
Abstract
Castleman's disease is a primary infectious disease of the lymph node that causes local symptoms or a systemic inflammatory syndrome. Histopathology reveals a destroyed lymph node architecture that can range from hyaline-vascular disease to plasma-cell disease. Viral interleukin 6 (vIL-6) produced during the replication of human herpesvirus type 8 (HHV8) is the key driver of systemic inflammation and cellular proliferation. Stage progression of Castleman's disease results from switches between viral latency and lytic replication, and lymphatic and hematogenous spread. Multicentric plasma-cell disease in HIV-1 patients is associated with HHV8 infection. Polyclonal plasmablast proliferation escapes control in the germinal center with eventual malignant transformation into non-Hodgkin lymphoma. Surgery produces excellent results in unicentric disease, while multicentric disease responds to anti-CD20 therapy or IL-6 and chemotherapy. Lymphovascular endothelium and naive B cells are infectious reservoir-opening options for antiangiogenic and anti-CD19 strategies to enhance outcomes in patients with systemic disease.
Key Points
-
Castleman's disease is a rare primary disease of the lymph node, which undergoes universal architectural changes
-
Categories of unicentric versus multicentric disease, or hyaline-vascular disease versus plasma-cell disease, represent parts of the same pathogenetic spectrum
-
Infection by human herpesvirus type 8 drives systemic inflammation and cellular proliferation via viral interleukin-6 (vIL-6). Complementing HIV infection always induces multicentric plasma cell disease
-
Infection is propagated in two cellular compartments: lymphovascular endothelial cells and plasma cell precursors. They differ in proliferation dynamics and vIL-6 production resulting in pleiomorphic pathological and clinical presentations
-
Surgery cures unicentric disease, while multicentric disease needs systemic therapy
Similar content being viewed by others


Introduction
Castleman's disease was first described in a case report by Castleman and Towne1 in 1954, which was followed by a series in 19562. It is a unicentric or multicentric disease of the lymph node with or without polyclonal proliferation of B cells. Histopathology identifies a hyaline-vascular type, a plasma-cell variant or a mix of both.3 A number of high-quality reviews on Castleman's disease have been published.4,5,6,7,8,9,10,11,12 This Review discusses its stepwise disease progression as a consequence of chronic viral infection, which involves cytokine-driven switches between latency and replication in two reservoirs of differing cell proliferation dynamics.
Histopathology
Castleman's disease is characterized by significant architectural changes in all lymphatic compartments.Lymph nodes are enlarged and often transformed into scar-like areas.1,2 Consequently, a surgical sample of a diseased lymph node is most likely to provide the needed material for comprehensive histopathology while cytopathology is of limited value.13 Lymphoid follicles in Castleman's disease are increased in number and display fundamental abnormalities, the diagnosis of which has resulted in several redundant disease names such as follicular lympho-reticuloma,14 angiofollicular lymph node hyperplasia,15 angiomatous lymphoid hamartoma,16 benign giant lymphoma,17 and lymphoid or lymphnodal hamartoma;18 these terms should be superseded by the unifying term Castleman's disease. Two main histopathology variants have been identified (Figure 1 and Table 1): the hyaline-vascular type and the plasma-cell type. Frequent transitions between types have led to the identification of the mixed type that is reported in 15% of cases.3 The other major pathological classification scheme is that of unicentric versus multicentric disease. Keller et al.3 determined that unicentric disease is not equivalent to unifocal disease: “in each plasma-cell case, multiple discrete lymph nodes made up the clinically observed mass, unlike the single rounded mass found in most of the hyaline-vascular cases”, a finding confirmed later.11 In the absence of HIV infection, unicentric disease is most frequent, while HIV-positive patients invariably present with multicentric disease.

Hyaline-vascular Castleman's disease with a | eosinophilic hyaline scars and b | onion skin lesions. Plasma-cell variant Castleman's disease with c | hyperplastic germinal centers and d | sheets of mature plasma cells and paracortical plasma cell proliferation filling the sinusoids. Representative markers in plasma-cell variant Castleman's disease showing e | tight clusters of dendritic cells in the follicle centers with no meshwork expansion and f | presence of human herpesvirus type 8-infected plasmablasts in the vicinity of the germinal centers.
B-cell proliferation in Castleman's disease is varied and is initially polyclonal in origin.19,20 A key difference between hyaline-vascular disease and plasma-cell disease relates to the follicular dendritic cell network. This network is normal in plasma-cell disease, while it is expanded or disrupted in hyaline-vascular disease with multiple tight collections of follicular dendritic cells.21 Morphological findings in hyaline-vascular disease are close to pathognomonic. However, it is difficult to differentiate the plasma-cell disease from other conditions such as reactive changes in autoimmune disease, Castleman's disease-like changes associated with HIV infection, plasmacytic lymphoma, marginal zone lymphoma with plasmacytoid differentiation, plasmacytoma, and plasmablastic lymphoma. Under such circumstances, clinical information becomes crucial to establish the diagnosis: the coincidence of hyalinized germinal centers together with an intense interfollicular plasmacytic hyperplasia in a HIV-positive patient with accompanying fever, lymphadenopathy, splenomegaly and cytopenia is highly suggestive of multicentric Castleman's disease.10 Indeed, the lymph nodes of these individuals frequently contain Kaposi lesions, supporting the diagnosis.22 Detection of human herpesvirus type 8 (HHV8) latency-associated nuclear antigen (LANA) is crucial23 because multicentric disease is invariably associated with a positive HHV8 status. Absence of HHV8 virtually excludes HIV positive-related Castleman's disease.24,25 Up-to-date reviews on classification11 and viral association26 provide more detail on these issues.
Classification and clinical staging
The classification of Castleman's disease has historically been based on clinical presentation (unicentric disease versus multicentric disease) and histopathogenic type (hyaline-vascular disease versus plasma-cell disease).3,11 This defines unicentric hyaline-vascular disease and multicentric plasma-cell disease; however, significant overlap of these disease features establishes diagnosis of a mixed type in 15% of patients. Many patients are assigned to a type on the basis of predominant clinical findings rather than total concordance with either end of the spectrum. Coexistence of hyaline-vascular disease and plasma-cell disease has been identified in most patients in whom multiple lymph nodes have been examined in vivo or on autopsy.27 This fosters the argument of Keller et al.3 that hyaline vascular features might represent scars of plasma-cell disease and thus reflect the evolution of Castleman's disease over time rather than represent absolute categories of the disease.
Unicentric and multicentric disease are hence part of a continuous spectrum ranging from unifocal disease (one lymph node), and unicentric disease (one main lymph node surrounded by a group of satellites),2,3 to multicentric disease (multiple groups of lymph nodes in multiple locations).11 Accordingly, unicentric disease based in a solid organ, such as the spleen, is also identified in draining locoregional lymph nodes.22
A model of progressive viral infection
HHV8 (also called Kaposi sarcoma-associated herpesvirus) is a rhadinovirus.28 A large genome enables these viruses to modulate and evade the immune control of their hosts with frequent transitions between latent and replicating lytic viral disease.28 This transition explains the clinical variability between unapparent, smoldering, and progressive disease, focal and systemic illness, and benign scarring and malignant transformation. We consider Castleman's disease as a model case of this pleiomorphic process of adaptation and immune evasion, which depends on host factors and is deeply influenced by complementing viral infections.
