of FLAG-AR and the knockdown of PRMT5. *P<0.05; **P<0.01; and ***P<0.001.
Recently, a PRMT5-specific small molecule inhibitor Compound 5 (named here as BLL3.3) has been identified.31 To determine whether inhibition of PRMT5 by BLL3.3 can recapitulate the effect of PRMT5 knockdown in prostate cancer cells, we treated LNCaP cells with BLL3.3, and observed that the growth of LNCaP cells and the expression of AR were significantly inhibited (Supplementary Figures S4a and b). No inhibitory effect was observed when DU145 and RWPE-1 cells were similarly treated with BLL3.3 (Supplementary Figures S4c and d). These results provide additional evidence that the enzymatic activity of PRMT5 is required for AR expression and cell growth in prostate cancer cells.
AR is an epigenetic target of PRMT5 in prostate cancer cells
To determine how PRMT5 regulates AR expression, we examined the effect of PRMT5 knockdown on AR transcription by performing quantitative real-time PCR (qRT-PCR), and observed that transient knockdown of PRMT5 decreased the mRNA level of AR by ~50% (Figure 2a). As PRMT5 may regulate AR transcription epigenetically or indirectly via the regulation of AR transcriptional regulators, we examined the effect of PRMT5 knockdown on the AR-Luciferase reporter gene (AR-Luc) activity, and observed that PRMT5 knockdown had no impact on the AR-Luc activity (Figure 2b). This result suggests that a native chromatin status is required for the downregulation of AR by PRMT5 knockdown. Thus it is likely through epigenetic control of AR transcription. Indeed, the symmetric dimethylation status of H4R3 was significantly enriched on the proximal promoter region of the AR gene when compared with H3R8 and H2AR3 (Figure 2c), despite that all three antibodies can efficiently immunoprecipitate histones H4, H3 and H2A (Supplementary Figure 5). Knockdown of PRMT5 exhibited a greater inhibitory effect on the methylation status of H4R3 (Figure 2d), but a lesser effect on H3R8 and H2AR3 (Supplementary Figure S6). Consistent with this, knockdown of PRMT5 reduced the binding of PRMT5 to the proximal promoter region of the AR gene (Figure 2e), and decreased the level of H4R3me2s on the AR promoter region (Figure 2f). Further, treatment of LNCaP cells with the PRMT5 inhibitor BLL3.3 also decreased the level of AR and H4R3me2s (Supplementary Figure S4b). Taken together, these results demonstrate that PRMT5 epigenetically activates AR transcription by symmetrically dimethylating H4R3.

Epigenetic activation of AR transcription by PRMT5 in LNCaP cells. (a) Transient knockdown of PRMT5 (KD) reduced AR mRNA level when compared with SC. (b) Transient knockdown of PRMT5 had no effect on the AR-luciferase reporter gene (AR-Luc) activity. (c) Enrichment of H4R3me2s, but not H3R8me2s and H2AR3me2s, on the proximal promoter region of the AR gene in LNCaP cells. (d) Transient knockdown of PRMT5 reduced symmetric dimethylation of H4R3 (H4R3me2s). (e) Knockdown of PRMT5 induced by doxycycline (Dox+) reduced PRMT5 binding to the proximal promoter region of the AR gene when compared with cells without Dox (Dox−). (f) Knockdown of PRMT5 induced by doxycycline (Dox+) reduced the enrichment of H4R3me2s on the proximal promoter region of the AR gene when compared with cells without Dox (Dox−).
PRMT5 interacts with Sp1 and Brg1 on the AR promoter
To determine how PRMT5 is recruited to the AR promoter, we examined whether PRMT5 interacts with Sp1, the major and only well-characterized transcription factor that positively regulates AR transcription in prostate cancer cells.32, 33 Indeed, Sp1 was co-immunoprecipitated with PRMT5 from LNCaP cells (Figure 3a). Because both H3R8me2s and H4R3me2s are associated with the activation of target gene expression when PRMT5 is associated with the ATP-dependent chromatin-remodeling enzyme Brg1,34, 35 we performed co-immunoprecipitation and found that Brg1 was also co-immunoprecipitated with PRMT5 from LNCaP cells (Figure 3b). To substantiate this finding, we established a Dox-inducible Sp1 knockdown cell line (LNCaP-shSp1) and confirmed that knockdown of Sp1 indeed repressed AR expression (Figure 3d). Significantly, knockdown of Sp1 in this cell line not only abolished the binding of Sp1 to the proximal promoter region of the AR gene (Figure 3d), but also abolished the binding of PRMT5 (Figure 3e) as well as reduced the binding of Brg1 to the same region (Figure 3f). These results together suggest that Sp1, PRMT5 and Brg1 form a complex on the AR proximal promoter region to activate AR transcription.

