
Key Points
-
The complement system is an important part of the innate immune system that participates in tissue injury in a number of inflammatory and autoimmune diseases.
-
Polymorphisms in genes that encode complement proteins involved in activation or regulation of the system are recognized as triggers of diseases such as atypical haemolytic uraemic syndrome and age-related macular degeneration.
-
Modulation of complement activation could be beneficial in reducing tissue injury or even preventing the onset of such diseases.
-
Currently, only a few agents that are known to inhibit complement activation are approved for clinical use for narrow indications. These include an antibody to C5 (eculizumab) aimed at the treatment of paroxysmal nocturnal haemoglobinuria, Serping1 concentrate as a replacement therapy in hereditary angioedema and pooled immunoglobulin G preparations for intravenous use in some autoimmune diseases.
-
Recent literature shows that there is a growing interest in developing new and potent agents that target complement proteins and products of activation. Some examples include humanized monoclonal antibodies to complement proteins, activation products or receptors, synthetic molecules that target complement proteins or receptors, constructs of naturally occurring complement regulators and receptors modified to enhance tissue-directed activity and prolong half-life.
-
Many new agents have been tested in in vitro and in vivo animal models, generally with encouraging results. Some agents have reached clinical trials for indications such as hereditary angioedema, age-related macular degeneration, rheumatoid arthritis and psoriasis.
-
Because of a better understanding of the role of complement in many pathological states, targeting its activation holds promise as an innovative way to approach therapeutic intervention in many patients.
Abstract
The complement system is an essential component of innate immunity that has been more recently recognized as an unexpected player in various pathological states. These include age-related macular degeneration, atypical haemolytic uraemic syndrome, allergy, foetal loss, and axonal and myelin degradation after trauma. Its importance has also been recognized in physiological processes including haematopoietic stem cell homing to the bone marrow, liver regeneration and modulation of adaptive immune responses. Although the complement system has long been known to be involved in autoimmune and inflammatory diseases, few agents that target the complement system are currently approved for clinical use. However, renewed interest in modulating this system in various pathological conditions has emerged, and several agents are now in development.
Similar content being viewed by others
Main
The complement system is comprised of more than 30 fluid phase and cell-associated proteins that act in synergy when needed, to promote inflammation and damage invaders such as microbes or foreign cells1,2,3. Because of this capacity for tissue damage, there are many regulatory proteins that control complement activation and thereby downregulate complement-mediated damage. Individuals lacking complement proteins are rare, although polymorphisms of the proteins leading to altered functional activity as well as abnormalities of the complement control proteins are common. This is demonstrated by the recent discovery of strong associations between polymorphisms in genes encoding complement components and regulatory proteins, and diseases such as atypical haemolytic uraemic syndrome and age-related macular degeneration (AMD)4. A major function of complement is to direct damage to cells, microbes and tissues that are identified as abnormal by a specific antibody. The development of antibodies to one's own tissues, as occurs in many autoimmune diseases, may also lead to tissue-directed inflammation and tissue destruction due to complement activation.
Although the importance of complement in the development of inflammation and in tissue damage in autoimmune diseases has been known for decades5, until recently, little attention has been directed at developing pharmaceuticals that interrupt or dampen complement-mediated responses. The aim of this Review is to discuss some of the disorders in which complement is proven or thought to have a crucial pathogenic role and highlight the various therapeutic complement-targeted approaches that have been proposed or are in development. We focus only on those agents that show promise in clinical settings. More detailed information on some agents that may attract less attention today is provided by past reviews6,7,8.
Complement activation pathways
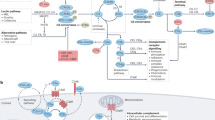
The complement proteins are important elements of the innate immune system that act in a cascade-like system to induce their physiological effects. Three main pathways for activation are recognized: the classical, alternative and lectin pathways (Fig. 1).

Classical pathway: antibody binding to antigen allows for the noncovalent binding of the three-subunit component C1 (composed of C1q and two chains of each C1r and C1s). C1q binds to the Fc part of immunoglobulin, and activation of C1 leads to cleavage of the C1r and C1s chains with resulting protease activity. Activated C1 cleaves C4 and C2 into large (C4b and C2a) and small (C4a and C2b) fragments, and the small fragments are released. C4b binds covalently to the target surface and C2a coordinates with C4b to form an enzymatic complex termed a C3 convertase with the ability to cleave the two-chain molecule C3. C3 is cleaved into a large fragment, C3b, which interacts with the target surface, and a small fragment, C3a, which possesses pro-inflammatory properties. Deposition of C3b on the target surface induces the formation of a C5 convertase (C4b2a3b) which cleaves C5 into C5b and C5a. Again, the larger fragment C5b binds to the target surface, whereas the smaller fragment C5a is released and has potent pro-inflammatory properties. C6, C7, C8 and C9 bind serially to the target surface, C5b, to form the membrane attack complex (MAC; C5b–C9). Alternative pathway: C3 in circulation undergoes slow-rate hydrolysis which exposes its internal thioester group. It then interacts with factor B (B) which is cleaved by the circulating protease factor D (D) to form an initiation phase C3 convertase (iC3Bb). The smaller fragment of factor B, Ba, is released. This C3 convertase cleaves additional C3 into metastable C3b. The exposed thioester group in metastable C3b can interact with nearby microorganisms. Factor B binds to microorganism-bound C3b and is cleaved into Bb and Ba by factor D to lead to an amplification phase C3 convertase (C3bBb). This C3 convertase is stabilized by properdin (P), which increases its half-life, and cleaves additional C3 molecules. Deposition of further C3b molecules leads to the formation of a C5 convertase (C3b2BbP) that cleaves C5 into C5b and C5a. Deposition of C5b triggers the formation of the MAC. Lectin pathway: the lectin pathway is triggered by direct interaction of mannan-binding lectin (MBL) or ficolin with terminal carbohydrates found on microbial glycoproteins (mannose, N-acetyl-glucosamine and fucose). MBL-associated serine proteases (MASPs) (MASP2 having properties analogous to C1s) trigger cleavage of C4 and C2 to form a C3 convertase similar to that of the classical pathway (C4b2a) and activate complement in a manner similar to that seen during classical pathway activation. iC3, hydrolysed C3.
Classical-pathway activation. The classical pathway is usually triggered by the interaction of antigen and specific antibody of the correct isotype or subclass; immunoglobulin M (IgM), IgG3 and IgG1 are the most efficient activators of the classical pathway1,2,3. Certain microorganisms, DNA, C reactive protein, polyanionic molecules and apoptotic bodies can also directly activate the classical pathway. Complement activation leads to the generation of potent pro-inflammatory mediators such as complement proteins C3a and C5a, termed anaphylatoxins. Activation of the late-acting proteins leads to the deposition of the membrane attack complex (MAC; C5b–C9) on the target surface with penetration and polymerization of the terminal component, C9, into the lipid bilayer of a cell envelope. This generates 10 nM pores that cause death of certain target cells and microorganisms through disruption of the target membrane9 (Fig. 1).
Alternative pathway activation. A far more ancient pathway of complement activation is termed the alternative pathway which, unlike the classical pathway, does not rely on antibodies for pathogen recognition1,2,3. The alternative pathway is initiated by the slow hydrolysis of circulating C3, which exposes an internal thioester group, a phenomenon referred to as 'C3 tickover'. Binding of the alternative-pathway-specific proteins factor B, factor D and properdin to hydrolysed C3 or to the complement cleavage fragment C3b leads to further activation of C3. Cleaved C3 in the form of C3b can then interact with polysaccharides or proteins on the surface of microorganisms or endotoxins (bacterial lipopolysaccharides) to initiate alternative-pathway activation and generate the MAC, as occurs during classical pathway activation10 (Fig. 1). Other activators of the alternative pathway include IgG and IgA immune complexes. Recent evidence suggests that properdin, in addition to stabilizing the C3 convertase in the alternative pathway, can initiate alternative-pathway activation on apoptotic cells and certain microorganisms through direct interaction with specific molecular patterns11.
Lectin pathway activation. A third pathway of complement activation, named the lectin pathway, was identified about 20 years ago12. It is triggered by the binding of a C-type lectin, mannan-binding lectin (MBL; also known as mannose-binding lectin) or a related series of proteins termed ficolins (L-ficolin, H-ficolin and M-ficolin)13, to terminal sugars as expressed on glycoproteins or envelope polysaccharides found on the surface of microorganisms. MBL and the ficolins are structurally related to C1q. In the circulation, these proteins associate with MBL-associated serine protease 1 (MASP1), MASP2 and MASP3, and a truncated form of MASP2 termed MAP19. Binding of the MBL–MASP complex to the target organism leads to C4 cleavage by MASP2 and C2 cleavage by MASP1 and MASP2, resulting in the generation of a C3 convertase similar to that of the classical pathway13 (Fig. 1).
Because all three complement activation pathways merge with C3b deposition on a target and C3b is an initiating factor of the alternative pathway, complement activation can be initiated by the classical or lectin pathway and amplified by the alternative pathway. Furthermore, complement activation can occur in vivo in the absence of either C4 or C2 through the so-called bypass pathways14. Therefore, complement activation in vivo cannot simply be viewed as the sole result of any one of the three recognized pathways. Under certain pathological conditions, it is likely that two or even all three complement activation pathways are solicited to induce inflammation and tissue damage. However, in most cases it seems that the alternative pathway is responsible for most of the tissue damage10.