HHV8
The prominent role of HHV8 in the pathogenesis of Castleman's disease is undisputed.12,29,30,31 HHV8 infection is ubiquitous in HIV-positive Castleman's disease, and present in the majority of HIV-negative patients with multicentric disease. Mechanisms of molecular viral pathogenesis are well understood on the cellular level, but less so on the level of tissues, organs and the whole body (Figure 2). HHV8 was found to be causally related to Kaposi sarcoma in HIV-positive and HIV-negative patients in the mid 1990s.25,32 The target of HHV8 infection in Kaposi sarcoma is vascular endothelial cells, which are subsequently transformed into the abundant spindle cells embedded in fibrovascular stroma typically containing hyalinized granules.33 The infection is mostly latent and viral replication occurs at a low level.31 HHV8 infection is also associated with primary effusion lymphomas or body-cavity-based lymphomas.31,34 These are B-cell-derived lymphomas that are often coinfected with Epstein-Barr virus (EBV).35

Circles correspond to the number of observed patients. Arrow width corresponds to mean IL-6 levels (the wider the arrow, the higher the IL-6 level). HHV8 particle numbers correspond to virus status. HIV particle numbers correspond to prevalence of infection status. Abbreviations: HHV8, human herpesvirus type 8; IL-6, interleukin 6.
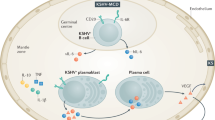
Kaposi sarcoma and Castleman's disease share a number of features: both conditions are inflammatory, neoplastic, related to HHV8 infection, associated with hyalinizing processes, produce a fibrovascular stroma, and are often associated with HIV infection. In HIV-positive patients with Castleman's disease, the lymph nodes are often also involved by Kaposi sarcoma,22 which usually affects endothelial cells.33 It has been hypothesized that the association of Castleman's disease and Kaposi sarcoma in a single lymph node is due to lytic HHV8 infection of B-lymphoid cells. This exposes susceptible endothelial cells to extreme HHV8 viral loads resulting in the formation of Kaposi sarcoma tumorlets in multicentric Castleman's disease-related lymph nodes.22 In our opinion, the pathogenic pathway might work both ways. Double immunostaining with antibodies directed towards latent nuclear antigen-1 (LNA-1) and a viral analog of interleukin 6 (vIL-6; encoded by HHV8 ORFK2)36,37,38 in both multicentric Castleman's disease and Kaposi sarcoma identifies a further important similarity: both HHV8-infected vascular cells and HHV8-infected lymphoid cells serve as infective reservoirs.39 In Castleman's disease, specific double immunostaining for HHV8 products LNA-1 and vIL-6 identifies infected lymphovascular epithelial cells, some of which exclusively express LNA-1 (corresponding to latency), while some coexpress LNA-1 and vIL-6.40 Expression of vIL-6 is induced by the lytic program of viral replication.40,41 The lytic cycle of HHV8 in lymphovascular cells leads to destruction of these cells resulting in a hyaline scar, while production of vIL-6 causes neo-angiogenesis with formation of penetrating vessels via basic fibroblast growth factor and VEGF.42 The result is an expansion of the lymphovascular epithelial cell reservoir of HHV8-infected cells (Table 1). In addition, the chemotactic effect of human IL-6 leads to cluster formation of plasmablasts around small vessels (Figure 3).

In hyaline-vascular disease the virus is latent in the VEC and vIL-6 production occurs at a low level. In plasma-cell disease there is crossinfection of significant numbers of perivascular plasmablasts inducing massive viral replication resulting in high levels of vIL-6 and vIL-6-induced bFGF and VEGF. The multiple arrows in plasma-cell disease correspond to the exponential increase in IL-6 with an increase in HHV8 replication. Abbreviations: bFGF, basic fibroblast growth factor; HHV8, human herpesvirus type 8; IL-6, interleukin 6; VEC, vascular epithelial cell; vIL-6, viral IL-6.
IL-6
The IL-6-like cytokines activate the glycoprotein 13 chain, which results in Janus kinase-mediated activation of the signal transducers and activators of transcription (STATs).43 Alternatively, IL-6 activates the mitogen-activated protein kinase (MAPK) cascade.44 IL-6 production has a triple role in Castleman's disease: it induces B-cell proliferation, mediates inflammatory clinical symptoms, and induces VEGF and angiogenesis.45,46 Germinal centers of hyperplastic lymph nodes produce large quantities of IL-6 and removal of such lymph nodes results in clinical improvement and a decrease in IL-6 levels.47 Subsequent dissection of the lymph node revealed increased IL-6 expression in interfollicular areas (other cytokines are absent), whereas elevated IL-6 gene expression inside follicles only occurs in the localized form of the disease associated with systemic manifestations.47 Administration of antibodies directed towards IL-648 or the IL-6 receptor49 alleviate systemic symptoms in patients with Castleman's disease. It is unclear whether the IL-6 excess predominantly derives from human or viral sources (IL-6 versus vIL-6) as expression can be independent of HIV status.30 However, it is known that vIL-6 activates angiogenesis and hematopoiesis pathways in a similar manner to human IL-6 (hIL-6),50 and increases expression of hIL-6.51 In contrast to human IL-6, the effects of vIL-6 are not dependent on, but are enhanced by, the gp80 path of the IL-6 receptor.52 Thus, vIL-6 is a 'pirate' cytokine with optimized and unregulated IL-6 functions, and is the key mediator for expansion of both infective compartments in Castleman's disease: plasma cell precursors and lymphovascular endothelial cells (Figure 3).
B-cell populations
B-cell immunity depends on specialized lymphoid compartments containing distinct B-cell populations.53 Maturation pathways of the extrafollicular T-zone generate plasma cell precursors with unmutated immunoglobulin genes. Activated B cells are primed in the germinal center to become memory cells or plasma cells with a mutated immunoglobulin gene.54 The mantle zone around the germinal center contains mostly naive B cells that secrete high levels of IgD and are prepared for mitosis. These cells are hypothesized to be the origin of mantle-cell lymphoma.55 By contrast, B cells of the splenic marginal zone are IgM-high, IgD-low and express CD27.56 In Castleman's disease, HHV8 virus particles are predominantly, but not solely, identified in the mantle zone of the lymph node.6 In lymphoid cells, vIL-6 is expressed around lymphoid follicles and within the follicular dendritic reticulum network. Detection of IL-6 in apoptotic bodies suggests that a complete lytic cycle is achieved in a subset of CD138− lymphoid cells.39 HHV8-positive plasmablasts express interferon-regulatory factor 4 (IRF4, also called MUM1) and B-lymphocyte-induced maturation protein 1 (BLIMP1). They are negative for paired box protein 5 (PAX5) and CD30, and positive for the proliferation marker Ki67.10 The expression of CD20 is not universal. In unicentric Castleman's disease HHV8 virus infection might be restricted to CD19+ cells, while it might involve the T-cell population in multicentric disease.57
The target cell for HHV8 infection is a plasma cell precursor or plasmablast at the stage of non-mutated immunoglobulin production, and infection seems to occur in the mantle zone of the lymph node or around vascular structures of the lymph nodes. Here, plasma cell precursors cluster around the vessels, possibly as a result of chemotaxis induced by IL-6 or vIL-6.
Lymphovascular epithelial cells and B cells are involved at entirely different levels of intensity, in terms of the lytic cycles of HHV8 infection in Castleman's disease. For the lymphovascular epithelial cell population, viral replication results in cell death and the slow formation of neovasculature corresponding to the moderate mitotic potential of these cells. By contrast, the vIL-6 driven expansion in the plasmablast population implies exponential growth of a cell population with high mitotic potential for which IL-6 is a natural key proliferogenic factor. The critical difference between the processes in these two cell populations is the mitotic potential of the two compartments.