PRMT5 interacts with Sp1 and Brg1 on the proximal promoter region of the AR gene in LNCaP cells. (a) Co-immunoprecipitation of Sp1 with PRMT5. (b) Co-immunoprecipitation of Brg1 with PRMT5. (c) Knockdown of Sp1 induced by doxycycline (Dox+) reduced AR expression in Dox-inducible stable cell line LNCaP-shSp1. (d–f) Dox-induced knockdown of Sp1 reduced the binding of Sp1, PRMT5 and Brg1 to the same proximal promoter region of the AR gene. *P<0.05 and ***P<0.001.
PRMT5 is overexpressed in human prostate cancer tissues and correlates with AR expression
Next, we examined the expression level of PRMT5 in a human prostate cancer tissue microarray (TMA) consisting of 32 benign prostatic hyperplasia (BPH) tissues and 40 prostate cancer tissues (20 with Gleason score 6 and 20 with Gleason score⩾7), and found that PRMT5 expression was significantly higher in prostate cancer tissues than BPH tissues (Figure 4a). Although there is no statistically significant difference in the expression scores between prostate cancer tissues with Gleason score 6 and those with Gleason score 7 and above, 60% of prostate cancer tissues with Gleason score 7 and above showed moderate to high expression (total expression score 40–60) of PRMT5 whereas 40% of prostate cancer tissues Gleason score 6 had similar expression of PRMT5. Because PRMT5 subcellular localization appears to be an important determinant of cell fate,36, 37 we compared the expression level of PRMT5 in both the cytoplasm and the nucleus and observed that some cells showed more nuclear or cytoplasmic localization of PRMT5. However, there was no significant difference in PRMT5 subcellular localization in either BPH tissues or prostate cancer tissues (Supplementary Figure S7). To analyze the correlation between AR and PRMT5 expression, we examined the expression of AR from the same TMA. In fact, PRMT5 expression in the nucleus correlated positively with AR expression in prostate tissues (Figures 4b and c). We also retrieved data from Oncomine that have >60 cases in each study, and found that PRMT5 expression correlated with AR at the transcript level in prostate cancer tissues (Figure 4d). Thus, it is likely that nuclear-localized PRMT5 may activate AR transcription in prostate tissues.

PRMT5 expression correlates positively with AR expression in prostate cancer. (a) Shown are representative immunohistochemistry staining images (magnification × 400) of PRMT5 in benign tissue (N5), Gleason 6 prostate cancer tissue (6T1) and Gleason 7 prostate cancer tissue (7T8). The total expression score of PRMT5 is significantly higher in prostate cancer tissues (PCa) when compared with BPH. Scale bar, 30 μm. (b) PRMT5 expression correlates positively with AR expression at the protein level in the same TMA from a. (c) Representative images of PRMT5 and AR expression from serial sections of prostate cancer tissues. The upper panels show higher expression of both PRMT5 and AR in the nucleus and the lower panels show weaker expression of both PRMT5 and AR in the nucleus. Scale bar, 30 μm. (d) PRMT5 expression correlates positively with AR expression at the transcript level. The data were retrieved from Oncomine database.
PRMT5 knockdown inhibits AR expression and suppresses the growth of xenograft tumors in mice
To determine whether PRMT5 expression is necessary for the growth of xenograft tumors in mice, we used Dox-inducible stable cell lines expressing PRMT5 shRNA (LNCaP-shPRMT5) or SC (LNCaP-SC) to establish xenograft tumors in nude mice. As shown in Figure 5a, knockdown of PRMT5 completely suppressed the growth of LNCaP xenograft tumors. In fact, tumor growth in 8 out of 10 Dox-treated mice were completely suppressed. There was no significant difference in the growth of tumors derived from LNCaP-SC regardless of the Dox status (Figure 5b). The expression level of PRMT5 and AR was also downregulated in Dox-treated residual tumor nodules derived from LNCaP-shPRMT5 when compared with Dox-untreated (Figure 5c). Similar expression of PRMT5 and AR was observed in SC control tumors regardless of the Dox status (Figure 5d). These results demonstrate that PRMT5 is required for the growth of xenograft tumors in mice.