In addition to being the component at which all three activation pathways merge, C3 is a central molecule in the complement system as its cleavage products mediate numerous biological activities (Box 1).
Complement regulation
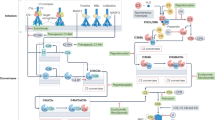
Although complement functions to damage microorganisms or abnormal cells, complement activation can also damage host tissues due to the generation of potent pro-inflammatory molecules, such as the anaphylatoxins and chemotaxins, and the formation of the MAC15. It must therefore be tightly controlled under normal physiological conditions to maintain homeostatic balance. To achieve this, many fluid-phase and cell membrane-bound regulators exist, often with overlapping functions (Fig. 2).

Fluid-phase regulators: the C1 inhibitor serping1 (also known as C1inh) serves as a substrate for the serine proteases C1r and C1s and forms two serping1–C1r–C1s complexes for full C1 inactivation. Serping1 also inactivates mannan-binding lectin (MBL)-associated protease 1 (MASP1) and MASP2 in the lectin pathway, which are structurally and functionally related to C1r and C1s. C4b and C3b (and hydrolysed C3) are inactivated by proteolytic cleavage by factor I, which requires co-factor binding to C4b and C3b; the co-factors are C4b-binding protein (C4BP) for C4b and factor H for C3b. C4BP and factor H further accelerate the decay of the classical and alternative pathway C3 convertases C4b2a and C3bBb, respectively. Anaphylatoxin peptides C3a and C5a are inactivated by carboxypeptidase N (CPN), which removes a terminal arginine residue to reduce their pro-inflammatory activities. Regulatory proteins such as S-protein and clusterin bind the C5b–C7 complex to prevent insertion in the cell membrane and interfere with C9 polymerization. Cell membrane-bound regulators: complement receptor 1 (CR1; also known as CD35), expressed on erythrocytes, phagocytes, B cells and subsets of T cells, acts as a co-factor for factor I-mediated cleavage of both C4b and C3b. It also has decay-accelerating activity towards the C3 convertases of all 3 activation pathways. Membrane co-factor protein (MCP; also known as CD46) is expressed on most cells and has cofactor activity for factor I-mediated cleavage of C4b and C3b. Decay-accelerating factor (DAF; also known as CD55) is also expressed on most cells and accelerates the decay of C3 convertases of all 3 activation pathways. CD59 (also known as protectin) interferes with C9 binding and polymerization. Present on macrophages and Kupffer cells, complement regulator of the immunoglobulin superfamily (CRIg) binds C3b and blocks both the C3 and C5 convertases of the alternative pathway. C2 receptor inhibitor trispanning (CRIT) binds C2 and prevents its cleavage by C1s, thus interfering with the generation of the classical-pathway C3 convertase.
Fluid-phase regulators. The only circulating inhibitor of C1, termed Serping1 (also known as C1inh) is a serine protease inhibitor16. Serping1 prevents C1 autoactivation in the fluid phase and also prevents initiation of classical-pathway activation on antigen–antibody complexes when the antibody has low antigen affinity17 or interacts weakly with C1q18. Serping1 also inactivates MASP1 and MASP2 of the lectin pathway. Moreover, Serping1 may regulate the alternative pathway, possibly through an interaction with C3b19,20. Serping1 also regulates the kinin and fibrinolytic pathways by interactions with activated factor XI, activated factor XII, kallikrein, tissue-type plasminogen activator and, to a lesser extent, plasmin16.
Proteolytic inactivation of the activated complement components C4 and C3 is carried out by factor I, a circulating serine protease that requires either C4b-binding protein (C4BP), and factor-H21 as co-factors for inactivation of C4b, and C3b and iC3b, respectively. Factor H is the main plasma regulator of the alternative pathway and accelerates the decay of the C3 convertase. Binding sites within the factor H molecule allow interaction with glycosaminoglycans and sulphated polysaccharides for complement regulation on cell surfaces and in the extracellular matrix. Factor H-like protein 1 (FHL1), a truncated form of factor H, has factor I co-factor and decay-accelerating activities towards C3b and the alternative-pathway C3 convertase. Because of its ability to downregulate complement function, many microorganisms have evolved structures to specifically bind factor H to their surface in an active form3,22. Inactivation of C3a and C5a is provided by carboxypeptidase N. S protein (also known as vitronectin) and clusterin (also known as apolipoprotein J) control the deposition of the MAC1.
Cell membrane-bound regulators. Complement is also regulated on cell surfaces1,2,3,23. Complement receptor 1 (CR1; also known as CD35) acts as a co-factor for factor I-mediated cleavage of C4b and C3b, an activity that is shared by the membrane co-factor protein (MCP; also known as CD46). CR1 also displays decay-accelerating activity towards the C3 convertases of all three complement activation pathways, as does decay-accelerating factor (DAF; also known as CD55). Further regulation of the MAC is provided by CD59. Two additional cell surface regulators have been recently identified. Complement regulator of the immunoglobulin superfamily (CRIg)24 and C2 receptor inhibitor trispanning (CRIT)25 have specific regulatory activities towards the alternative and classical pathways, respectively.
Complement receptors
Upon activation, proteolytic products of complement activation are recognized by specific receptors on cell surfaces that control cellular function3. For example, receptors specific to either the globular (gC1qR) or collagen portion of C1q (C1qRp and cC1qR) are present on phagocytes and promote phagocytosis of C1q-coated particles. cC1qR, which is identical to calreticulin, is also termed the collectin receptor as it binds MBL and surfactant protein A (SPA). CR1 has binding sites that reportedly recognize C1q, C4b, C3b and iC3b. It is the principal immune adherence receptor on erythrocytes, allowing binding and bloodstream clearance of complement-coated immune complexes. Complement receptor 2 (CR2; also known as CD21) is mainly expressed on B cells and follicular dendritic cells. It recognizes degradation fragments of C3, such as C3dg and C3d, which are produced by further degradation of iC3b (Box 1). CR2, complexed with CD19 and CD81, lowers the activation threshold of B cells upon interaction with specific antigen coated with C3d fragments. It is also important in transferring antigen into lymphoid follicles. CR2 on follicular dendritic cells helps retain antigen in germinal centres of lymphoid organs and allows maintenance of immunological memory26. Complement receptor 3 (known as integrin aM (ITGAM) and CD11bCD18) is an adhesion molecule expressed on phagocytes that interacts with iC3b-coated target organisms to promote their elimination by phagocytosis. Complement receptor 4 (known as integrin β2 (ITB2) and CD11cCD18) is a related adhesion molecule on phagocytes that exerts a function similar to ITGAM and is present mainly on dendritic cells. Receptors specific for C3a (C3aR) and C5a (C5aR; also known as CD88) are present on various inflammatory cells and smooth muscle cells to promote an inflammatory reaction. They are also present on subsets of T cells and dendritic cells to regulate the adaptive immune response27. G protein-coupled receptor 77 (GPR77; also known as C5L2), recognizes C5a and its inactivated form, C5a desArg, but seems to downregulate rather than promote an inflammatory response28. CRIg binds C3b and iC3b and is involved in clearance from the bloodstream and phagocytosis of complement-coated particles by macrophages29.
Roles of complement
Activation of complement serves three key physiological roles2. First, complement controls infection by helping to eliminate microorganisms. Complement causes lysis of target organisms through insertion of the MAC into the cell membrane. It also facilitates elimination of microorganisms by opsonization and by promoting local inflammation with recruitment of phagocytes. Second, complement is involved in waste disposal. Complement activation on immune complexes promotes their solubilization and clearance from the bloodstream by transport on erythrocytes through an interaction with CR1. C1q interacts with the membranes of apoptotic cells to mediate their efficient phagocytosis and destruction. Finally, complement is involved in the development of adaptive immune responses. Indeed, recent observations suggest that products of complement activation interact with receptors on dendritic cells, B cells and T cells, to regulate the immune response to an antigen, promote the formation of specific antibodies and maintain immunological memory26,27. It is therefore important to consider these roles when designing agents that interfere with complement activation. Targeting of certain complement molecules might interfere with important physiological roles and induce potential deleterious effects, especially if long-term treatment is warranted.
Diseases associated with complement
Complement activation. Complement activation has been shown to occur in many pathological states, including renal, vascular, neurological, allergic and infectious disorders1,10,30,31 (Box 2). Although complement is clearly activated in subsets of patients with these diseases, because of the lack of effective complement inhibitors, its precise role is often unknown and animal models must be relied upon to determine the contribution of the complement proteins. In some cases, complement is activated systemically whereas in many others, complement activation occurs locally at the site of tissue injury. The triggering of complement activation by microorganisms during sepsis is thought to have a role in systemic inflammatory response syndrome32. Often, complement participates in disease by being activated by an abnormal antibody reacting against a self antigen. For example, in systemic lupus erythematosus, antibodies reacting with various intracellular and extracellular components form immune complexes that activate complement, causing tissue injury in the kidneys, skin or other tissues33. Complement activation by autoantibodies is also seen in various renal, dermatological, neurological and rheumatological diseases1,3. Interestingly, molecules that undergo pathological alteration, such as prion proteins34 and β-amyloid, can activate complement and cause tissue injury. The β-amyloid peptide, found in senile plaques in Alzheimer's disease, can bind C1q and activate the classical pathway35. Ischaemia–reperfusion injury — as occurs in stroke, coronary bypass surgery, acute myocardial infarction, bowel infarction and organ transplantation — has an important complement activation component. Upon reperfusion, ischaemic organs are subjected to an inflammatory reaction in which complement is activated. Complement activation, as detected by C4d staining in biopsies, is now viewed as an important marker of acute antibody-mediated rejection of organ allografts36. Recent studies show that complement synthesis and activation within the allograft contributes to the rejection process by regulating the local alloimmune response37.