Anatomy and virus-based pathogenetic pathway
HHV8 infection targets two cellular compartments: the vascular endothelial cells and plasma precursor cells (Figure 3). Replication rate in vascular endothelial cells is as low as one copy per cell, resulting in low vIL-6 production. Perivascular chemotaxis and proliferation of plasmablasts results in apoptosis or cell lysis of both cell compartments with formation of the radial scars of hyaline-vascular disease. This follows temporary lymph-node swelling and eventual low grade systemic symptoms. Crossinfection of significant numbers of perivascular plasmablasts induces massive viral replication, which produces high levels of vIL-6 with paracrine stimulation of plasmablast proliferation. This amplifies the local target-cell population followed by enhanced viral infection, lysis, and vIL-6 production. The outcome is an exponential escalation of IL-6 secretion into the systemic circulation causing a systemic inflammatory syndrome. The transition of latency into aggressive replication occurs stochastically, explaining the coexistence of different forms of Castleman's disease in one patient.27
Unicentric plasma-cell disease consists of a cluster of lymph nodes rather than a single lymph node.11 This suggests that the infectious agent, as well as IL-6, is transported to the target cells primarily via the locoregional lymphovascular drainage system. The systemic rise of IL-6 eventually causes multifocal expansion of the vulnerable plasmablast pool resulting in extraregional multicentric disease. In HIV-negative patients, unicentric and multicentric Castleman's disease is observed in the absence of HHV8 infection, but with different clinical, pathological and laboratory findings. In HIV-negative patients with multicentric Castleman's disease, HHV8 infection is detected in about 50% of cases, while in unicentric Castleman's disease HHV8 infection has been detected in only a minority of patients (K.-M. Schulte & N. Talat, unpublished data).
We propose an anatomical model of disease progression initiated by primary viral infection at any level, most frequently involving the upper respiratory tract. The lymphatic fluid leaving the infected tissue is filtered in the locoregional draining 'sentinel' lymph node, which develops a lymphovascular endothelial infection provoking a T-cell-mediated immune response. Clinically, this initial infection presents as unicentric disease associated with local symptoms and is essentially curable by surgical excision. Eventually, lymphovascular endothelial containment of the virus in this sentinel lymph node is lost, engaging plasmablasts in a lytic viral infection. This causes B-cell infection and lymphatic transport into the surrounding lymph node and eventually into the blood, generating multicentric disease. At this stage symptoms are systemic and prognosis is poor. Disease progression corresponds to the biphasic progression of solid organ cancer with initial lymphatic and secondary blood-borne propagation. The induction process in the absence of HHV8 infection remains elusive, but causation by an unidentified viral agent with lower pathogenicity seems likely. Such a pathogen might also cause associated pathologies including 'paraneoplastic pemphigoid', predominantly identified in cases of Castleman's disease without HHV8 infection.
Concomitant viral infections
In Castleman's disease, HHV8 infects monotypic, but not polyclonal, naive B cells and is associated with a range of lymphoproliferative disorders, including microlymphomas and plasmablastic lymphomas.58 These observations add a second layer of insight into infection with HHV8; transformation into B-cell lymphomas is observed in diseases ranging from primary effusion lymphomas to the plasma-cell type of Castleman's disease. The context appears to be defined by the co-infecting agent, such as HIV or EBV.12 In humans, LANA is critical to induce B-cell hyperplasia and lymphoma.59 HHV8 is specifically associated with a variant of multicentric Castleman's disease in which lambda light-chain restricted, HHV8-positive plasmablasts localize in B-cell follicle mantle zones and, in some cases, form microscopic lymphomas.23 Animal models have helped to elucidate mechanisms of viral complementation. Rhesus rhadinovirus 17577 is a close phylogenetic relative of HHV8. In macaques previously infected with simian immunodeficiency virus, infection with rhesus rhadinovirus 17577 induces a phenotypically similar disease to human multicentric Castleman's disease in HIV-positive patients.60,61,62 Both Kaposi sarcoma and multicentric Castleman's disease are rare in HIV-2 patients and a first case of the latter was only recently reported.63 Complementary effects of viral co-infection are hence specific to HHV-8-specific CD8+ T cells rather than related to defects in the T-cell repertoire.64 HHV8-negative Castleman's disease does not exist in HIV-positive patients, which indicates that strong selection pressures related to HIV factors promote HHV8 infection. A molecular candidate is the transactivating HIV protein Tat that is capable of suppressing immunity and apoptosis. Tat synergizes with angiogenic factors and upregulates IL-6 expression by activating the IL-6 promoter.65 Overall, viral infection defines three subtypes of Castleman's disease: absence of HHV8 and HIV infection with excellent prognosis (unicentric Castleman's disease), presence of HHV8 and HIV infection with poor prognosis (multicentric Castleman's disease), and an intermediate disease subtype associated with HHV8 infection only.
Essential clinical staging
The purpose of staging is to assign risk and prognosis, and to guide treatment decisions from initial presentation to long-term therapy. Practical staging of Castleman's disease needs to address three key clinical questions. Firstly, what is the extent of disease? Imaging by CT, MRI, or PET will clarify whether the disease is unicentric or multicentric. It will also reveal the eventual transition of the cells into solid forms of malignancy, such as lymphoma. Evaluation of the blood smear, differential analysis of blood cell counts, and an eventual bone marrow or effusion aspirate will demonstrate non-solid malignancy. Secondly, what is the histopathological type? Surgical biopsy of an accessible lymph node will classify the patient as having hyaline-vascular, plasma-cell or mixed type Castleman's disease. Finally, what is the viral etiology? Blood-based tests for HIV-1 antibody, HIV-2 antibody, and the viral load will clarify the HIV-status. Serology for EBV and PCR-based EBV detection may be useful in suspected primary effusion lymphoma. Immunostaining of lymphatic tissue for LANA, vIL-6, and eventually hIL-6 will clarify HHV8 association. Where available, information about HHV8 viral load may also help guide clinical decisions.
The choice of imaging
The main purpose of imaging in Castleman's disease is to facilitate disease staging, that is to clarify if the disease is unicentric or multicentric (a second vital purpose is to define surgical access and resectability). The best modality seems to be CT with analysis of the anatomical regions harboring the majority of the lesions, including the neck, chest, abdomen and pelvis.2,66,67 Mediastinal lymphadenopathy is frequently observed and easily identifiable on chest x-rays or CTs. No unique features allow safe differentiation from lymphoma, but the presence of hypervascularity is generally indicative of Castleman's disease.68,69 Calcification might be observed. The combination of mediastinal lesions with reticular or nodular interstitial lung changes, representing acute reticulonodular interstitial pneumonitis, might indicate Castleman's disease in HIV-positive patients.70 18F-fluorodeoxyglucose (FDG)-PET or PET-CT might identify multifocality.71,72
Clinical presentation
Clinical symptoms fall into four categories. First, local compression effects due to enlarged lymph nodes or extranodal Castleman's disease, with a mean lymph node size of 5.4 cm in unicentric disease. The mean size of the lymph nodes in multicentric disease is considerably smaller. Second, systemic B-cell-related symptoms due to cytokine activation with fever, weight loss, night sweats, weakness, and fatigue increase with tumor burden and histopathological type. Third, fluid retention in individuals with multicentric disease may be pronounced with edema, ascites, pleural or pericardial effusions, and seizures. Many such patients display multifocal lymphadenopathy and hepatosplenomegaly. Finally, symptoms of complications or associated diseases of Castleman's disease, such as lymphoma, paraneoplastic pemphigus, HIV, or polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes (POEMS) syndrome. Manifestations are nonspecific and the diagnosis is made by histopathology. No combination of symptoms safely differentiates Castleman's disease from a longer list of differential diagnoses.