Knockdown of PRMT5 suppresses the growth of xenograft tumors in mice. (a) LNCaP-shPRMT5 cells were implanted subcutaneously into the right lower flanks of 10 nude mice per group, and the tumor growth was monitored twice weekly in Dox-treated (Dox+) and untreated (Dox−) mice. (b) Similar experiment was performed as described in a for LNCaP-SC cell line. (c and d) Representative images showing inhibition of PRMT5 and AR expression in Dox-treated tumor nodules. No effect on PRMT5 and AR expression in xenograft tumors derived from LNCaP-SC was observed. Scale bar: 10 μm.
Discussion
AR signaling is a critical determinant of prostate cancer development and progression. Many studies have characterized how AR transcriptional activity is modulated by its co-activators and co-repressors.19, 21, 24 However, how the transcription of AR itself is regulated, particularly at the epigenetic level, remains poorly understood. Here, we provide evidence showing that PRMT5 is a novel epigenetic activator of AR transcription in prostate cancer. First, knockdown of PRMT5 or inhibition of PRMT5 by a small molecule inhibitor specifically inhibited the growth of prostate cancer cells in an AR-dependent manner. Second, knockdown of PRMT5 specifically inhibited AR transcription. Third, PRMT5 binds to the proximal promoter region of the AR gene along with Sp1 and Brg1. Fourth, H4R3me2s is highly enriched on the proximal promoter region of the AR gene. Fifth, PRMT5 is highly expressed in prostate cancer tissues and its expression correlates positively with AR expression at both mRNA and protein levels. Finally, depletion of PRMT5 expression completely suppressed the growth of LNCaP xenograft tumors in mice by downregulating AR expression.
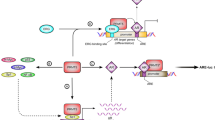
Transcriptional regulation of gene expression is a tightly regulated process that involves the participation of multiple transcriptional regulatory proteins such as transcription factors, co-activators and co-repressors as well as chromatin-remodeling enzymes. Consistent with the fact that Sp1 is the major and well-characterized transcription factor that activates AR transcription in prostate cancer cells,33, 38 we indeed confirmed that Sp1 binds to the AR promoter and regulates AR expression in LNCaP cells. Because PRMT5 interacts with Sp1 and Brg1 and because Sp1 knockdown also reduces the binding of PRMT5 to the AR promoter, we suggest that Sp1 may recruit PRMT5 to the AR promoter. Interestingly, Brg1, an ATP-dependent chromatin remodeler,39 was also recruited to the AR promoter through its interaction with PRMT5. This finding suggests that PRMT5-mediated H4R3 dimethylation could also activate transcription of target genes such as AR when Brg1 is recruited to the promoters (Figure 6), though PRMT5 generally represses transcription of target genes. Interestingly, PRMT5-mediated H3R8 dimethylation is also involved in transcriptional activation of target genes when Brg1 is recruited to the target gene promoters.34, 35 Although this manuscript was in preparation, a recent report showed that PRMT5 can dimethylate H4R3 and H3R8 to regulate the expression of the protein kinase FLT3 in acute myeloid leukemia cells via two distinct pathways.40 Thus, dimethylation of either H3R8 or H4R3 by PRMT5 may permit ATP-dependent chromatin remodeling, leading to activation or repression of target gene transcription. Given that PRMT5 and Brg1 also cooperate to repress transcription of target genes41, 42, 43 and that AR transcription is subjected to the regulation of DNA methylation and histone lysine methylation,44 it is likely that AR transcription is subjected to a high order of epigenetic regulation. Future studies to gain insight into the epigenetic regulation of AR may offer new opportunities to develop novel targeting strategies to inhibit or even eliminate AR expression. Because PRMT5 may exhibit an opposite role in the cytoplasm and nucleus in cells,9, 25 it remains to be determined whether cytoplasmic- and nuclear-localized PRMT5 may have distinct effects on the transcription of AR.

Proposed model for epigenetic activation of AR transcription by PRMT5.
The present finding has significant clinical implications due to the central role of AR in prostate cancer development and progression. Our findings here, together with a previous study showing that PRMT5 may form a complex with MEP50 and AR to modulate the transcriptional activity of AR,45 raise an interesting possibility that targeting PRMT5 may have a dual effect on both the expression and activity of AR. Thus, PRMT5 may be an ideal target for development of novel therapeutics. As radiotherapy in combination with adjuvant ADT is the current standard treatment for locally advanced prostate cancer, combining radiotherapy with PRMT5 targeting may be an alternative approach. Perhaps targeting AR expression by inhibiting PRMT5 may avoid some adverse effects often seen with ADT. It is worth noting that PRMT5 also regulates the expression of AR in the CRPC line C4-2. As AR reactivation is the major mechanism underlying the development of CRPC13, 14 and the resistance to the next-generation anti-androgen therapy,17, 18 targeting PRMT5 alone or in combination with other AR-targeting agents may exhibit a better treatment efficacy than the existing treatments. Given that two small molecule inhibitors of PRMT5 have been developed,31, 40, 46 preclinical evaluation of these inhibitors alone or in combination with radiotherapy or other AR-targeting agents may lead to the development of novel therapeutic approaches for prostate cancer treatment.