Defective complement regulation. Complement regulation is known to be defective in various disorders (Table 1), the most carefully studied example being the Serping1 deficiency that is associated with hereditary angioedema (HAE)38. Paradoxically, it is the regulatory activities of Serping1 towards the kinin-generating system and not the complement system that is considered to be key in HAE. Defects in factor H, the main regulator of the alternative pathway, are associated with type II membranoproliferative glomerulonephritis, atypical haemolytic uraemic syndrome (aHUS), AMD (the leading cause of blindness in the elderly in developed countries) and recently with HELLP (haemolytic anaemia, elevated liver enzymes and low platelets, occurring during pregnancy) syndrome21,39. Defects in MCP are also associated with aHUS40. Paroxysmal nocturnal haemoglobinuria (PNH) is a rare haematological condition that, among other manifestations, is associated with severe haemolytic anaemia41. This is caused by absence of CD59 expression in erythrocytes. It is noteworthy that PNH is not caused by an inherited specific deficiency in CD59, but rather by a mutation in the glycosyl phosphatidyl inositol cell membrane anchor originating in haematopoietic progenitor cells that is associated with improper membrane binding of 20 cell membrane proteins, including the complement regulators CD59 and DAF. Erythrocyte lysis results from improper complement regulation during events of complement activation (for example, infection, trauma, episodes of stress or normal C3 tickover).
Complement deficiency. Because of its crucial role in defence against microorganisms, defects in many of the complement proteins, although generally rare, may be associated with increased susceptibility to infection42,43. Deficiency in many of the classical and alternative-pathway-specific components is associated with recurrent bacterial infections involving encapsulated Gram positive or Gram negative organisms. Strikingly, defects in components of the MAC (C5–C9) are strongly associated with neisserial infections, highlighting the importance of the MAC in eliminating Neisseria bacteria. Defects in properdin and factor H are also associated with neisserial infections. The only exception to the rarity of complement component deficiency is a deficiency of MBL, a protein that is usually present in the circulation at a concentration of approximately 2 μg per ml44,45. It is estimated that approximately 5% of the general population has a homozygous mutation that leads to near total MBL absence, whereas around 30% of the population carries one of a number of heterozygous mutations that results in MBL levels that are approximately 10% that of normal values. MBL deficiency is associated with increased susceptibility to various upper respiratory tract infections, particularly during childhood. Moreover, MBL deficiency leads to increased susceptibility to infection in patients with co-existing immunodeficiency. Deficiencies in classical pathway-specific components C1, C4 and C2, in addition to susceptibility to bacterial infection, are associated with increased prevalence of autoimmune disease, especially systemic lupus erythematosus33.
It is now known that direct complement activation is one of the main clearance mechanisms of apoptotic bodies. It is thought that defective classical-pathway activation leads to the persistence of apoptotic cells in the circulation, exposure of intracellular antigens on apoptotic blebs and the generation of autoantibodies with subsequent pathogenic effects. Genetic defects and polymorphisms in genes that encode complement proteins, such as factor I, factor B, C3 and factor H-like protein 1, have been associated with aHUS, suggesting that it is a disease of defective alternative-pathway activation and regulation46.
It is noteworthy that acquired complement deficiencies also lead to disease. For example, C3 nephritic factor, an autoantibody directed against the alternative-pathway C3 convertase, stabilizes the convertase and promotes uncontrolled complement activation. It is associated with type II membranoproliferative glomerulonephritis and partial lipodystrophy47. An autoantibody against C1q is thought to cause hypocomplementemic urticarial vasculitic syndrome and is seen in some patients with systemic lupus erythematosus who present with nephritis. Autoantibodies directed against Serping1, as seen in some patients, often with B cell or other malignancies or rheumatological disorders, cause angioedema by interfering with the regulatory function of Serping1 (Ref. 48). Similarly, autoantibodies to factor H have been shown to cause aHUS49.
Currently used complement inhibitors
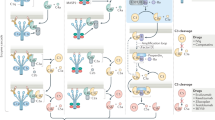
Given that the complement proteins and regulators were identified, isolated, sequenced and in some cases synthesized decades ago and that the complement enzymes have been studied in detail, it is perhaps surprising that only three agents that regulate or inhibit complement function are approved for clinical use (Table 2). Only one of these agents, eculizumab (Soliris; Alexion), targets specific complement proteins, whereas Serping1 and intravenous immunoglobulin (IVIg) not only show complement-regulating activity but also target physiological systems other than complement. Figure 3 shows sites of action of complement inhibitors.

Agents that control complement activation, currently in use or in development, target various steps in the complement activation cascade. Some are based on naturally occurring regulatory proteins (soluble complement receptor 1 (sCR1), soluble membrane co-factor protein (MCP), soluble decay-accelerating factor (sDAF), soluble CD59 (sCD59), Serping1 and factor H (fH)–CR2 construct), whereas others have been developed to target specific proteins (antibodies or synthetic peptides such as antibodies specific for C5 and C5a, compstatin and C5a receptor (C5aR) antagonists). In many cases IgG Fc fragment proteins have been fused to MCP and DAF to slow the rate of metabolism. Intravenous immunoglobulin (IVIg) action on the complement system is well documented but may not be the mechanism by which effective therapy for autoimmune disorders might be achieved.
C5-specific antibody. Eculizumab is a monoclonal antibody specific for C5 that effectively inhibits C5a generation and MAC formation50. This antibody was humanized to contain the mouse complementary determining regions that react with human C5, combined with a human IgG2–IgG4 hybrid that neither binds Fc receptors nor activates complement. It has been shown to be safe in patients with rheumatoid arthritis, systemic lupus erythematosus, myocardial infarction and membranous nephritis, as well as those who have undergone coronary artery bypass surgery. Eculizumab is currently approved for the treatment of PNH41,50, in which it markedly reduces intravascular haemolysis, the need for transfusion support and patient fatigue41. Despite an increased risk of neisserial and possibly other infections, patients treated with eculizumab so far have not shown markedly increased infectious complications50. A single-chain version of the C5-specific humanized monoclonal antibody, termed pexelizumab, is short-acting and was tested in acute myocardial complications following ischaemic heart disease. A meta-analysis reported no increased benefit of C5-specific antibody treatment despite a 26% decrease in the risk of death following coronary artery bypass surgery51. Interestingly, eculizumab was recently shown to be efficient in preventing acute antibody-mediated rejection of kidney allografts as an adjunct to standard immunosuppressive therapy52,53. Furthermore, eculizumab was shown to be effective in treating atypical haemolytic uraemic syndrome in two patients, one of whom had mutated complement regulatory proteins and one of whom did not54,55. In addition, a case of transfusion-dependent cold agglutinin disease successfully treated with eculizumab was recently reported56. Eculizumab will certainly be exploited for the treatment of other diseases with a complement activation component such as AMD and asthma, for which a nebulized formulation of eculizumab was reported to be under development57.
C1 inhibitor. Concentrates of Serping1 isolated from human plasma have been used for many years in Europe for the treatment of HAE. HAE is an autosomal dominant disease due to partial deficiency in Serping1 and uncontrolled kinin system activation leading to generation of the potent vasoactive mediator bradykinin16,48. Recently, a Serping1 concentrate termed Cinryze (Viro Pharma) was approved in the United States for the prophylaxis of HAE in adolescents and adults58. It is known that supraphysiological levels of this protein may downregulate complement activation, but there have been few systematic studies of its use in diseases other than HAE. To date, Serping1 concentrates have been used in humans for the treatment of sepsis. However, despite improvement in renal and multiple organ function, no survival benefit was noted59. Complement activation by tissue-specific antibody is thought to be involved in both hyperacute rejection in xenotransplantation models and acute humoral rejection of organ allografts, and Serping1 was shown to be effective in preventing complement-mediated tissue damage in a number of animal models of transplantation60. It was also shown to reduce capillary leakage in recipients of lung transplants61. Ischaemia–reperfusion injury, as occurs in myocardial infarction requiring coronary artery bypass surgery, presents with an inflammatory reaction in which complement activation participates. Clinical studies of the use of Serping1 in myocardial infarct have suggested improvement in various cardiac parameters, as well as reduction in infarct size, warranting further studies62,63,64.
The recent findings that the carbohydrate moieties of Serping1 can interact with E-selectin and P-selectin on leukocytes and endothelial cells, and extracellular matrix proteins such as type IV collagen and laminin, suggest that Serping1 could be useful in a number of inflammatory diseases60. Now that the purified protein is available in large quantities and is approved for use in patients with HAE, it is likely to be tested for efficacy in a number of inflammatory conditions. A challenge with the use of purified Serping1 is its short half-life. The current preparations are approved for intravenous administration and must be given every 3–4 days in HAE. Sustaining elevated plasma levels of Serping1 may prove difficult, and it is therefore unlikely to be used for chronic conditions.