As a hard and fast rule, unicentric hyaline-vascular Castleman's disease mostly causes local compression symptoms, while patients with the multicentric plasma-cell variant typically present with systemic symptoms. It is key, however, to carefully examine these patients in order to exclude diseases with a significantly overlapping presentation ranging from viral infection, to rheumatic or vasculitic disease.4 In this respect, a timely lymph-node biopsy is of the utmost importance.
Castleman's disease and HIV infection
HIV infection does not cause Castleman's disease. It is, however, a permissive factor and is associated with typical epidemiological and clinical features of Castleman's disease as well as a marked enhancement in disease progression.5,8 Oksenhendler10 has recently proposed the following clinical indicators to identify Castleman's disease in HIV-positive individuals: fever, diffuse lymphadenopathy, splenomegaly, severe cytopenia, high serum C-reactive protein, high HHV8 DNA levels in peripheral blood mononuclear cells, intense plasmocytosis in the lymph nodes or bone marrow, nasal obstruction, respiratory symptoms, Kaposi sarcoma lesions, prior identical episode with spontaneous resolution, positive direct agglutination test (Coombs test), and hemophagocytic syndrome. Patients with Castleman's disease may become systemically severely ill from unusual pulmonary complications, such as acute reticulonodular interstitial pneumonitis, and develop acute respiratory distress syndrome.73 Administration of therapy specific for Castleman's disease is generally sufficient to treat these complications.
Immune reconstitution inflammatory syndrome74 might explain the observation that some patients develop Castleman's disease within weeks of starting highly active antiretroviral therapy.75 Seemingly, a degree of immune competence is necessary to establish the signs and symptoms of Castleman's disease. This coincides with the observation that CD4 counts and HIV viral titers are not correlated or are inversely correlated with the development of Castleman's disease.5 A review5 of 25 studies including 84 HIV-positive cases coincides with clinical findings outlined by Oksehendler. Focal pathological changes involving Kaposi sarcoma were found in 72% of the patients and were in the respiratory tract of 34% of patients.
Associated conditions
Kaposi sarcoma
Kaposi sarcoma was identified in a minority of Castleman's disease cases without reported HIV-positive status and in about 50% of all reported HIV-positive patients. It is exclusively associated with a positive HHV8 status.23,76
Non-Hodgkin lymphoma
The high incidence of HHV8-associated non-Hodgkin lymphoma,77 supports the idea of clonal expansion following polyclonal activation.10 Non-Hodgkin lymphoma has been reported in <5% of Castleman's disease cases without reported HIV-positive status, and in <20% of those who are HIV positive. A prospective cohort study of 60 HIV-positive patients with Castleman's disease found a 15-fold increase in lymphomas in comparison with HIV-negative patients. These included HHV8-positive and EBV-positive primary-effusion lymphoma, HHV8-positive and EBV-negative visceral large-cell non-Hodgkin lymphoma, and plasmablastic lymphoma/leukemia.77 The latter is a large B-cell lymphoma, which is different from classic plasmablastic lymphoma. It originates from naive B cells with IgM expression at the stage preceding immunoglobulin hypermutation.78 HHV8 infection is frequently observed in HIV-related solid immunoblastic/plasmablastic diffuse large B-cell lymphoma.79 Ki76-staining might differentiate benign from malignant B-cell proliferation.80 Overall, the association of Castleman's disease with Hodgkin lymphoma remains elusive.77,82,83,84,85,86,87
Paraneoplastic pemphigus
Paraneoplastic pemphigus presents as debilitating chronic blistering of the skin and mucous membranes, including oral mucositis and cutaneous lichenoid lesions. It is associated with lymphoproliferative neoplasms. Autoantibodies against plakin are useful diagnostic markers.88
POEMS syndrome
POEMS syndrome is a plasma-cell dyscrasia with excess levels of VEGF.89 More than 95% of patients have one or more monoclonal lambda sclerotic plasmacytomas or bone marrow infiltration.89 Biopsy of an osteosclerotic lesion or a lymph node typical of Castleman's disease are considered essential to establish the diagnosis and may be guided by FDG-PET.90 HHV8 infection is strongly associated with POEMS syndrome,91 but is not essential for the establishment of this condition; there are at least seven reported cases of POEMS in the absence of HHV8 infection.91,92,93
Follicular dendritic cell tumors
Follicular dendritic cell tumors originate from the dendritic cell network of the lymph node21 and may be either polyclonal or clonal.94,95,96 Such tumors can become malignant and form dendritic cell sarcomas in either the lymph node or the skin.97,98 This cancer is not associated with HHV8 infection.99 However, it is of note that dendritic cell sarcomas appear in the context of paraneoplastic pemphigus,100 a condition that has only been reported in HHV8-negative patients. This observation supports the hypothesis that a hitherto unidentified virus is responsible for driving Castleman's disease and associated diseases in these patients.
Treatment modalities—outcomes
Once a diagnosis of Castleman's disease has been established, it is of vital importance to determine whether a patient has unicentric or multicentric disease. The former responds to local approaches, while the latter mostly requires systemic therapy. Prospective, randomized data are sparse and evidence has mainly been derived from small series; as such, the following paragraphs should be read with this important caveat in mind.
Unicentric disease
Unicentric Castleman's disease responds to local ablation therapy. Careful preoperative staging is mandatory to prevent the predictable failure of local therapy in systemic disease. Active surveillance is often considered as an option for patients with asymptomatic unicentric disease. Surgery, in terms of local resection, is the option of choice for unicentric disease when it is unlikely to inflict locoregional complications. Frequently involved and readily accessible sites include cervical, axillary, and inguinal lymph-node stations, which should be cleared by a systematic lymph-node dissection. The appropriate set-up is paramount: an experienced surgeon, free-resection margins at reasonable cost as opposed to circumferential R1-excision, systematic locoregional excision as opposed to excision of the targeted lymph node is needed to ensure proper histopathological staging. This might identify otherwise unrecognized transgression into systemic disease indicating a risk of relapse and the need for systemic treatment. Neck, mediastinal, abdominal, pelvic and retroperitoneal sites account for the majority of unicentric locations. At present, such lesions can be targeted with low morbidity using open or endoscopic techniques. Radiotherapy is preferable in cases when anatomy or patient-related features render safe or complete excision unlikely.101,102
Multicentric disease in HIV-negative patients
Multicentric disease is almost invariably symptomatic, and so therapy is indicated and is generally agreed to improve outcome. Experience in dealing with HIV-negative patients with Castleman's disease before the availability of specific chemotherapy and immunomodulatory agents shows that persistent or worsening symptoms and secondary complications are strongly associated with untreated disease.103 Therapy aims to cure the underlying infection such as HHV8, to modify IL-6-related cytokine escalation, to reduce the proliferation of B cells, to debulk tumor mass, or to compensate for the immunodeficiency resulting from the loss of lymph-node functions.
Antiviral therapy directed at non-HIV viruses
Antiviral therapy might improve outcomes in HHV8-positive patients. Potential agents include ganciclovir, valganciclovir, or foscarnet. The efficacy of valganciclovir to suppress HHV8 replication was documented in a prospective randomized trial.104 There is no evidence that EBV-directed therapy has any role in treating Castleman's disease.