Materials and methods
Cell lines and culture
Prostate cancer cell lines LNCaP, DU145, and PC-3 as well as RWPE-1 cells were purchased from ATCC (Manassas, VA, USA) and C4-2 cells were purchased from M.D. Anderson Cancer Center (Houston, TX, USA). All frozen stock received were immediately expanded and aliquots were prepared and stored in liquid nitrogen for future use, and cells were maintained for no longer than 3 months as described previously.30, 47 Cell line authentication was performed by IDEXX BioResearch (IMPACT I). The establishment of stable cell lines was described previously.26, 30
Plasmid construction
The pLKO-Tet-On plasmid for expressing shRNA was obtained from Addgene (Cambridge, MA, USA),48 and the two shRNA sequences that target 5′-GCCCAGTTTGAGATGCCTTAT-3′ (#1577) and 5′-CCCATCCTCTTCCCTATTAAG-3′ (#1832) for PRMT5 knockdown and that target 5′-CCACTCCTTCAGCCCTTATTA-3′ (#2310) for Sp1 knockdown were selected for constructing pLKO-Tet-On-shPRMT5 and pLKO-Tet-On-shSp1 as described previously.30 The pLKO-Tet-On-SC and pFLAG-CMV-AR were constructed before.30 The AR promoter luciferase reporter gene construct and the PSA promoter luciferase reporter gene construct were kindly provided by Dr Donald Tindall. pFLAG-CMV-AR was made by subcloning the AR cDNA into pFLAG-CMV vector. All plasmids were confirmed by DNA sequencing.
Cell proliferation assay
The cell proliferation assay was performed using MTT reagent (Sigma, St Louis, MO, USA). For transient transfection experiments, LNCaP, DU145 or PC-3 cells (4 × 103) were seeded in 48-well plates for 24 h, and then transiently transfected with pLKO-Tet-On-shPRMT5 (#1577) or the SC control using FuGENE HD or FuGENE 6 (Promega, Madison, WI, USA) for 96 h after the transfection. For MTT analysis, cell medium was removed and 70 μl of MTT solution (0.5 mg/ml) was added into each well and incubated at 37 °C for 4 h. At the end of incubation, MTT solution was removed and 130 μl of DMSO was added into each well and incubated at 37 °C for another 10 min. The plates were then read at 560 nm with TECAN Microplate Reader (TECAN, Mannedorf, Switzerland). For LNCaP, DU145, C4-2 and RWPE-1 stable cell lines, similar procedure was followed except that Dox was added at 1 μg/ml to induce PRMT5 knockdown during culture. At least three independent experiments were performed and the mean±s.d. was presented. Student’s t-test was performed to determine the statistical significance. The effect of PRMT5 inhibitor BLL3.3 on the growth of LNCaP, DU145 and RWPE-1 cells was similarly determined by MTT.
Soft-agar growth assay
The soft-agar growth assay to measure anchorage-independent proliferation of LNCaP cells was performed by using the 96-well plate format as described previously.49 Briefly, LNCaP cells were transfected with pLKO-Tet-On-shPRMT5 (#1577) or pLKO-Tet-On-SC for 24 h, and then 2.5 × 103 cells were added into the middle layer agar. Dox was added into each layer of soft agar at 1 μg/ml to induce the expression of shRNAs. The plates were incubated at 37 °C, 5% CO2 for 7 days. To quantify the colony-formation efficiency, 16 μl of AlamerBlue Cell Viability Reagent (Invitrogen, Carlsbad, CA, USA) was added into each well and incubated at 37 °C for another 4 h. Fluorescence intensity was measured at 570EX nm/600EM nm using Multi-Mode Microplate Reader (BioTek, Winooski, VT, USA). Experiments were performed in triplicate, and results from three independent experiments were analyzed and presented as mean±s.d. Student’s t-test was used to determine the statistical significance.