IVIg. IVIg is prepared from the plasma of thousands of blood donors and contains a high content of polyspecific IgG. It is given at a high dose (1–2 g per kg) and is approved for the treatment of autoimmune diseases such as Kawasaki disease and idiopathic thrombocytopenic purpura65. However, it has also shown considerable effectiveness in a number of other neurological, haematological, dermatological and rheumatological autoimmune diseases66. Its mechanism of action in therapeutic situations is unclear. However, IVIg inhibits complement deposition on targets, particularly when activation is triggered by antibodies through the classical pathway. It is thought that, because IgG is an excellent acceptor of activated complement proteins, IVIg acts to intercept the activated complement proteins C3b and C4b before they have an opportunity to bind to targets67. Complement inhibition is reported to be its mechanism of action in dermatomyositis68. Furthermore, it was recently proposed that IVIg acts as a scavenger for anaphylatoxins C3a and C5a, thereby reducing the inflammatory reaction induced by complement activation69. Interestingly, it has recently been shown in a mouse model that IVIg treatment, through its proposed complement-modulating activity, could significantly reduce infarct size following cerebral ischaemia–reperfusion as seen in patients who have had a stroke70. It must be stressed that IVIg has been shown to inhibit the action of many cytokines and that its use in humans is not aimed at inhibiting complement activation per se but rather at controlling autoimmune phenomena based on empirical observations.
Agents under development
Given the increasing recognition of the part that complement plays in many diseases, a number of promising agents are currently under development, some of which are being tested in clinical trials (Table 2).
Complement regulatory proteins. Approximately two decades ago, investigators succeeded in synthesizing a soluble form of CR1 lacking the intracellular and transmembrane portions of the molecule (termed sCR1 or TP10)71. CR1 is an attractive molecule for complement regulation as it possesses both factor I co-factor activity and decay-accelerating activity, and inhibits proteins and convertases of all complement pathways. sCR1 was extensively tested in many animal models of disease and was reported to be effective8. However, despite efficient complement inhibition in patients undergoing cardiac surgery on cardiopulmonary bypass, improvement of primary clinical end points was observed in some male patients, but not in female patients72. Promising results were seen in adults undergoing lung transplantation and children undergoing cardiopulmonary bypass surgery73. However, sCR1 has never become part of the clinical armamentarium, even after modification of its structure to add a sialyl Lewis X sugar to target E-selectin expressed on endothelial cells in inflammatory sites57. The reasons for this are not entirely clear, but one reason might be that this extended rod-shaped molecule has a short half-life in the circulation. Failed recent studies in a non-human primate model of cerebral ischaemia–reperfusion injury showed that successful investigations of sCR1 and sCR1–sialyl Lewis X in small animal models may not always translate into clinical success74. It is noteworthy that sCR1, when linked to blood group antigens, could be used in transfusion medicine to prevent or treat complement-induced immune haemolysis75. A truncated version of sCR1, termed APT070, was shown to reduce post-ischaemic damage of kidney grafts and prolong graft survival in rats76. However, it has not yet entered clinical trials53.
Other naturally occurring complement regulatory proteins have also been synthesized for use as therapeutic agents. For example, soluble forms of MCP, DAF and CD59 have been produced and shown to be effective in blocking complement activation and improving the outcome in several disease models6. However, these agents show lower potency than the parent molecules. A fusion protein containing both MCP and DAF, called complement activity blocker 2 was generated and shown to be effective in animal models. To extend the half-life of soluble forms of complement protein inhibitors, genes encoding regulatory molecules are fused with the gene encoding the Fc portion of IgG. Examples include both MCP and DAF, which show increased bioavailability77. A rodent-specific complement regulatory protein, CRRY, has both factor I cofactor activity and decay-accelerating activity and was produced by recombinant technology as a soluble complement regulator. Again, this agent, fused with IgG Fc to extend its half-life, was shown to be efficient in a number of animal models of disease78,79. CRRY is immunogenic in humans and is unlikely to become a major agent for human use.
An inhibitor of the alternative pathway with similar function, containing the binding site for C3 breakdown products of CR2 and the amino-terminal portion of factor H containing its C3 inhibitory activity (termed TT30) was constructed80. This inhibitor targets sites of complement-mediated inflammation in which C3 breakdown products are already deposited. It was shown to efficiently block complement activation and to protect against intestinal ischaemia–reperfusion injury in mice. The human forms of the proteins would not be immunogenic in man. TT30 is in preclinical development for use in AMD, aHUS and autoimmune diseases. Another fusion protein containing the C3d-binding site of CR2 and a humanized antibody fragment directed against factor B (termed TA106) is also in preclinical development for AMD, aHUS and asthma. However, despite encouraging results in animal models, these agents have not yet entered clinical investigation.
With the discovery of a new complement receptor or regulatory molecules of the alternative pathway, termed CRIg, a soluble fusion protein containing the Fc portion of IgG was created and shown to reverse inflammation and bone destruction in a murine model of arthritis81. This agent is likely to be explored further as a potential therapeutic agent. As noted above, an approach to improving the potency of soluble complement regulatory molecules is to fuse them with CR2. This allows targeting of cells already opsonized with C3 fragments and further regulation of complement activity. This has been carried out for DAF, CD59 and CRRY with demonstration of complement regulatory activity at inflammatory sites82,83. Whereas CR2 has only limited complement regulatory activity, when attached to antigen it shows impressive B cell-activating activity. As such, it is an important bridge between innate and adaptive immunity. C3d-coated antigens recognized by B cells elicit more potent specific antibody responses than free antigen and allow better maintenance of immunological memory. Vaccination with gene constructs of specific antigens from infectious agents coupled to multiple copies of C3d have been reported84,85. Results varying from increased antibody response to elimination of the antibody response were observed, depending on the model used. This reflects differences in immune responses elicited by different antigens despite a common C3d tag.
Another interesting approach is to couple antigen to an antibody directed against CR2. In mice and cynomolgous monkeys, this approach triggers strong responses to antigen, suggesting potential utility in vaccine development86.
Monoclonal antibodies. In addition to the C5-specific antibody eculizumab, several other antibodies to complement proteins have been developed. The advantages of such antibodies are their extended half-life compared with other molecules and the possibility of large-scale production. Interestingly, an antibody to C5 that recognizes the C5a epitope within C5, without blocking C5 cleavage during activation, was generated87. This antibody (known as TNX-558) blocks C5a interaction with the C5a receptor CD88, thereby interfering with the inflammatory response but without affecting complement activation on a target pathogen per se57,87. Other antibodies are also in development for potential applications in human disease57. Of these, antibodies blocking the activities of factor D (termed TNX-234), factor B and properdin of the alternative pathway may prove useful. TNX-234 is now undergoing Phase I clinical trials for AMD88. Factor B-specific antibodies were shown to be quite efficient in reducing tissue damage and inflammation in mouse models of ischaemia–reperfusion injury89, traumatic brain injury90 and asthma91, among others. Using phage display technology, an antibody that specifically binds C3b was recently produced. It was shown to block factor B and C5 binding to C3b, thereby demonstrating potent alternative-pathway-modulating activity92. This antibody, developed by Genentech, is likely to be explored as a therapeutic agent. Furthermore, an antibody to the C5a receptor called neutrazumab is under development for the treatment of rheumatoid arthritis and stroke57.
Small molecules. Recombinant technology using RNA nucleotides can be used to construct molecules that block complement function. The effective oligonucleotides are formed by recombinant technology, and a sequential panning procedure is used to enrich for molecules that bind increasingly well to the target binding site. Nucleotides are chosen with bonds that are difficult to cleave by plasma RNAses. These 'aptamers' are small and can be potent. An aptamer inhibitor of human C5 was developed one decade ago93. It has recently been pegylated to improve its pharmacodynamic properties and seems to be aimed at clinical development for coronary artery bypass surgery and AMD57.
Using combinatorial phage-display library screening, a 13-amino-acid cyclic peptide was found to specifically bind C3 and prevent its cleavage94. This peptide was named compstatin and seems to interfere with complement activation by blocking the access of C3 to the C3 convertases95. Compstatin is specific to primate C3 but has been shown to be a potent blocker of complement activation in some animal models of disease, such as xenograft rejection, extracorporeal circulation and heparin–protamine complex-induced inflammation, which is thought to occur in heart surgery. A compstatin derivative, termed POT-4, has completed Phase I clinical trials for AMD and is expected to enter Phase II clinical trials by the end of 2009.