Modulation of the cytokine network
The nonspecific, immune-suppressive effects of corticosteroids might reduce lymph node size, clinical symptoms, and inflammatory markers in HIV-negative patients with Castleman's disease. However, their short-lived effect and significant side effects preclude their use beyond a transient setting. The monoclonal antibody tocilizumab blocks the human IL-6 receptor and antagonizes the crucial role of IL-6 in disease propagation and manifestation. Accordingly, this agent also reduces the size of involved lymph nodes.105
Cytostatic therapy
Thalidomide can be used with considerable success in HIV-negative patients with Castleman's disease, although the multiple biological effects related to use of this agent are incompletely understood.106,107 In the context of treating Castleman's disease, the teratogenic side effects of thalidomide are of secondary concern. Chemotherapy with cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP) is frequently employed in the treatment of systemic disease. Cyclophosphamide, vinblastine and etoposide can also be used as single agents.
Multicentric disease in HIV-positive patients
HIV therapy alters the spontaneous evolution of Castelman's disease. Treatment outcomes in HIV-positive patients with Castleman's disease have recently been reviewed.5,10 At present, multimodal therapy achieves similar outcomes in multicentric HIV-positive patients with Castleman's disease in comparison with HIV-negative patients. Single-agent therapy rapidly resolves acute clinical and laboratory findings. Etoposide given at a single intravenous dose (120–150 mg/m2) followed by weekly oral maintenance therapy (100–120 mg/m2) provides good disease control.108 However, bone marrow suppression and oncogenic effects make this regimen unsuitable for long-term use.
Rituximab, an anti-CD20 monoclonal antibody, achieves high response rates in HIV-positive patients with Castleman's disease. A phase II trial using rituximab in 21 patients achieved 2-year overall survival and disease-free survival rates of 95% (95% CI 86–100%) and 79% (95% CI 49–100%), respectively.109 Plasma acute-phase proteins, immunoglobulins, and HHV8 viral load decreased following initiation of therapy. The prospective, open-label, ANRS 117 CastlemaB Trial demonstrated sustained remission in 24 chemotherapy-dependent patients using rituximab.110 Disease was stable under chemotherapy, sustained remission was observed in more than 90% of patients at 2 months and the overall survival rate was also above 90%, and patients tolerated rituximab remarkably well. Moderate infections accounted for the majority of adverse effects. Both studies identified reactivation of formerly controlled Kaposi sarcoma.109,110 As a caveat to these findings, recent reports identified a total failure of rituximab therapy resulting in rapid death,111,112 indicating that it is not safe as single agent, specifically in aggressive disease.
The combination of immunotherapy and chemotherapy, termed immunochemotherapy, might enhance safety or efficacy. A case series using a mix of anthracycline-based chemotherapy and rituximab in HIV-positive patients with Castleman's disease has shown good response rates.113 Polychemotherapy with CHOP has induced long-term remission, but the target group needs to be more precisely defined.5 Vinblastine has induced life-threatening drug interactions with ongoing antiretroviral treatment.114
Potential new treatment pathways
Immunotherapy with anti-CD20 antibodies depletes mature B cells, but not pre-B cells, certain peripheral B-cell subpopulations, most antibody-producing cells, or B-cell malignancies. Mature B cells can also be targeted with a monoclonal antibody to CD19.115 Indeed, the CD19+ cell population can remain unaffected following treatment with monoclonal anti-CD20 antibody.116 In unicentric disease, the infected cell population may be restricted to CD19+ cells,57 encouraging the consideration of monoclonal anti-CD19 antibody therapy in systemic disease.
Treatment failure can also be interpreted in the context of the two-compartment model of cellular infection. Latent HHV8 infection of the lymphovascular endothelial compartment escapes the effects of a complete eradication of the B-cell compartment, and infection there might manifest as latent antigen expression only. However, the alternative pathway of very low-copy viral replication is a likely origin of recurrent disease. There is little experience in how to target a latent viral reservoir in endothelial cells. Long-term therapy with antiviral-based analogs might be an option. The complex and targeted antiangiogenic effects of thalidomide on the endothelial compartment have not yet been fully elucidated,116,117 but the drug seems to be a viable treatment option given its effectiveness in other hematological malignancies.117 The evidence supports administration of rituximab followed by thalidomide for maintenance, which has produced complete regression in some cases.118
Bortezomib also targets the endovascular compartment and reversibly inhibits the 26S proteasome, which normally functions to degrade ubiquitinated proteins. This leads to cell-cycle arrest, apoptosis, and inhibition of angiogenesis. A specific feature of this agent is the inhibition of nuclear factor (NF)-kappaB. Bortezomib has been shown to reduce proinflammatory cytokine levels and transfusion dependency in a patient with multicentric Castleman's disease.117,119
Future therapy might target both cellular compartments in a sequential manner. Anti-CD19 therapy would comprehensively destroy the lymphatic compartment of Castleman's disease (the proliferating plasmablast and B-cell precursor pool) without damaging the stem-cell population (the pro-B cell of the bone marrow). Thalidomide or bortezomib could then contain the latent HHV8 infection, which causes inflammatory and angiogenic disease propagation in the lymphovascular compartment.
Conclusions
Castleman's disease is a paradigm of chronic viral infection manifesting as solid lymphatic disease. It exemplifies the potential of rhadinoviruses, with their large genomes, to sustain chronic disease through skillful immune evasion and low-copy replication. Multiple swaps between latency and lytic cycle affecting two fundamentally different cell pools are ectopically enhanced by secretion of the viral cytokine vIL-6, which becomes the pacemaker not only of viral replication itself but of the ever growing target-cell pool for viral infection. Co-infection with HIV transforms local disease into systemic disease on the basis of effective molecular inter-viral cooperation in the presence of HIV infection. The sustained presence of HHV8 infection can lead to specific lymphatic conditions due to the transforming potential shared by human rhadinoviruses. While initial and local disease can be effectively treated by the ablation of the effective focus, systemic modalities are needed to control regional and extra regional spread. Multi modal approaches targeting both infected cellular reservoirs, that is B cells and endothelial cells, seem more promising than attempts to contain infection in either compartment alone. Chronic rhadinoviral infection is overwhelmingly common, and 50 years of research into Castleman's disease has provided essential contributions to the understanding and therapy of related human disease.
Review criteria
We searched the NCBI PubMed database in September 2009 using the search term “Castleman's disease” without further constraints. This identified 1,791 articles in English, French, Spanish, and German. We read the abstracts of all articles and studied the full text of all cited references. These were also searched for further leads using the Thompson ISI Web of Science software.
References
Castleman, B. & Towne, V. W. Case records of the Massachusetts General Hospital; weekly clinicopathological exercises; founded by Richard C. Cabot. N. Engl. J. Med. 251, 396–400 (1954).
Castleman, B., Iverson, L. & Menendez, V. P. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer 9, 822–830 (1956).
Keller, A. R., Hochholzer, L. & Castleman, B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer 29, 670–683 (1972).
Dispenzieri, A. Castleman disease. Cancer Treat. Res. 142, 293–330 (2008).
Mylona, E. E. et al. Multicentric Castleman's disease in HIV infection: a systematic review of the literature. AIDS Rev. 10, 25–35 (2008).
Casper, C. The aetiology and management of Castleman disease at 50 years: translating pathophysiology to patient care. Br. J. Haematol. 129, 3–17 (2005).