qRT-PCR and western blotting
To determine the effect of PRMT5 knockdown on AR expression, PRMT5 were transiently or stably knocked down in LNCaP cells for 96 h, and total RNA was isolated using TRIzol Reagent (Thermo Fisher Scientific, Waltham, MA, USA). One microgram of total RNA was used for reverse transcription using the High Capacity cDNA Reverse Transcription Kit (Promega) according to manufacturer’s instruction. The qRT-PCR analysis of AR or AR target genes (PSA, KLK2, TMPRSS2) was performed as described previously.30 Antibodies against AR (SC-816, Santa Cruz, CA, USA), PRMT5 (07-405, Millipore, Billerica, MA, USA), PSA (1984-1, Epitomics, Burlingame, CA, USA), FLAG (Sigma, F-1804), Sp1 (ab13370, Abcam, Cambridge, MA, USA), H4R3me2s (Abcam, ab5823), H3R8me2s (Abcam, ab130740), H2AR3me2s (Abcam, ab22397), and Brg1 (Abcam, ab110641) were used for western blotting analysis.
Chromatin immunoprecipitation assay
The LNCaP stable cell line or parental cells were cultured in the presence or absence of Dox (1 μg/ml) for 96 h. At the end of induction, 270 μl of 37% formaldehyde was added into each dish and incubated at room temperature for 10 min. Then 1 ml of 1.25 M glycine was added to stop the cross-linking reaction. Cells were then harvested, resuspended in 1 ml of immunoprecipitation buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% TritonX-100, 0.5% NP-40, 0.5 mM DTT, and protease and phosphatase inhibitors), and finally sonicated (Branson Sonifier250set, Wilmington, NC, USA) to prepare sheared chromatin. Antibodies against PRMT5 (Millipore, 07-405), Sp1 (Santa Cruz, SC7824), Brg1 (Abcam, ab110641), H4R3me2s (Abcam, ab5823), H3R8me2s (Abcam, ab130740), H2AR3me2s (Abcam, ab22397) and IgG (Santa Cruz, SC2027) were used to immunoprecipiate protein-DNA complexes for isolation of PCR-ready DNA using the Fast ChIP protocol described previously.50 The co-immunoprecipitated proximal promoter region of AR (−493 to −226) was quantified by qRT-PCR. Results were normalized to the IgG control and are presented as mean±s.d. from three independent experiments. Student’s t-test was used to determine the statistical significance.
Co-immunoprecipitation of PRMT5 with Sp1 and Brg1
Total cell lysate of LNCaP cells was prepared in immunoprecipitation buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% TritonX-100, 0.5% NP-40, 0.5 mM DTT, and protease and phosphatase inhibitors) for co-immunoprecipitation. Anti-PRMT5 antibody or IgG was used to immunoprecipitate PRMT5 from 500 μg of total lysate, and co-immunoprecipitated Sp1 and Brg1 was detected with Sp1 and Brg1 antibodies.
Expression of PRMT5 and AR and the analysis of their correlation in prostate cancer tissues
A TMA consisting of 32 BPH tissues and 40 prostate cancer tissues (20 with Gleason score 6 and 20 with Gleason score⩾7) was used for immunohistochemistry analysis of PRMT5 and AR expression. Briefly, paraffin section of the TMA was deparaffinized in xylene and rehydrated in graded ethanol, followed by inactivation of endogenous peroxidase activity in 3% hydrogen peroxide for 10 min. Antigen retrieval was performed by heating slides in 10 mM Tris-HCl (pH 10) for 30 min in microwave. After three washes with phosphate-buffered saline containing 0.1% Tween 20 (PBST), slides were blocked in 5% non-fat milk in PBST at room temperature for 1 h. The primary antibodies against PRMT5 or AR was incubated at 4 °C overnight, followed by three washes with PBST and incubation with HRP-conjugated anti-rabbit secondary antibodies (Amersham, Pittsburgh, PA, USA) at room temperature for 1 h. The signal was developed with diaminobenzidine for 10 min, and sections were counterstained with hematoxyline. The semi-quantification of PRMT5 and AR expression was performed as described previously with slight modifications.51 The intensity was scored as 0 (no expression), 1 (low expression), 2 (moderate expression) and 3 (high expression), and the percentage of cells showing the expression was scored ranging from 0 to 10 with 10 as the highest percentage (100%). The expression score for cytoplasmic- and nuclear-localized PRMT5 was respectively determined by the intensity score times the percentage (0–30), and the total expression score is the sum of the cytoplasmic and nuclear expression scores (0–60). The unpaired t-test was used to determine the statistical significance of the total mean expression score between BPH and prostate cancer tissues, and paired t-test was used to determine the difference in expression scores between cytoplasmic-localized PRMT5 and nuclear-localized PRMT5. The same semi-quantification method was used for AR expression in the nucleus.