Receptor antagonists. Upon complement activation, small peptides derived from C3 and C5 cleavage, C3a and C5a respectively, are generated. These have anaphylatoxin properties and are strong inducers of inflammation. They attract leukocytes to sites of inflammation, induce smooth muscle cell contraction and promote the release of vasoactive substances such as histamine and serotonin from mast cells15,28. However, cells other than those of haematopoietic origin, such as microglial cells in the nervous system and Kupffer cells in the liver, also respond to C5a28. High levels of C5a are found in various human diseases, including rheumatoid arthritis, ischaemia–reperfusion injury, asthma and allergy, atherosclerosis, graft rejection, glomerulonephritis and systemic lupus erythematosus28. Therefore, targeting C5a might prove useful in numerous diseases. Peptides with antagonistic properties toward the C5a receptor have been developed as potential therapeutic agents. Of these, a product termed PMX53 is a cyclic hexapeptide that exhibits strong inhibitory activity toward the C5a receptor CD88 but has a negligible effect on the activity of the C5a receptor C5L2. PMX53 was shown to be potent at inhibiting inflammation in animal models, and safe when administered orally and topically in patients with rheumatoid arthritis and psoriasis, respectively96. However, it was recently reported to be ineffective at reducing synovial inflammation in a double-blind, placebo-controlled study in patients with rheumatoid arthritis, despite the detection of biologically active levels in serum97. PMX53 has a short half-life of 70 minutes and is susceptible to proteolytic cleavage. Arana Therapeutics recently announced it had discontinued clinical development of PMX53 for AMD. However, preclinical studies for indications such as osteoarthritis are under way. Other peptidomimetics and organic molecules are being developed as C5a receptor antagonists, such as JPE-1375 and JSM-7717, which have yet to be tested in clinical studies57. Targeting of the C3a receptor has attracted only limited attention and agents have not entered clinical trials, although experiments in animals were carried out6. C3a–C3a receptor interactions are thought to be important in mediating airway hyper-responsiveness and causing mucus secretion98. This has recently also been shown in mice99,100 and in patients with allergic asthma101. However, the C3a receptor antagonists that are presently available require further development as on binding to the receptor they may exert some agonist activities102. Another potential application of C3a receptor antagonists could be in stem cell transplantation. A source of stem and progenitor haematopoietic cells for transplantation in some patients comes from treatment of donors with the growth factor granulocyte colony-stimulating factor (G-CSF), which induces the egress of stem and progenitor cells from the bone marrow microenvironment into blood, a process called mobilization. Some patients are 'poor mobilizers' under standard treatment modalities. Experiments in mice have shown that C3a–C3a receptor interactions within the bone marrow participate in stem and progenitor cell homing in the bone marrow103. Disruption of this interaction promotes cell mobilization. One might anticipate an advantage of C3a receptor antagonists in patients identified as poor mobilizers.
Animal proteins. Many molecules that activate complement but are not easily inhibited by the normal complement control proteins have been isolated. About a century ago, cobra venom factor (CVF) was isolated and shown to be a potent complement activator. CVF is a cobra analogue of C3 that acts like C3b in activating complement but it is not inhibited by the natural inhibitors, factors H and I104. Although CVF was used in a large number of animal models to demonstrate the role of complement in disease, the protein itself is not useful in treatment because its powerful immunogenicity prevents its continuing activity. It is noteworthy that CVF, when administered to some animals, can cause a systemic inflammatory reaction that leads to circulatory collapse and pulmonary shock105. Nevertheless, a recent resurgence of interest in CVF has led to the development of CVF–human C3 hybrid molecules that retain CVF complement-depleting activities106. With the creation of less immunogenic molecules, CVF–C3 hybrids, by depleting complement, might become interesting therapeutic agents in preventing reperfusion injury.
Microbial inhibitors. Several microorganisms have evolved complement evasion mechanisms for survival purposes. Some viruses and bacteria express unique molecules that regulate complement activation or its biological effects. Mechanisms through which microorganisms evade complement attack include: expression of molecules that inhibit complement proteins, convertases or receptors; recruitment of host complement regulatory proteins; proteolytic degradation of complement proteins; and expression of molecules that mimic human complement regulatory proteins107. Development of new complement-targeting therapeutics can be based on a thorough understanding and characterization of these microbial complement regulators. One such example is Staphylococcus aureus, which expresses multiple complement-regulating proteins. Chemotaxis inhibitory protein of S. aureus is an antagonist of the C5a receptor. Staphylococcal complement inhibitor binds classical and alternative pathway C3 convertases, thereby interfering with C5a generation. Extracellular fibrinogen-binding protein and extracellular complement-binding protein bind C3b and block cleavage of C3b-containing convertases, namely the alternative pathway C3 convertase and C5 convertases of all complement activation pathways108. Two soluble proteins, produced by variola (variola virus complement control protein) and vaccinia (smallpox inhibitor of complement enzymes) viruses, demonstrate complement-regulating activities. Both proteins are structurally related to the human regulator of complement activation protein family and possess factor I co-factor activity towards C3b and decay-accelerating activity towards C3 convertases107. They have been used in complement-dependent inflammatory response models109 and their mechanisms of action are being investigated110. Although these proteins are potentially immunogenic, a structural insight into their functions may help develop potential drug candidates for therapeutic purposes.
Replacement therapy. As noted, Serping1 replacement therapy has long been used in HAE48. Over the years, a number of patients have been treated with individual complement proteins to replace deficient proteins in acute situations. For example, C3-sufficient blood was used to treat persistent fever in a C3-deficient patient111. In animals, such replacement of an absent protein regularly induces antibody formation. Nevertheless, infusion of plasma-derived MBL was reported to be successful in treating one child with MBL deficiency and recurrent infections112. A recombinant form of MBL that retains activity was shown to be well tolerated and is presently in clinical trials in patients with MBL deficiency undergoing high-dose chemotherapy, progenitor and stem cell transplantation or liver transplantation113. However, the role of MBL deficiency in these clinical settings is still debated and its usefulness as a therapeutic agent awaits further clinical investigation114. Moreover, its use in patients with a complete deficiency could lead to antibody formation and an anaphylactic response. A recombinant factor H molecule (termed rhCFH) is currently under preclinical development for the treatment of AMD, aHUS and dense deposit disease (type II membranoproliferative glomerulonephritis), and possibly aimed at patients with factor H defects that lead to disease.
Conclusions
The role of complement in many autoimmune and inflammatory diseases has been known for decades. Many therapeutic agents used in the treatment of various diseases, such as captopril (an antihypertensive drug), prednisolone (a steroidal anti-inflammatory drug) and heparin (an anticoagulant) inhibit complement activation but cannot be used as complement inhibitors in therapy because of their lack of specific activity115. Agents that inhibit specific complement components are required for targeted efficacy. Surprisingly, only two agents are currently approved for clinical use for narrow indications (HAE and PNH), but disorders of complement regulation, such as aHUS and AMD, have led to a resurgence of interest in complement-directed therapeutic drugs. Unfortunately, early studies indicate that some complement-targeting drugs may not be as potent in clinical situations as expected from animal models. However, successful use of eculizumab in aHUS, antibody-mediated acute renal allograft rejection and cold agglutinin disease, although still anecdotal, indicate that targeting complement is a promising strategy in many clinical situations.
A major challenge in developing potent therapeutic complement inhibitors is the need to fully elucidate the exact role of complement in various immune-mediated diseases. As more than one activation pathway may participate in disease manifestations, complement modulation will either have to target multiple steps in the activation cascade or a common step that prevents the generation of harmful components. Moreover, the crucial role of complement in the prevention of infection and elimination of immune complexes and apoptotic cells needs to be preserved, while blocking tissue injury. Another challenge stems from the pharmaceutical criteria for ideal drug compounds (low molecular mass, high potency, selectivity, few adverse reactions, low production costs and oral administration), which make inhibitors of serine proteases (such as C1s, the MASPs and factor D) and anaphylatoxin receptors attractive drug candidates57. One might expect development of such components in the future. However, the remarkable example of eculizumab, which is used to treat PNH and reported to be effective in treating aHUS, acute antibody-mediated kidney allograft rejection and cold agglutinin disease, indicates that humanized monoclonal antibodies that target specific components of complement will increasingly be developed. Because of the increasingly recognized effects of complement activation in aHUS, AMD, ischaemia–reperfusion injury as occurs in cardiopulmonary bypass surgery, stroke, organ preservation before transplantation and transplant rejection, it is likely that these pathological states, as well as autoimmune diseases, will be subject to further clinical investigations.
In addition to agents that block complement activation, there is a benefit of replacing missing complement proteins in diseases such as HAE and aHUS due to factor H or factor I mutations. Large-scale production of concentrates from human plasma is costly, labour intensive and carries risks of viral transmission. However, it is possible to prepare the human proteins by recombinant technology, as demonstrated by the recombinant human Serping1 preparation rhucin (Table 1). The use of Pichia yeast strains that have been engineered to produce glycoproteins with human N-glycosylation patterns holds promise in large-scale production of complement proteins116,117.
Modulation of complement activation in specific disease states and the agent that is selected for use will vary according to the length of therapy required. In conditions such as myocardial infarction or stroke, short-term treatment is required and agents that effectively block complement activation will certainly be used. However, for chronic diseases such as autoimmune disorders, asthma and defects in complement regulation as seen in aHUS and AMD, more specific and targeted therapy that allows long-term use will probably be needed. One could expect fusion proteins that target sites of inflammation such as fH-CR2 constructs to be quite effective in these situations, but other complement-modulating agents might also be useful.