Dham, A. & Peterson, B. A. Castleman disease. Curr. Opin. Hematol. 14, 354–359 (2007).
Stebbing, J. et al. HIV-associated multicentric Castleman's disease. Am. J. Hematol. 83, 498–503 (2008).
Kessler, E. Multicentric giant lymph node hyperplasia. A report of seven cases. Cancer 56, 2446–2451 (1985).
Oksenhendler, E. HIV-associated multicentric Castleman disease. Curr. Opin. HIV AIDS 4, 16–21 (2009).
Cronin, D. M. & Warnke, R. A. Castleman disease: an update on classification and the spectrum of associated lesions. Adv. Anat. Pathol. 16, 236–246 (2009).
Hengge, U. R. et al. Update on Kaposi's sarcoma and other HHV8 associated diseases. Part 2: pathogenesis, Castleman's disease, and pleural effusion lymphoma. Lancet Infect. Dis. 2, 344–352 (2002).
Mallik, M. K., Kapila, K., Das, D. K., Haji, B. E. & Anim, J. T. Cytomorphology of hyaline-vascular Castleman's disease: a diagnostic challenge. Cytopathology 18, 168–174 (2007).
Zettergren, L. Probably neoplastic proliferation of lymphoid tissue (follicular lympho-reticuloma). Reports of four cases with a survey of literature. Acta Pathol. Microbiol. Scand. 51, 113–126 (1961).
Harrison, E. G. Jr & Bernatz, P. E. Angiofollicular mediastinal lymph-node hyperplasia resembling thymoma. Arch. Pathol. 75, 284–292 (1963).
Tung, K. S. & McCormack, L. J. Angiomatous lymphoid hamartoma. Report of five cases with a review of the literature. Cancer 20, 525–536 (1967).
Festen, C., Flendrig, J. A. & Schillings, P. H. [Giant lymphomas]. Ned. Tijdschr. Geneeskd 113, 1918–1919 (1969).
Abell, M. R. Lymphnodal hamartoma versus thymic choristoma of pulmonary hilum. AMA Arch. Pathol. 64, 584–588 (1957).
Al-Maghrabi, J., Kamel-Reid, S. & Bailey, D. Immunoglobulin and T-cell receptor gene rearrangement in Castleman's disease: molecular genetic analysis. Histopathology 48, 233–238 (2006).
Radaszkiewicz, T., Hansmann, M. L. & Lennert, K. Monoclonality and polyclonality of plasma cells in Castleman's disease of the plasma cell variant. Histopathology 14, 11–24 (1989).
Nguyen, D. T. et al. Castleman's disease. Differences in follicular dendritic network in the hyaline vascular and plasma cell variants. Histopathology 24, 437–443 (1994).
Naresh, K. N., Rice, A. J. & Bower, M. Lymph nodes involved by multicentric Castleman disease among HIV-positive individuals are often involved by Kaposi sarcoma. Am. J. Surg. Pathol. 32, 1006–1012 (2008).
Dupin, N. et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 95, 1406–1412 (2000).
Oksenhendler, E. et al. High levels of human herpesvirus 8 viral load, human interleukin-6, interleukin-10, and C reactive protein correlate with exacerbation of multicentric castleman disease in HIV-infected patients. Blood 96, 2069–2073 (2000).
Marcelin, A. G. et al. Relationship between the quantity of Kaposi sarcoma-associated herpesvirus (KSHV) in peripheral blood and effusion fluid samples and KSHV-associated disease. J. Infect. Dis. 196, 1163–1166 (2007).
Du, M. Q., Bacon, C. M. & Isaacson, P. G. Kaposi sarcoma-associated herpesvirus/human herpesvirus 8 and lymphoproliferative disorders. J. Clin. Pathol. 60, 1350–1357 (2007).
Ascoli, V. et al. Primary effusion lymphoma in HIV-infected patients with multicentric Castleman's disease. J. Pathol. 193, 200–209 (2001).
Westmoreland, S. V. & Mansfield, K. G. Comparative pathobiology of Kaposi sarcoma-associated herpesvirus and related primate rhadinoviruses. Comp. Med. 58, 31–42 (2008).
Hayashi, M. et al. Kaposi's sarcoma-associated herpesvirus infection in the lung in multicentric Castleman's disease. Intern. Med. 38, 279–282 (1999).
Komatsu, T., Barbera, A. J., Ballestas, M. E. & Kaye, K. M. The Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen. Viral Immunol. 14, 311–317 (2001).
Ablashi, D. V., Chatlynne, L. G., Whitman, J. E. Jr & Cesarman, E. Spectrum of Kaposi's sarcoma-associated herpesvirus, or human herpesvirus 8, diseases. Clin. Microbiol. Rev. 15, 439–464 (2002).
Chang, Y. et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266, 1865–1869 (1994).
Pyakurel, P. et al. Lymphatic and vascular origin of Kaposi's sarcoma spindle cells during tumor development. Int. J. Cancer 119, 1262–1267 (2006).
Cesarman, E., Chang, Y., Moore, P. S., Said, J. W. & Knowles, D. M. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332, 1186–1191 (1995).
Horenstein, M. G. et al. Epstein-Barr virus latent gene expression in primary effusion lymphomas containing Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8. Blood 90, 1186–1191 (1997).
Moore, P. S. & Chang, Y. Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N. Engl. J. Med. 332, 1181–1185 (1995).
Neipel, F. et al. Human herpesvirus 8 encodes a homolog of interleukin-6. J. Virol. 71, 839–842 (1997).
Nicholas, J. et al. Kaposi's sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat. Med. 3, 287–292 (1997).
Brousset, P., Cesarman, E., Meggetto, F., Lamant, L. & Delsol, G. Colocalization of the viral interleukin-6 with latent nuclear antigen-1 of human herpesvirus-8 in endothelial spindle cells of Kaposi's sarcoma and lymphoid cells of multicentric Castleman's disease. Hum. Pathol. 32, 95–100 (2001).
Parravicini, C. et al. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman's disease. Am. J. Pathol. 151, 1517–1522 (1997).
Abe, Y. et al. Distinct expression of Kaposi's sarcoma-associated herpesvirus-encoded proteins in Kaposi's sarcoma and multicentric Castleman's disease. Pathol. Int. 56, 617–624 (2006).
Foss, H. D. et al. Expression of vascular endothelial growth factor in lymphomas and Castleman's disease. J. Pathol. 183, 44–50 (1997).
Nishimoto, N. & Kishimoto, T. Interleukin 6: from bench to bedside. Nat. Clin. Pract. Rheumatol. 2, 619–626 (2006).
Heinrich, P. C. et al. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20 (2003).
Nishimoto, N. et al. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 112, 3959–3964 (2008).
Yoshizaki, K. et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman's disease. Blood 74, 1360–1367 (1989).
Leger-Ravet, M. B. et al. Interleukin-6 gene expression in Castleman's disease. Blood 78, 2923–2930 (1991).
Beck, J. T. et al. Brief report: alleviation of systemic manifestations of Castleman's disease by monoclonal anti-interleukin-6 antibody. N. Engl. J. Med. 330, 602–605 (1994).
Nishimoto, N. et al. Improvement in Castleman's disease by humanized anti-interleukin-6 receptor antibody therapy. Blood 95, 56–61 (2000).
Aoki, Y. et al. Angiogenesis and hematopoiesis induced by Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6. Blood 93, 4034–4043 (1999).