To determine the correlation between the expression of PRMT5 and AR in the nucleus in prostate tissues, their nuclear expression scores were used for Pearson’s analysis. To determine the correlation of PRMT5 and AR expression at the transcript level, we retrieved their expression data from 8 studies that have >60 tissues from Oncomine. The statistic Q was calculated to test the homogeneity of effect sizes across studies for each of the three methods (Pearson’s, Spearman’s and Kendall’s),52 and it was found that the effect sizes across studies were not homogeneous (all with P-value<1e−12). Therefore, we used a random-effects model for the meta-analysis of each method.53
Xenograft tumor growth in nude mice
Animal experiments were approved by the Purdue University Animal Care and Use Committee. Male athymic nude mice (5–7 week old) were purchased from Harlan Laboratories (Indianapolis, IN, USA), and 3 × 106 cells of established stable cell lines that inducibly express PRMT5 shRNA or SC were co-injected subcutaneously into the right lower flank of 20 mice with Matrigel (1:1 in volume). Assuming that PRMT5 knockdown can reduce tumor volume by 30% and that standard deviation within each group is about 25% of the mean tumor volume, a sample size of 10 male mice per group will have over 80% power to detect a 30% difference between the two groups at alpha level 0.05. Mice were randomly divided into two groups (10 mice/group) for each stable cell line by using Excel-based randomization method, and treated with Dox (1 mg/ml in drinking water) or without Dox (drinking water only). Tumor growth was monitored twice weekly, and tumor volume was calculated using ½ × L × W × H without using blinding method. At the end of experiments, tumors were resected and formalin fixed, and paraffin embedded. Immunohistochemistry analysis of PRMT5 and AR expression was similarly performed as described above. We used the following linear mixed model to model the j-th observed xenograft tumor volume of i-th mouse, that is, yij, assuming cubic polynomial growth of tumors over time,

Where, tj is the number of days after implantation for the j-th observation, Di indicates whether the i-th subject is under Dox. The random-effects are independent, and the errors of the same subject are assumed to follow a first-order continuous autoregressive model.
To evaluate the effects of Dox on the tumor growth, we are subject to test the H0: δ0=δ1=δ2=δ3=0 against Hα: at least one of δ0, δ1, δ2, δ3 is not zero.
We used the likelihood ratio test (χ2-test) to conduct the hypothesis tests.
For PRMT5 knockdown, the P-value is 1.9305 × 10−6. For SC, the P-value is 0.1670. Error bar, s.e.m.
References
Bedford M, Clarke S . Protein arginine methylation in mammals: who, what, and why. Mol Cell 2009; 33: 1–13.
Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, Pestka S . Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther 2007; 113: 50–87.
Cho EC, Zheng S, Munro S, Liu G, Carr SM, Moehlenbrink J et al. Arginine methylation controls growth regulation by E2F-1. EMBO J 2012; 31: 1785–1797.
Gu Z, Gao S, Zhang F, Wang Z, Ma W, Davis RE . Protein arginine methyltransferase 5 is essential for growth of lung cancer cells. Biochem J 2012; 446: 235–241.
Powers MA, Fay MM, Factor RE, Welm AL, Ullman KS . Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Res 2011; 71: 5579–5587.
Wang L, Pal S, Sif S . Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol Cell Biol 2008; 28: 6262–6277.
Wei TY, Juan CC, Hsia JY, Su LJ, Lee YC, Chou HY et al. PRMT5 is a potential oncoprotein that upregulates G1 cyclins/CDKs and the PI3K/AKT signaling cascade. Cancer Sci 2012; 103: 1640–1650.
Yan F, Alinari L, Lustberg ME, Martin LK, Cordero-Nieves HM, Banasavadi-Siddegowda Y et al. Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res 2014; 74: 1752–1765.
Karkhanis V, Hu YJ, Baiocchi RA, Imbalzano AN, Sif S . Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem Sci 2011; 36: 633–641.
Stopa N, Krebs JE, Shechter D . The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci 2015; 72: 2041–2059.
Siegel RL, Miller KD, Jemal A . Cancer statistics, 2015. CA Cancer J Clin 2015; 65: 5–29.
Dehm SM, Tindall DJ . Molecular regulation of androgen action in prostate cancer. J Cell Biochem 2006; 99: 333–344.
Dutt SS, Gao AC . Molecular mechanisms of castration-resistant prostate cancer progression. Future Oncol 2009; 5: 1403–1413.
Ryan CJ, Tindall DJ . Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. J Clin Oncol 2011; 29: 3651–3658.