The risks associated with modulation of complement activation are only speculative. Proteins of the complement system are present in the most primitive of animals and have great evolutionary stability. They clearly have an important role in host defence. Disease studies of rare patients with complement deficiency suggest that short-term inhibition of complement activity can be safe, particularly with the availability of modern antibiotics. Long-term inhibition, as may be required for the treatment of chronic conditions, might be far more difficult. Inhibition of different parts of the activation pathways may have different consequences, as complement not only participates in defence against microorganisms but also in the regulation of adaptive immunity, elimination of apoptotic bodies, regulation of self tolerance, haematopoietic stem and progenitor cell homing and tissue regeneration. Based on the information derived from genetic deficiencies, sustained complement inhibition may be difficult to achieve and may lead to infectious or autoimmune conditions. By contrast, C9 deficiency may have relatively minor consequences as C9 is the last protein in the complement activation cascade. Pore formation may occur at the level of C8, and a population in Japan with a genetic deficiency in C9 does relatively well clinically. Nevertheless, the use of Serping1 concentrates in HAE patients, eculizumab in PNH patients and high-dose IVIg in many patients with autoimmune disorders have not yet been associated with serious complications as might have been expected in modulating complement activity. As we further understand the role of the complement proteins in normal physiology and host defence, we may have a better understanding of the proteins that can be safely inhibited and those that cannot.

References
Wagner, E. Jiang, H. & Frank, M. M. in Clinical Diagnosis and Management by Laboratory Methods (ed. Henry, J. B.) 892–913 (W. B. Saunders, Philadelphia, 2001).
Walport, M. J. Complement. First of two parts. N. Engl. J. Med. 344, 1058–1066 (2001).
Speth, C., Prodinger, W. M., Würzner, R., Stoiber, H. & Dierich, M. P. in Fundamental Immunology (ed. Paul, W. E.) 1047–1078 (Lippincott Williams & Wilkins, Philadelphia, 2008).
Zipfel, P. F., Heinen, S., Józsi, M. & Skerka, C. Complement and diseases: defective alternative pathway control results in kidney and eye diseases. Mol. Immunol. 43, 97–106 (2006).
Frank, M. M. in The Human Complement System in Health and Disease (eds Volanakis, J. A. & Frank, M. M.) 1–8 (Marcel Dekker, New York, 1998).
Makrides, S. C. Therapeutic inhibition of the complement system. Pharmacol. Rev. 50, 59–87 (1998).
Wagner, E. & Frank, M. M. in The Human Complement System in Health and Disease (eds Volanakis, J. & Frank, M. M.) 527–546 (Marcel Dekker, New York, 1998).
Mollnes, T. E. & Kirschfink, M. Strategies of therapeutic complement inhibition. Mol. Immunol. 43, 107–121 (2006).
Cole, D. S. & Morgan, B. P. Beyond lysis: how complement influences cell fate. Clin. Sci. 104, 455–466 (2003).
Holers, V. M. The spectrum of complement alternative pathway-mediated diseases. Immunol. Rev. 223, 300–316 (2008). This review describes the alternative pathway of complement activation and its increasingly recognized role in many diseases.
Kemper, C. & Hourcade, D. E. Properdin: new roles in pattern recognition and target clearance. Mol. Immunol. 45, 4048–4056 (2008). This review presents experimental evidence of a role of properdin as a recognition molecule that directs alternative-pathway activation on microorganisms and apoptotic cells, allowing a better understanding of complement activation mechanisms.
Dommett, R. M., Klein, N. & Turner M. W. Mannose-binding lectin in innate immunity: past, present and future. Tissue Antigens 68, 193–209 (2006).
Thiel, S. Complement activating soluble pattern recognition molecules with collagen-like regions, mannan-binding lectin, ficolins and associated proteins. Mol. Immunol. 44, 3875–3888 (2007).
Atkinson, J. P. & Frank, M. M. Bypassing complement: evolutionary lessons and future implications. J. Clin. Invest. 116, 1215–1218 (2006). This commentary article describes the largely unrecognized complement-bypass pathways.
Markiewski, M. M. & Lambris, J. D. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 171, 715–727 (2007).
Cicardi, M., Zingale, L., Zanichelli, A., Pappalardo, E. & Cicardi, B. C1 inhibitor: molecular and clinical aspects. Springer Semin. Immun. 27, 286–298 (2005).
Chen, C.-H., Lam, C. F. & Boackle, R. J. C1 inhibitor removes the entire C1qr2s2 complex from anti-C1Q monoclonal antibodies with low binding affinities. Immunology 95, 648–654 (1998).
Chen, C.-H. & Boackle, R. J. A newly discovered function for C1 inhibitor, removal of the entire C1qr2s2 complex from immobilized human IgG subclasses. Clin. Immunol. Immunopathol. 87, 68–74 (1998).
Jiang, H., Wagner, E., Zhang, H. & Frank, M. M. Complement 1 inhibitor is a regulator of the complement alternative pathway. J. Exp. Med. 194, 1609–1616 (2001).
Nielsen, E. W. et al. Effect of supraphysiologic levels of C1-inhibitor on the classical, lectin and alternative pathways of complement. Mol. Immunol. 44, 1819–1826 (2007).
Rodriguez de Cordoba, S., Esparza-Gordillo, J., Goicoechea de Jorge, E., Lopez-Trascasa, M. & Sanchez-Corral, P. The human complement factor H: functional roles, genetic variations and disease associations. Mol. Immunol. 41, 355–367 (2004).
Jarva, H., Jokiranta, T. S., Würzner, R. & Meri, S. Complement resistance mechanisms of streptococci. Mol. Immunol. 40, 95–107 (2003).
Kim, D. D. & Song, W.-C. Membrane complement regulatory proteins. Clin. Immunol. 118, 127–136 (2006).
Wiesmann, C. et al. Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature 444, 217–220 (2006).
Inal, J. M. et al. Complement C2 receptor inhibitor trispanning: a novel human complement inhibitory receptor. J. Immunol. 174, 356–366 (2005).
Carroll, M. C. The complement system in regulation of adaptive immunity. Nature Immunol. 5, 981–986 (2004).
Kemper, C. & Atkinson, J. P. T-cell regulation: with complements from innate immunity. Nature Rev. Immunol. 7, 9–18 (2007).
Monk, P. N., Scola, A.-M., Madala, P. & Fairlie, D. P. Function, structure and therapeutic potential of complement C5a receptors. Br. J. Pharmacol. 152, 429–448 (2007).
Helmy, K. Y. et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 124, 915–927 (2006).
Sjöberg, A. P., Trouw, L. A. & Blom, A. M. Complement activation and inhibition: a delicate balance. Trends Immunol. 30, 83–90 (2009).
Walport M. J. Complement. Second of two parts. N. Engl. J. Med. 344, 1140–1144 (2001).
Ward, P. A. The dark side of C5a in sepsis. Nature Rev. Immunol. 4, 133–142 (2004).
Manderson, A. P., Botto, M. & Walport, M. J. The role of complement in the development of systemic lupus erythematosus. Annu. Rev. Immunol. 22, 431–456 (2004).
Mitchell, D. A., Kirby, L., Paulin, S. M., Villiers, C. L. & Sim, R. B. Prion protein activates and fixes complement directly via the classical pathway: implications for the mechanisms of scrapie agent propagation in lymphoid tissue. Mol. Immunol. 44, 2997–3004 (2007).
Bonifati, D. M. & Kishore, U. Role of complement in neurodegeneration and neuroinflammation. Mol. Immunol. 44, 999–1010 (2007).
Baldwin, W. M. III, Kasper, E. K., Zachary, A. A., Wasowska, B. A. & Rodriguez, E. R. Beyond C4d: other complement-related diagnostic approaches to antibody-mediated rejection. Am. J. Transplant. 4, 311–318 (2004).
Lin, T., Zhou, W. & Sacks, S. H. The role of complement and toll-like receptors in organ transplantation. Transplant. Int. 20, 481–489 (2007).
Cugno, M., Zanichelli, A., Foieni, F., Caccia, S. & Cicardi, M. C1 inhibitor deficiency and angioedema: molecular mehanisms and clinical progress. Trends Mol. Med. 15, 69–78 (2009).
Fang, C. J., Richards, A., Liszewski, M. K., Kavanagh, D. & Atkinson, J. P. Advances in understanding of pathogenesis of aHUS and HELLP. Br. J. Haematol. 143, 336–348 (2008).
Dragon-Durey, M.-A. & Fremeaux-Bacchi, V. Atypical haemolytic uraemic syndrome and mutations in complement regulator genes. Springer Semin. Immun. 27, 359–374 (2005).
Parker, C. J. Eculizumab for paroxysmal nocturnal haemoglobinuria. Lancet 373, 759–767 (2009).
Wagner, E. & Frank, M. M. in Medical Immunology (eds Parslow, T. G., Stites, D. P., Terr, A. I. & Imboden, J. B.) 341–348 (McGraw-Hill, New York, 2001).
Sullivan, K. E. & Winkelstein J. A. in Immunologic Disorders in Infants and Children (eds Stiehm, E. R., Ochs, H. D. & Winkelstein, J. A.) 652–684 (Elsevier Saunders, Philadelphia, 2004). This chapter describes the clinical impact of all of the known complement deficiency states in humans for a better understanding of the impact of inhibiting specific complement components, especially in a chronic manner.
Garred, P., Larsen, F., Madsen, H. O & Koch, C. Mannose-binding lectin deficiency — revisited. Mol. Immunol. 40, 73–84 (2003).
Eisen, D. P. & Minchinton, R. M. Impact of mannose-binding lectin on susceptibility to infectious diseases. Clin. Infect. Dis. 37, 1996–1505 (2003).
Jokiranta, T. S. et al. Where next with atypical hemolytic uremic syndrome? Mol. Immunol. 44, 3889–3900 (2007).