Mori, Y. et al. Human herpesvirus 8-encoded interleukin-6 homologue (viral IL-6) induces endogenous human IL-6 secretion. J. Med. Virol. 61, 332–335 (2000).
Hu, F. & Nicholas, J. Signal transduction by human herpesvirus 8 viral interleukin-6 (vIL-6) is modulated by the nonsignaling gp80 subunit of the IL-6 receptor complex and is distinct from signaling induced by human IL-6. J. Virol. 80, 10874–10878 (2006).
Hardy, R. R. & Hayakawa, K. B cell development pathways. Annu. Rev. Immunol. 19, 595–621 (2001).
MacLennan, I. C. Germinal centers. Annu. Rev. Immunol. 12, 117–139 (1994).
Frater, J. L. & Hsi, E. D. Properties of the mantle cell and mantle cell lymphoma. Curr. Opin. Hematol. 9, 56–62 (2002).
Dono, M. et al. The human marginal zone B cell. Ann. N. Y. Acad. Sci. 987, 117–124 (2003).
Kikuta, H. et al. Tropism of human herpesvirus 8 for peripheral blood lymphocytes in patients with Castleman's disease. Br. J. Haematol. 99, 790–793 (1997).
Du, M. Q. et al. Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in Castleman disease and associated lymphoproliferative disorders. Blood 97, 2130–2136 (2001).
Fakhari, F. D. et al. The latency-associated nuclear antigen of Kaposi sarcoma-associated herpesvirus induces B cell hyperplasia and lymphoma. J. Clin. Invest. 116, 735–742 (2006).
Wong, S. W. et al. Induction of B cell hyperplasia in simian immunodeficiency virus-infected rhesus macaques with the simian homologue of Kaposi's sarcoma-associated herpesvirus. J. Exp. Med. 190, 827–840 (1999).
Estep, R. D., Powers, M. F., Yen, B. K., Li, H. & Wong, S. W. Construction of an infectious rhesus rhadinovirus bacterial artificial chromosome for the analysis of Kaposi's sarcoma-associated herpesvirus-related disease development. J. Virol. 81, 2957–2969 (2007).
O'Connor, C. M. & Kedes, D. H. Rhesus monkey rhadinovirus: a model for the study of KSHV. Curr. Top. Microbiol. Immunol. 312, 43–69 (2007).
Bouvresse, S. et al. The first reported case and management of multicentric Castleman's disease associated with Kaposi's sarcoma in an HIV-2-infected patient. AIDS 21, 1492–1494 (2007).
Guihot, A. et al. Multicentric Castleman disease is associated with polyfunctional effector memory HHV-8-specific CD8+ T cells. Blood 111, 1387–1395 (2008).
Romani, B., Engelbrecht, S. & Glashoff, R. H. Functions of Tat: the versatile protein of human immunodeficiency virus type 1. J. Gen. Virol. 91, 1–12 (2009).
Zhou, L. P. et al. Imaging findings of Castleman disease of the abdomen and pelvis. Abdom. Imaging 33, 482–488 (2008).
Meador, T. L. & McLarney, J. K. CT features of Castleman disease of the abdomen and pelvis. AJR Am. J. Roentgenol. 175, 115–118 (2000).
Ferrozzi, F. et al. Focal Castleman disease of the lung: MRI findings. Clin. Imaging 25, 400–402 (2001).
Yamashita, Y. et al. Radiological presentations of Castleman's disease. Comput. Med. Imaging Graph. 17, 107–117 (1993).
Guihot, A. et al. Pulmonary manifestations of multicentric Castleman's disease in HIV infection: a clinical, biological and radiological study. Eur. Respir. J. 26, 118–125 (2005).
Enomoto, K. et al. Unicentric and multicentric Castleman's disease. Br. J. Radiol. 80, e24–e26 (2007).
Halac, M. et al. PET/CT findings in a multicentric form of Castleman's disease. Hell. J. Nucl. Med. 10, 172–174 (2007).
Guihot, A. et al. Thoracic radiographic and CT findings of multicentric Castleman disease in HIV-infected patients. J. Thorac. Imaging 22, 207–211 (2007).
Dhasmana, D. J., Dheda, K., Ravn, P., Wilkinson, R. J. & Meintje, G. Immune reconstitution inflammatory syndrome in HIV-infected patients receiving antiretroviral therapy: pathogenesis, clinical manifestations and management. Drugs 68, 191–208 (2008).
Zietz, C. Bogner, J. R., Goebel, F. D. & Löhrs, U. An unusual cluster of cases of Castleman's disease during highly active antiretroviral therapy for AIDS. N. Engl. J. Med. 340, 1923–1924 (1999).
Bacon, C. M. et al. Pathology of bone marrow in human herpes virus-8 (HHV8)-associated multicentric Castleman disease. Br. J. Haematol. 127, 585–591 (2004).
Oksenhendler, E. et al. High incidence of Kaposi sarcoma-associated herpesvirus-related non-Hodgkin lymphoma in patients with HIV infection and multicentric Castleman disease. Blood 99, 2331–2336 (2002).
Isaacson, P., Campo, G. E. & Harris, N. Large B-cell lymphoma arising in HHV8-associated multicentric Castleman disease. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue, Eds C. E. Swerdlow et al., 258–259, (WHO, Lyon 2008).
Deloose, S. T. et al. High incidence of Kaposi sarcoma-associated herpesvirus infection in HIV-related solid immunoblastic/plasmablastic diffuse large B-cell lymphoma. Leukemia 19, 851–855 (2005).
Bryant, R. J., Banks, P. M. & O'Malley, D. P. Ki67 staining pattern as a diagnostic tool in the evaluation of lymphoproliferative disorders. Histopathology 48, 505–515 (2006).
Maheswaran, P. R., Ramsay, A. D., Norton, A. J. & Roche, W. R. Hodgkin's disease presenting with the histological features of Castleman's disease. Histopathology 18, 249–253 (1991).
Saletti, P. et al. Hodgkin's and Castleman's disease in a patient with systemic mastocytosis. Ann. Hematol. 78, 97–100 (1999).
Zanetto, U., Pagani, F. P. & Pérez, C. Interfollicular Hodgkin's lymphoma and Castleman's disease. Histopathology 48, 317–319 (2006).
Frank, D. K., Charney, D. & Kashani, A. Plasma cell variant of Castleman's disease occurring concurrently with Hodgkin's disease in the neck. Head Neck 23, 166–169 (2001).
Jegalian, A. G., Facchetti, F. & Jaffe, E. S. Plasmacytoid dendritic cells: physiologic roles and pathologic states. Adv. Anat. Pathol. 16, 392–404 (2009).
Pellet, C. & Lebbe, C. Kaposi's disease and HHV8: a new model of virus-induced tumorigenesis [French]. Pathol. Biol. (Paris) 48, 529–532 (2000).
Zarate-Osorno, A., Medeiros, L. J., Danon, A. D. & Neiman, R. S. Hodgkin's disease with coexistent Castleman-like histologic features. A report of three cases. Arch. Pathol. Lab. Med. 118, 270–274 (1994).
Nikolskaia, O. V., Nousari, C. H. & Anhalt, G. J. Paraneoplastic pemphigus in association with Castleman's disease. Br. J. Dermatol. 149, 1143–1151 (2003).
Dispenzieri, A. POEMS syndrome. Blood Rev. 21, 285–299 (2007).
Alberti, M. A. et al. 18F-FDG PET/CT in the evaluation of POEMS syndrome. Eur. J. Radiol. doi: 10.1016/j.ejrad.2009.06.004.