Chandrasekar T, Yang JC, Gao AC, Evans CP . Targeting molecular resistance in castration-resistant prostate cancer. BMC Med 2015; 13: 206.
Guo Z, Qiu Y . A new trick of an old molecule: androgen receptor splice variants taking the stage?!. Int J Biol Sci 2011; 7: 815–822.
Giacinti S, Bassanelli M, Aschelter AM, Milano A, Roberto M, Marchetti P . Resistance to abiraterone in castration-resistant prostate cancer: a review of the literature. Anticancer Res 2014; 34: 6265–6269.
Nakazawa M, Antonarakis ES, Luo J . Androgen receptor splice variants in the era of enzalutamide and abiraterone. Hormone Cancer 2014; 5: 265–273.
Agoulnik IU, Weigel NL . Androgen receptor coactivators and prostate cancer. Adv Exp Med Biol 2008; 617: 245–255.
Cai C, Yuan X, Balk SP . Androgen receptor epigenetics. Translat Androl Urol 2013; 2: 148–157.
Chmelar R, Buchanan G, Need EF, Tilley W, Greenberg NM . Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int J Cancer 2007; 120: 719–733.
Gao L, Alumkal J . Epigenetic regulation of androgen receptor signaling in prostate cancer. Epigenetics 2010; 5: 100–104.
Jeronimo C, Bastian PJ, Bjartell A, Carbone GM, Catto JW, Clark SJ et al. Epigenetics in prostate cancer: biologic and clinical relevance. Eur Urol 2011; 60: 753–766.
Wang L, Hsu CL, Chang C . Androgen receptor corepressors: an overview. Prostate 2005; 63: 117–130.
Gu Z, Li Y, Lee P, Liu T, Wan C, Wang Z . Protein arginine methyltransferase 5 functions in opposite ways in the cytoplasm and nucleus of prostate cancer cells. PLoS One 2012; 7: e44033.
Zhang HT, Zhang D, Zha ZG, Hu CD . Transcriptional activation of PRMT5 by NF-Y is required for cell growth and negatively regulated by the PKC/c-Fos signaling in prostate cancer cells. Biochim Biophys Acta 2014; 1839: 1330–1340.
Tilley WD, Wilson CM, Marcelli M, McPhaul MJ . Androgen receptor gene expression in human prostate carcinoma cell lines. Cancer Res 1990; 50: 5382–5386.
Bello D, Webber MM, Kleinman HK, Wartinger DD, Rhim JS . Androgen responsive adult human prostatic epithelial cell lines immortalized by human papillomavirus 18. Carcinogenesis 1997; 18: 1215–1223.
Mirochnik Y, Veliceasa D, Williams L, Maxwell K, Yemelyanov A, Budunova I et al. Androgen receptor drives cellular senescence. PLoS One 2012; 7: e31052.
Hsu CC, Hu CD . Transcriptional activity of c-Jun is critical for the suppression of AR function. Mol Cell Endocrinol 2013; 372: 12–22.
Alinari L, Mahasenan KV, Yan F, Karkhanis V, Chung JH, Smith EM et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015; 125: 2530–2543.
Faber PW, van Rooij HC, Schipper HJ, Brinkmann AO, Trapman J . Two different, overlapping pathways of transcription initiation are active on the TATA-less human androgen receptor promoter. The role of Sp1. J Biol Chem 1993; 268: 9296–9301.
Tilley WD, Marcelli M, McPhaul MJ . Expression of the human androgen receptor gene utilizes a common promoter in diverse human tissues and cell lines. J Biol Chem 1990; 265: 13776–13781.
Dacwag CS, Ohkawa Y, Pal S, Sif S, Imbalzano AN . The protein arginine methyltransferase Prmt5 is required for myogenesis because it facilitates ATP-dependent chromatin remodeling. Mol Cell Biol 2007; 27: 384–394.
LeBlanc SE, Konda S, Wu Q, Hu YJ, Oslowski CM, Sif S et al. Protein arginine methyltransferase 5 (Prmt5) promotes gene expression of peroxisome proliferator-activated receptor gamma2 (PPARgamma2) and its target genes during adipogenesis. Mol Endocrinol 2012; 26: 583–597.
Tee WW, Pardo M, Theunissen TW, Yu L, Choudhary JS, Hajkova P et al. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev 2010; 24: 2772–2777.
Teng Y, Girvan AC, Casson LK, Pierce Jr WM, Qian M, Thomas SD et al. AS1411 alters the localization of a complex containing protein arginine methyltransferase 5 and nucleolin. Cancer Res 2007; 67: 10491–10500.