Trouw, L. A., Roos, A. & Daha, M. R. Autoantibodies to complement components. Mol. Immunol. 38, 199–206 (2001).
Agostoni, A. et al. Hereditary and acquired angioedema: proceedings of the third C1 inhibitor deficiency workshop and beyond. J. Allerg. Clin. Immunol. 114, S51–S131 (2004). This publication provides a comprehensive description of hereditary and acquired angioedema due to C1 inhibitor deficiency, including basic and clinical aspects of the disease.
Dragon-Durey, M. A. et al. Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 16, 555–563 (2005).
Rother, R. P., Rollins, S. A., Mojcik, C. F., Brodsky, R. A. & Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nature Biotech. 25, 1256–1264 (2007). This review describes the development of the C5-specific antibody currently approved for the treatment of PNH and results of experimental and clinical studies that have led to its use in a clinical setting.
Testa, L. et al. Pexelizumab in ischemic heart disease: a systematic review and meta-analysis on 15,196 patients. J. Thorac. Cardiovasc. Surg. 136, 884–893 (2008).
Locke, J. E. et al. The use of antibody to complement protein C5 for salvage treatment of severe antibody-mediated rejection. Am. J. Transplant. 9, 23123–23125 (2009).
Stegall, M. et al. Prevention of acute humoral rejection with C5 inhibition [abstract]. American Society of Transplantation Winter Symposium. (Banff, Alberta, Canada, 2009).
Nürnberger, J. et al. Eculizumab for atypical hemolytic-uremic syndrome. N. Engl. J. Med. 360, 542–544 (2009).
Gruppo R. A. & Rother, R. P. Eculizumab for congenital atypical hemolytic-uremic syndrome. N. Engl. J. Med. 360, 544–546 (2009).
Dührsen, U. & Philipp, T. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood 113, 3885–3886 (2009).
Ricklin, D. & Lambris, J. D. Complement-targeted therapeutics. Nature Biotech. 25, 1265–1275 (2008). An excellent review of complement-targeting agents with an emphasis on pharmacological development.
Epstein, T. G. & Bernstein, J. A. Current and emerging management options for hereditary angioedema. Drugs 68, 2561–2573 (2008).
Caliezi, C. et al. C1-inhibitor in patients with severe sepsis and septic shock: beneficial effect on renal dysfunction. Crit. Care Med. 30, 1722–1728 (2002).
Davis, A. E. III, Mejia, P. & Lu, F. Biological activities of C1 inhibitor Mol. Immunol. 45, 4057–4063 (2008).
Struber, M. et al. C1-esterase inhibitor in graft failure after lung transplantation. Intensive Care Med. 25, 1315–1318 (1999).
Thielmann, M. et al. Administration of C1-esterase inhibitor during emergency coronary artery bypasses surgery in acute ST-elevation myocardial infarction. Eur. J. Cardiothorac. Surg. 30, 285–293 (2006).
Fattouch, K. et al. Beneficial effects of C1 esterase inhibitor in ST-elevation myocardial infarction in patients who underwent surgical reperfusion: a randomised double-blind study. Eur. J. Cardiothorac. Surg. 32, 326–332 (2007).
Baig, K. et al. Complement factor 1 inhibitor improves cardiopulmonary function in neonatal cardiopulmonary bypass. Ann. Thoracic Cardiovasc. Surg. 84, 1477–1482, (2007).
Asghar, S. S. & Pasch, M. C. Therapeutic inhibition of the complement system. Y2K update. Front. Biosci. 5, e63–e81 (2000).
Jolles, S., Sewell, W. A. C. & Misbah, S. A. Clinical uses of intravenous immunoglobulin. Clin. Exp. Immunol. 142, 1–11 (2005).
Frank, M. M., Miletic, V. D. & Jiang, H. Immunoglobulin in the control of complement activation. Immunol. Res. 22, 137–146 (2000).
Basta, M. & Dalakas, M. C. High-dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J. Clin. Invest. 94, 1729–1735 (1994).
Basta, M. Ambivalent effect of immunoglobulins on the complement system: activation versus inhibition. Mol. Immunol. 45, 4073–4079 (2008).
Arumugan, T. V. et al. Intravenous immunoglobulin (IVIG) protects the brain against experimental stroke by preventing complement-mediated neuronal cell death. Proc. Natl Acad. Sci. USA 104, 14104–14109 (2008).
Weisman, H. F. et al. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science 249, 146–151 (1990). This paper describes the design of a complement-modulating agent based on a known complement regulator and proves its efficacy in a murine model of myocardial infarction.
Lazar, H. L. et al. Soluble human complement receptor 1 limits ischemic damage in cardiac surgery patients at high risk requiring cardiopulmonary bypass. Circulation 110 (suppl. II), 274–279 (2004).
Li, J. S., Jaggers, J. & Anderson, P. A. The use of TP10, soluble complement receptor 1, in cardiopulmonary bypass. Expert Rev. Cardiovasc. Ther. 4, 649–654 (2006).
Mocco, J. et al. Preclinical evaluation of the neuroprotective effect of soluble complement receptor type 1 in a nonhuman primate model of reperfused stroke. J. Neurosurg. 105, 595–601 (2006).
Yazdanbakhsh, K. Development of complement therapeutics for inhibition of immune-mediated red cell destruction. Transfusion 45, S122–S129 (2005).
Patel, H., Smith, A. G., Sacks, S. H. & Zhou, W. Therapeutic strategy with a membrane-localizing complement regulator to increase the number of usable donor organs after prolonged cold storage. J. Am. Soc. Nephrol. 17, 1102–1111 (2006).
Williams, A. S., Linton, S. M. & Morgan B. P. Coupling complement regulators to immunoglobulin domains generates effective anti-complement reagents with extended half-life in vivo. Clin. Exp. Immunol. 129, 198–207 (2002).
Leinhase, I. et al. Pharmacological complement inhibition at the C3 convertase level promotes neuronal survival, neuroprotective intracerebral gene expression, and neurological outcome after traumatic brain injury. Exp. Neurol. 199, 454–464 (2006).
Hepburn, N. J. et al. Prevention of experimental autoimmune myasthenia gravis by rat Crry-Ig: a model agent for long-term complement inhibition in vivo. Mol. Immunol. 45, 395–405 (2008).
Huang, Y., Qiao, F., Atkinson, C., Holers, V. M. & Tomlinson, S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J. Immunol. 181, 8068–8076 (2008).
Katschke, K. Jr et al. A novel inhibitor of the alternative pathway of complement reverses inflammation and bone destruction in experimental arthritis. J. Exp. Med. 204, 1319–1325 (2007).
Song, H. et al. Complement receptor 2-mediated targeting of complement inhibitors to sites of complement activation. J. Clin. Invest. 111, 1875–1885 (2003).
Atkinson, C., Qiao, F., Song, H., Gilkeson, G. S. & Tomlinson, S. Low-dose targeted complement inhibition protects against renal disease and other manifestations of autoimmune disease in MRL/lpr mice. J. Immunol. 180, 1231–1238 (2008).
Bergmann-Leitner, E. S., Leitner, W. W. & Tsokos, G. C. Complement 3d: from molecular adjuvant to target of immune escape mechanisms. Clin. Immunol. 121, 177–185 (2006).
Bergmann-Leitner, E. S. et al. C3d-defined complement receptor-binding peptide p28 conjugated to circumsporozoite protein provides protection against Plasmodium berghei. Vaccine 25, 7732–7736 (2007).
Whipple, E. C. et al. Low doses of antigen coupled to anti-CR2 mAbs induce rapid and enduring IgG immune responses in mice and cynomolgous monkeys. Mol. Immunol. 44, 377–388 (2007).
Sprong, T. et al. Inhibition of C5a-induced inflammation with preserved C5b-9-mediated bactericidal activity in a human whole blood model of meningococcal sepsis. Blood 102, 3702–3710 (2003).
Holmer, M. Genentech makes its first ever acquisition. Nature Biotech. 25, 4–5 (2007).
Thurman, J. M. et al. Treatment with an inhibitory monoclonal antibody to mouse factor B protects mice from induction of apoptosis and renal ischemia/reperfusion injury. J. Am. Soc. Nephrol. 17, 707–715 (2006).
Leinhase, I. et al. Inhibition of the alternative complement activation pathway in traumatic brain injury by a monoclonal anti-factor B antibody: a randomized placebo-controlled study in mice. J. Neuroinflamm. 4, 13–25 (2007).
Taube, C. et al. Factor B of the alternative complement pathway regulates development of airway hyperresponsiveness and inflammation. Proc. Natl Acad. Sci. USA 103, 8084–8089 (2003).
Katschke, K. J. et al. Structural and functional analysis of a C3b-specific antibody that selectively inhibits the alternative pathway of complement. J. Biol. Chem. 284, 10473–10479 (2009).
Biesecker, G., Dihel, L., Enney, K. & Bendele, R. A. Derivation of RNA aptamer inhibitors of human complement C5. Immunopharmacology 42, 219–230 (1999).
Holland, M. C., Morikis, D. & Lambris, J. D. Synthetic small-molecule complement inhibitors. Curr. Opin. Investig. Drugs 5, 1164–1173 (2004).
Janssen, B. J., Halff, E. F., Lambris, J. D. & Gros, P. Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition, J. Biol. Chem. 282, 29241–29247 (2007).