Belec, L. et al. Human herpesvirus 8 infection in patients with POEMS syndrome-associated multicentric Castleman's disease. Blood 93, 3643–3653 (1999).
Ganti, A. K., Pipinos, I., Culcea, E., Armitage, J. O. & Tarantolo, S. Successful hematopoietic stem-cell transplantation in multicentric Castleman disease complicated by POEMS syndrome. Am. J. Hematol. 79, 206–210 (2005).
Obermoser, G., Larcher, C., Sheldon, J. A., Sepp, N. & Zelger, B. Absence of human herpesvirus-8 in glomeruloid haemangiomas associated with POEMS syndrome and Castleman's disease. Br. J. Dermatol. 148, 1276–1278 (2003).
Cokelaere, K., Debiec-Rychter, M., De Wolf-Peeters, C., Hagemeijer, A. & Sciot, R. Hyaline vascular Castleman's disease with HMGIC rearrangement in follicular dendritic cells: molecular evidence of mesenchymal tumorigenesis. Am. J. Surg. Pathol. 26, 662–669 (2002).
Pauwels, P. et al. A chromosomal abnormality in hyaline vascular Castleman's disease: evidence for clonal proliferation of dysplastic stromal cells. Am. J. Surg. Pathol. 24, 882–888 (2000).
Chan, J. K., Tsang, W. Y. & Ng, C. S. Follicular dendritic cell tumor and vascular neoplasm complicating hyaline-vascular Castleman's disease. Am. J. Surg. Pathol. 18, 517–525 (1994).
Chan, A. C. et al. Development of follicular dendritic cell sarcoma in hyaline-vascular Castleman's disease of the nasopharynx: tracing its evolution by sequential biopsies. Histopathology 38, 510–518 (2001).
Kazakov, D. V., Morrisson, C., Plaza, J. A., Michal, M. & Suster, S. Sarcoma arising in hyaline-vascular castleman disease of skin and subcutis. Am. J. Dermatopathol. 27, 327–332 (2005).
Nayler, S. J., Taylor, L. & Cooper, K. HHV-8 is not associated with follicular dendritic cell tumours. Mol. Pathol. 51, 168–170 (1998).
Marzano, A. V. et al. Paraneoplastic pemphigus associated with follicular dendritic cell sarcoma and Castleman disease. Br. J. Dermatol. 153, 214–215 (2005).
Hsieh, M. L., Quint, L. E., Faust, J. M. & Turner, J. E. Enhancing mediastinal mass at MR: Castleman disease. Magn. Reson. Imaging 11, 599–601 (1993).
Mallik, A. A., Katchy, K. C. & Clotan, N. Solitary intracranial Castleman's disease, plasma cell variant: a case report. Med. Princ. Pract. 16, 226–229 (2007).
Gaba, A. R., Stein, R. S., Sweet, D. L. & Variakojis, D. Multicentric giant lymph node hyperplasia. Am. J. Clin. Pathol. 69, 86–90 (1978).
Casper, C. et al. Valganciclovir for suppression of human herpesvirus-8 replication: a randomized, double-blind, placebo-controlled, crossover trial. J. Infect. Dis. 198, 23–30 (2008).
Matsuyama, M. et al. Anti-interleukin-6 receptor antibody (tocilizumab) treatment of multicentric Castleman's disease. Intern. Med. 46, 771–774 (2007).
Starkey, C. R., Joste, N. E. & Lee, F. C. Near-total resolution of multicentric Castleman disease by prolonged treatment with thalidomide. Am. J. Hematol. 81, 303–304 (2006).
Lee, F. C. & Merchant, S. H. Alleviation of systemic manifestations of multicentric Castleman's disease by thalidomide. Am. J. Hematol. 73, 48–53 (2003).
Scott, D., Cabral, L. & Harrington, W. J. Jr. Treatment of HIV-associated multicentric Castleman's disease with oral etoposide. Am. J. Hematol. 66, 148–150 (2001).
Bower, M. et al. Brief communication: rituximab in HIV-associated multicentric Castleman disease. Ann. Intern. Med. 147, 836–839 (2007).
Gérard, L. et al. Prospective study of rituximab in chemotherapy-dependent human immunodeficiency virus associated multicentric Castleman's disease: ANRS 117 CastlemaB Trial. J. Clin. Oncol. 25, 3350–3356 (2007).
Buchler, T. et al. Rituximab failure in fulminant multicentric HIV/human herpesvirus 8-associated Castleman's disease with multiorgan failure: report of two cases. AIDS 22, 1685–1687 (2008).
Marcelin, A. G. et al. Rituximab therapy for HIV-associated Castleman disease. Blood 102, 2786–2788 (2003).
Bestawros, A., Michel, R., Séguin, C. & Routy, J. P. Multicentric Castleman's disease treated with combination chemotherapy and rituximab in four HIV-positive men: a case series. Am. J. Hematol. 83, 508–511 (2008).
Kotb, R. et al. Life-threatening interaction between antiretroviral therapy and vinblastine in HIV-associated multicentric Castleman's disease. Eur. J. Haematol. 76, 269–271 (2006).
Tedder, T. F. CD19: a promising B cell target for rheumatoid arthritis. Nat. Rev. Rheumatol. 5, 572–577 (2009).
Therapontos, C., Erskine, L., Gardner, E. R., Figg, W. D. & Vargesson, N. Thalidomide induces limb defects by preventing angiogenic outgrowth during early limb formation. Proc. Natl Acad. Sci. USA 106, 8573–8578 (2009).
Li, W. W., Hutnik, M. & Gehr, G. Antiangiogenesis in haematological malignancies. Br. J. Haematol. 143, 622–631 (2008).
Stary, G. et al. Complete regression of HIV-associated multicentric Castleman disease treated with rituximab and thalidomide. AIDS 22, 1232–1234 (2008).
Hess, G., Wagner, V., Kreft, A., Heussel, C. P. & Huber, C. Effects of bortezomib on pro-inflammatory cytokine levels and transfusion dependency in a patient with multicentric Castleman disease. Br. J. Haematol. 134, 544–545 (2006).
Acknowledgements
The authors are most grateful to Dr. Salvador Diaz-Cano, Director of Pathology, King's College Hospital London for the pathological and immunohistochemical images (Figure 1) as well as his contribution to discussions about histopathology features.
Author information
Authors and Affiliations
Contributions
K.-M. Schulte wrote the first draft of the manuscript and K.-M. Schulte and N. Talat provided different aspects of the literature review. Both contributed equally to the editing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Schulte, KM., Talat, N. Castleman's disease—a two compartment model of HHV8 infection. Nat Rev Clin Oncol 7, 533–543 (2010). https://doi.org/10.1038/nrclinonc.2010.103
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrclinonc.2010.103
This article is cited by
-
The role of IL-6 in host defence against infections: immunobiology and clinical implications
Nature Reviews Rheumatology (2017)
-
HHV8-related lymphoid proliferations: a broad spectrum of lesions from reactive lymphoid hyperplasia to overt lymphoma
Modern Pathology (2017)
-
Monoclonality and cytogenetic abnormalities in hyaline vascular Castleman disease
Modern Pathology (2014)
-
Inhibitor of differentiation 1 is a candidate prognostic marker in multicentric Castleman’s disease
Annals of Hematology (2014)
-
Ill-Defined Germinal Centers and Severely Reduced Plasma Cells are Histological Hallmarks of Lymphadenopathy in Patients with Common Variable Immunodeficiency
Journal of Clinical Immunology (2014)
