
Abstract
The development of therapies and vaccines for human hepatropic pathogens requires robust model systems that enable the study of host-pathogen interactions. However, in vitro liver models of infection typically use either hepatoma cell lines that exhibit aberrant physiology or primary human hepatocytes in culture conditions in which they rapidly lose their hepatic phenotype. To achieve stable and robust in vitro primary human hepatocyte models, we developed micropatterned cocultures (MPCCs), which consist of primary human hepatocytes organized into 2D islands that are surrounded by supportive fibroblast cells. By using this system, which can be established over a period of days, and maintained over multiple weeks, we demonstrate how to recapitulate in vitro hepatic life cycles for the hepatitis B and C viruses and the Plasmodium pathogens P. falciparum and P. vivax. The MPCC platform can be used to uncover aspects of host-pathogen interactions, and it has the potential to be used for drug and vaccine development.
Similar content being viewed by others

Introduction
Liver pathogens collectively mount an enormous global burden on human health; hepatitis B virus (HBV) chronically infects the livers of >250 million people worldwide, hepatitis C virus (HCV) chronically infects the livers of 130–170 million more, and the Plasmodium protozoan underlying malaria matures asymptomatically within the liver before causing cyclic fever in the blood stages during its infection of over 250 million individuals globally. These two viruses exhibit very distinct genome structures and life cycles, yet HCV and HBV both use parenteral transmission, after which they establish chronic infection in the hepatocyte, the main parenchymal cell type of the liver. Chronic infection in a subset of patients leads to fibrosis and ultimately end-stage liver diseases such as cirrhosis and hepatocellular carcinoma1,2. Malaria is transmitted by Plasmodium sporozoites after they are injected into a human host via a mosquito vector. These uninucleate sporozoites invade hepatocytes, where they establish exoerythrocytic forms (EEFs) that mature and multiply to form schizonts, which eventually release thousands of pathogenic merozoites into the blood. Merozoites invade erythrocytes and lead to the major clinical symptoms, signs and pathology of malaria. What is shared by all three of these pathogens is their exclusive dependence on the human hepatocyte host environment for a portion of their life cycles. The hepatocyte is a normally quiescent, polarized, specialized, species-specific cell type that is uniquely susceptible to infections. Capturing its phenotype ex vivo using engineering methods to manipulate its microenvironment has been beneficial for understanding human metabolism and toxicity. Here, we discuss the origins and evolution of our engineered MPCC microliver platform (Fig. 1), in the context of other existing in vitro liver model systems. We outline the specific requirements of an experimental platform suitable for the study of diverse pathogens, and we describe how the MPCC has been adapted to serve as an infection model for DNA and RNA viruses, as well as for acute and relapsing parasitic infections.

Left, fabrication of micropatterned collagen islands inside wells of a 24- or 96-well plate. Center, MPCCs are formed by sequential seeding of primary human hepatocytes onto collagen islands and 3T3-J2 fibroblasts onto intervening space around islands. Top right, quality control of MPCCs using integrated functional readouts. Bottom right, infection of MPCCs with various hepatotropic pathogens under different experimental perturbations to discover new biology and/or screen compounds for drug development.
There have been considerable strides made in the clinical management of hepatotropic pathogens in recent years. For example, a safe and effective HBV vaccine exists, and yet incomplete immunization of at-risk individuals allows disease burden to grow, and current antivirals for HBV do not typically cure chronically infected patients. Furthermore, although direct-acting HCV antivirals have emerged with marked cure rates across a variety of genotypes, prophylactic options for HCV remain unavailable3. For malaria, drug resistance is emerging, only a few drugs target liver-stage parasites, and only one licensed drug eliminates the dormant hypnozoite form of P. vivax, which causes clinical relapses4,5,6. Collectively, there are strong motivations for the development of improved vaccines and therapeutic interventions. Achieving a more complete understanding of the biology of HBV, HCV and the Plasmodium pathogens, and their pathogenesis within human hosts, will drive improvements in the clinic. However, because of the narrow species tropism of HBV and HCV, the only robust animal model is the chimpanzee, which is costly and often inaccessible. Notably, forward progress is being made in the development of liver-humanized mouse models of hepatitis B, hepatitis C and malaria, yet—to date—these tools are still generally restricted to a small number of research laboratories7,8, and their reproducibility and reliability need to be further demonstrated. As such, the majority of research programs tackling these diseases typically use in vitro models of the liver9.
Conventional in vitro liver models
Over the past two decades, a variety of in vitro liver models have been developed and used in the study of basic hepatocyte function and metabolism, in addition to their application in establishing platforms for investigating hepatropic infections. Some examples of these models include whole organs, wedge biopsies, liver slices, microsomes, cell lines and primary hepatocytes (either in suspension or in adherent culture)10,11,12,13, several of which use confluent hepatocyte monolayers and extracellular matrix (ECM) manipulations (such as collagen and Matrigel)11,14,15. However, as with most in vitro models, each system is subject to a range of limitations with respect to the faithful recapitulation of human liver biology, as described below.
Whole livers, liver slices and biopsies. Whole livers, biopsies and slices retain in vivo cytoarchitecture and liver-specific hepatocyte-stromal interactions, yet they have restricted viability (usually <48 h, although some reports demonstrate limited hepatic functions for up to 10 d (ref. 16)), they require complex logistics to work with successfully and they do not allow medium- to high-throughput screening (HTS) because of donor organ shortages. Microsomes are used in HTS to identify enzymes involved in drug metabolism but lack the gene expression and cellular machinery required for investigating complex cellular responses. There have been some advances in culture of liver slices in microfluidic devices to prolong their lifetime17; even this short-term extension of viability (<72 h) still precludes the possibility of prolonged drug dosing over several weeks. Furthermore, each experiment using slices is, by necessity, from a different liver donor, which can make data difficult to interpret across experiments because of donor-to-donor variability in responses. Slices also do not allow investigators to create culture platforms on-demand from the 'bottom-up' using cryopreserved cells from the same donor, which can be customized for specific applications and at the level of throughput required.