Köhl, J. Drug evaluation: the C5a receptor antagonist PMX-53. Curr. Opin. Mol. Ther. 8, 529–538 (2006).
Vergunst, C. E. et al. Blocking the receptor for C5a in patients with rheumatoid arthritis does not reduce synovial inflammation. Rheumatol. 46, 1773–1778 (2007).
Marom, Z., Shelhamer, J., Berger, M., Frank, M. M. & Kaliner, M. Anaphylatoxin C3a enhances mucous glycoprotein release from human airways in vitro. J. Exp. Med. 161, 657–668 (1985).
Drouin, S. M., Corry, D. B., Hollman, T. J., Kildsgaard, J. & Wetsel, R. A. Absence of the complement anaphylatoxin C3a receptor suppresses Th2 effector functions in a murine model of pulmonary allergy. J. Immunol. 169, 5926–5933 (2002).
Drouin, S. M., Corry, D. B., Kildsgaard, J. & Wetsel, R. A. Cutting edge: the absence of C3 demonstrates a role for complement in Th2 effector functions in a murine model of pulmonary allergy. J. Immunol. 67, 4141–4144 (2001).
Wills-Karp, M. Complement activation pathways. A bridge between innate and adaptive immune responses in asthma. Proc. Am. Thorac. Soc. 4, 247–251 (2007). This review describes the experimental evidence of the dual role of complement in asthma through anaphylatoxins C3a and C5a.
Mathieu, M.-C. et al. The C3a receptor antagonist SB 290157 has agonist activity. Immunol. Lett. 100, 139–145 (2005).
Ratajczak, J. et al. Mobilization studies in mice deficient in either C3 or C3a receptor (C3aR) reveal a novel role for complement in retention of hematopoietic stem/progenitor cells in bone marrow. Blood 103, 2071–2078 (2004).
von Zabern, I. in Activators and Inhibitors of Complement (ed. Sim, R. B.) 127–135 (Kluwer Academic Publishers., Dordrecht, 1993).
Younger, J. G. et al. Systemic and lung physiological changes in rats after intravascular activation of complement. J. Appl. Physiol. 90, 2289–2295 (2001).
Fritzinger, D. C. et al. Functional characterization of human C3/cobra venom factor hybrid proteins for therapeutic complement depletion. Mol. Immunol. 33, 105–116 (2009).
Lambris, J. D., Ricklin, D. & Geisbrecht, B. V. Complement evasion by human pathogens. Nature Rev. Microbiol. 6, 132–142 (2008). This review provides detailed information on the mechanisms used by microorganisms to avoid complement-mediated elimination that could be exploited as complement therapeutics.
Jongerius, I. et al. Staphylococcal complement evasion by various convertase-blocking molecules. J. Exp. Med. 204, 2461–2471 (2007).
Thorgersen, E. B. et al. Inhibition of complement and CD14 attenuates the Escherichia coli-induced inflammatory response in porcine whole blood. Infect. Immun. 77, 725–732 (2008).
Liszewski, M. K. et al. Smallpox inhibitor of complement enzymes (SPICE): regulation of complement activation on cells and mechanisms of its cellular attachment. J. Immunol. 181, 4199–4207 (2009). This recent article provides an improved understanding of the mechanism of action of complement inhibitors expressed by smallpox viruses, which is invaluable to the design of novel complement-targeting agents.
Osofsky, S. G., Thompson, B. H., Lint, T. F. & Gewurz, H. Hereditary deficiency of the third component of complement in a child with fever, skin rash, and arthralgias: response to transfusion of whole blood. J. Pediatr. 90, 180–186 (1977).
Valdimarsson, H. Infusion of plasma-derived mannan-binding lectin (MBL) into MBL-deficient humans. Biochem. Soc. Trans. 31, 768–769 (2003).
Petersen, K. A. et al. Phase I safety, tolerability, and pharmacokinetic study of recombinant human mannan-binding lectin. J. Clin. Immunol. 26, 465–475 (2006).
Casanova, J.-L. & Abel, L. Human mannose-binding lectin in immunity: friend, foe, or both? J. Exp. Med. 199, 1295–1299 (2004).
Bureeva, S., Andia-Pravdivy, J. & Kaplun, A. Drug design using the example of the complement system inhibitor's development. Drugs Discov. Today 10, 1535–1542 (2005).
Hamilton, S. R. et al. Humanization of yeast to produce complex terminally sialylated glycoproteins. Science 313, 1441–1443 (2006). This study shows that the pichia yeast system can be used to massively produce glycoproteins bearing terminal sugars as expressed in humans, possibly paving the way to large-scale, cost-efficient production of recombinant human complement proteins.
Beinrohr, L., Dobó, J., Zavódszky, P. & Gál, P. C1, MBL-MASPs and C1 inhibitor: novel approaches for targeting complement-mediated inflammation. Trends Mol. Med. 14, 511–521 (2008).
Trapp, R. G., Fletcher, M., Forristal, J. & West, C. D. C4 binding protein deficiency in a patient with atypical Behcet's disease. J. Rheumatol. 14, 135–138 (1987).
Pickering, M. C. & Cook, H. T. Translational mini-review series on complement factor H: renal diseases associated with complement factor H: novel insights from humans and animals. Clin. Exp. Immunol. 151, 210–230 (2008).
Reis, E. S., Falcao, D. A. & Isaac, L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand. J. Immunol. 63, 155–168 (2006).
Herbert, A. et al. Structure shows glycosaminoglycan- and protein-recognition site in factor H is perturbed by age-related macular degeneration-linked SNP. J. Biol. Chem. 282, 18960–18968 (2007).
Kavanagh, D, Richards, A. & Atkinson, J. P. Complement regulatory genes and haemolytic uremic syndromes. Annu. Rev. Med. 59, 293–309 (2008).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Related links
DATABASES
Entrez Gene
http://www.ncbi.nlm.nih.gov/gene
OMIM
http://www.ncbi.nlm.nih.gov/omim
Glossary
- Age-related macular degeneration
-
The principal cause of blindness in the elderly in industrialized countries. It is characterized by a loss of retinal pigment epithelium and photoreceptors, and deposits of proteins and lipids (drusen) or neovascularization.
- Innate immune system
-
Innate immunity is said to be nonspecific, but many of its components, including complement, recognize pathogen associated molecular patterns (PAMPs) for initiation of a defence reaction.
- Anaphylatoxins
-
Small peptides derived from C3 and C5 cleavage that promote inflammation by inducing leukocyte chemotaxis and increasing vascular permeability, among other activities.
- Kinin pathway
-
Triggers the generation of bradykinin, a potent vasoactive peptide, upon activation of the contact system of the coagulation cascade.
- Decay-accelerating activity
-
Shortening of the half-lives of convertases of the complement activation cascade by CR1 and DAF by accelerated dissociation of their components.
- Immunological memory
-
The ability of the immune system to recognize and respond rapidly and vigorously to a specific antigen with which it has previously had contact.
- Opsonization
-
Coating of an organism with complement fragments that leads to recognition by specific receptors on phagocytes and elimination by phagocytosis.
- Systemic lupus erythematosus
-
A prototypical autoimmune disease characterized by the presence of antibodies directed against intracellular antigens (for example, DNA) that form immune complexes causing inflammation at sites of deposition such as the skin, joints and kidneys.
- Ischaemia–reperfusion injury
-
A condition that arises upon restoration of blood flow in an organ deprived of oxygen supply.
- Hereditary angioedema
-
Condition that is manifested by recurrent episodes of skin swelling and abdominal pain; may be fatal if swelling affects the larynx.
- Atypical haemolytic uraemic syndrome
-
Renal pathology manifested by a triad of microangiopathic haemolytic anaemia, thrombocytopaenia and acute renal failure. It can be sporadic or familial.
- Hyperacute rejection
-
This occurs within minutes of revascularization of the organ graft and is caused by complement-activating pre-formed antibodies reacting against endothelial cells. By contrast, acute rejection occurs within the first weeks following transplantation because of elicited antibodies.
- Cell homing
-
The process by which haematopoietic stem and progenitor cells are contained within the bone marrow and travel to this site when transplanted.
- Regulator of complement activation protein family
-
These include factor H, C4BP, DAF, MCP, CR1 and CR2. They share a common structure consisting of a number of repetitive units of 60 amino acids called short consensus repeats, or complement control protein modules, which contain binding sites for activated complement proteins.
Rights and permissions
About this article
Cite this article
Wagner, E., Frank, M. Therapeutic potential of complement modulation. Nat Rev Drug Discov 9, 43–56 (2010). https://doi.org/10.1038/nrd3011
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd3011
This article is cited by
-
Induced neural stem cell grafts exert neuroprotection through an interaction between Crry and Akt in a mouse model of closed head injury
Stem Cell Research & Therapy (2021)
-
Antibody Response to SARS-CoV-2 is Associated with Long-term Clinical Outcome in Patients with COVID-19: a Longitudinal Study
Journal of Clinical Immunology (2021)
-
The involvement of C5a in the progression of experimental arthritis with Porphyromonas gingivalis infection in SKG mice
Arthritis Research & Therapy (2018)
-
Induced neural stem cell-derived astrocytes modulate complement activation and mediate neuroprotection following closed head injury
Cell Death & Disease (2018)
-
Pharmacologic Complement Inhibition in Clinical Transplantation
Current Transplantation Reports (2017)