Hay CW, Hunter I, MacKenzie A, McEwan IJ . An Sp1 modulated regulatory region unique to higher primates regulates human androgen receptor promoter activity in prostate cancer cells. PLoS One 2015; 10: e0139990.
Hargreaves DC, Crabtree GR . ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res 2011; 21: 396–420.
Tarighat SS, Santhanam R, Frankhouser D, Radomska HS, Lai H, Anghelina M et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 2015; 30: 789–799.
Pal S, Yun R, Datta A, Lacomis L, Erdjument-Bromage H, Kumar J et al. mSin3A/histone deacetylase 2- and PRMT5-containing Brg1 complex is involved in transcriptional repression of the Myc target gene cad. Mol Cell Biol 2003; 23: 7475–7487.
Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S . Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol 2004; 24: 9630–9645.
Seth-Vollenweider T, Joshi S, Dhawan P, Sif S, Christakos S . Novel mechanism of negative regulation of 1,25-dihydroxyvitamin D3-induced 25-hydroxyvitamin D3 24-hydroxylase (Cyp24a1) Transcription: epigenetic modification involving cross-talk between protein-arginine methyltransferase 5 and the SWI/SNF complex. J Biol Chem 2014; 289: 33958–33970.
Liu C, Wang C, Wang K, Liu L, Shen Q, Yan K et al. SMYD3 as an oncogenic driver in prostate cancer by stimulation of androgen receptor transcription. J Natl Cancer Institute 2013; 105: 1719–1728.
Hosohata K, Li P, Hosohata Y, Qin J, Roeder RG, Wang Z . Purification and identification of a novel complex which is involved in androgen receptor-dependent transcription. Mol Cell Biol 2003; 23: 7019–7029.
Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 2015; 11: 432–437.
Deng X, Elzey BD, Poulson JM, Morrison WB, Ko SC, Hahn NM et al. Ionizing radiation induces neuroendocrine differentiation of prostate cancer cells in vitro in vivo and in prostate cancer patients. Am J Cancer Res 2011; 1: 834–844.
Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 2009; 8: 498–504.
Ke N, Albers A, Claassen G, Yu DH, Chatterton JE, Hu X et al. One-week 96-well soft agar growth assay for cancer target validation. Biotechniques 2004; 36: 826–828.
Nelson JD, Denisenko O, Sova P, Bomsztyk K . Fast chromatin immunoprecipitation assay. Nucleic Acids Res 2006; 34: e2.
Wang J, Place RF, Huang V, Wang X, Noonan EJ, Magyar CE et al. Prognostic value and function of KLF4 in prostate cancer: RNAa and vector-mediated overexpression identify KLF4 as an inhibitor of tumor cell growth and migration. Cancer Res 2010; 70: 10182–10191.
Hedges LV, Olkin I . Statiscal Methods for Meta-Analysis. Academic Press: Orlando, FL, USA, 1985.
Hedges LV, Vevea JL . Fixed- and random-effects models in meta-analysis. Psychol Methods 1998; 3: 486–504.
Acknowledgements
This study was partially supported by grants from U.S. Army Medical Research Acquisition Activity, Prostate Cancer Research Program (PC11190, PC120512, and PC150697) to C-D Hu, and Purdue University Center for Cancer Research Small Grants. DNA sequencing and animal experiments were conducted in Genomic Core facility and the Biological Evaluation Shared Resource facility, respectively, supported by NCI CCSG CA23168 to Purdue University Center for Cancer Research. Genbao Shao and Huan-Tian Zhang were visiting scholars supported by the China Scholarship Council.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Deng, X., Shao, G., Zhang, HT. et al. Protein arginine methyltransferase 5 functions as an epigenetic activator of the androgen receptor to promote prostate cancer cell growth. Oncogene 36, 1223–1231 (2017). https://doi.org/10.1038/onc.2016.287
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.287
This article is cited by
-
PRMT5-mediated homologous recombination repair is essential to maintain genomic integrity of neural progenitor cells
Cellular and Molecular Life Sciences (2024)
-
PRMT5 is a therapeutic target in choroidal neovascularization
Scientific Reports (2023)
-
The cancer testis antigen TDRD1 regulates prostate cancer proliferation by associating with the snRNP biogenesis machinery
Oncogene (2023)
-
USP7- and PRMT5-dependent G3BP2 stabilization drives de novo lipogenesis and tumorigenesis of HNSC
Cell Death & Disease (2023)
-
The PRMT5-LSD1 axis confers Slug dual transcriptional activities and promotes breast cancer progression
Journal of Experimental & Clinical Cancer Research (2022)