Cell lines. Hepatoma-derived cell lines and immortalized hepatocytes provide inexpensive and plentiful cell sources for experimentation, yet they are known to display abnormal liver-specific functions such as uncontrolled proliferation, dysregulated gene expression, altered host responses to infection, inadequate drug metabolism capacity, dysfunctional mitochondria and abnormal endocytic functions18,19. The HepaRG hepatoma-derived cell line differentiates spontaneously into hepatocyte-like and cholangiocyte-like cells after several weeks of culture in vitro, and it displays higher liver functions relative to HepG2 and other cell lines20. The utility of HepaRG in assessing drug-mediated CYP450 enzyme induction, drug-transporter interactions and drug clearance as a complementary tool to primary human hepatocytes has been well described18,21,22,23,24. However, the sensitivity for detecting toxicity of drugs was significantly lower in HepaRG than in primary human hepatocytes (16% versus 44%)18. Furthermore, as with all cell lines, HepaRG cells provide information on drug behavior in a single liver donor. Thus, primary human hepatocytes are still needed to obtain data from multiple donors in a more physiological (i.e., nontransformed) context.
Primary hepatocytes. Isolated primary hepatocytes are considered the 'gold standard' for probing hepatic functions because they are untransformed and relatively simple to use; however, these cells require adherence to ECMs to survive for longer than a few hours11. Furthermore, in conventional adherent formats that require seeding of primary hepatocytes at very high densities to provide fully confluent monolayers on collagen, hepatic functions rapidly decline over a handful of days, which makes these cultures only suitable for short-term (<48 h) culture and investigations12,15,25. Longer studies require the supplementation of culture medium with soluble factors that prolong culture survival but can influence hepatocyte differentiation, and/or the use of ECM sandwich or 3D culture that maintains hepatocyte morphology and function to a greater extent than simple monolayer cultures on collagen15,26. In contrast to hepatocytes in monolayer configuration, hepatocytes in sandwich cultures re-establish polarity, express functional transporters in culture, and constitute a useful tool for studying hepatobiliary transport in vivo15,26,27. However, historically, sandwich cultures have been shown to display a decline in key liver functions and have been challenging to miniaturize, and thus they are likely to be more useful for only a subset of acute phenomena (hours to days)25,28,29. Next-generation culture models apply engineering methods to in vitro culture systems11,29,30. The MPCC that is the subject of this protocol is one such model that maintains high and stable hepatic functions, as well as polarity, for weeks in culture28,31,32.
Stem cell–derived hepatocytes. In spite of the improvement in several hepatic model systems, those discussed above all rely on nonrenewable sources (e.g., primary tissue availability). Cryopreservation has certainly enhanced the capacity for such work, yet renewable sources are still desirable for establishing large-scale, automation-compatible platforms and studies on isogenic backgrounds. To this end, advances in induced pluripotency and directed differentiation protocols have led Duncan and colleagues32 and several other groups33 to produce hepatocyte-like cells (iHLCs) from numerous genetic backgrounds; however, such cells do not yet exhibit a fully mature adult phenotype33, and the protocols have been proven to be challenging to miniaturize. Recent progress has been made to produce iHLCs in 96-well formats34, but these efforts still require normalization strategies to account for stochastic well-to-well variations in differentiation efficiencies. Nonetheless, similar to their primary human hepatocyte counterparts, micropatterning strategies can be used to further mature stem cell–derived human hepatocyte-like cells. Notably, Khetani, Berger, and colleagues adhered iHLCs onto collagen-coated domains optimized via micropatterning, surrounded these islands with 3T3-J2 mouse embryonic fibroblasts and then added a Matrigel overlay (i.e., they combined the MPCC platform with an ECM sandwich technique). In this context, the MPCC platform can further mature human iHLCs toward adult primary human hepatocytes and maintain functions for at least 4 weeks in vitro relative to conventional pure iHLC monolayers35. These so-called 'iMPCCs' produced high levels of albumin and urea, maintained major CYP450 and phase II drug metabolism enzyme activity, and were sensitive to effects of drugs (induction, toxicity) with in vivo–relevant trends. Moreover, the capacity for the MPCC assay to be populated with iPS-derived cells offers the potential to model rare genotypes, as well as access to an expandable cell population, although—despite improvements in hepatocyte-derivation protocols—iPS-derived hepatocyte-like cells remain an imperfect proxy for adult cells33,36, and they often require substantial culture expertise. In a separate undertaking, we have identified small-molecule 'maturins' that promote the maturation of iHLCs toward a more adult phenotype36. Ongoing efforts seek to isolate the signaling pathways responsible for these outcomes, as well as to apply the small molecules in our 2D micropatterned platform, as well as 'on chip' in perfused 3D formats that link multiple lineages of iPS-derived cell types37.
Utility of in vitro hepatic model systems in the study of hepatropic pathogens
The establishment of this plethora of model human liver systems opened the door for their use in the study of hepatropic pathogens, particularly in the case of those that exclusively infect human hepatocytes. As discussed above, considerable work is also ongoing to improve existing liver-humanized mouse models, but thus far the application of these models to studies of pathogen infections has been limited to a narrow portion of the research community, and thus this section is focused on the utility of in vitro models in this context. The central design criteria for an ideal model of human hepatropic infections include the expression of entry receptors to permit initial infection, host factors that support replication and longevity to support the entire life cycle of the pathogen in question (days for HBV and HCV, and in the case of Plasmodium parasites, ∼7–10 d for the acute phase and ∼21 d for relapse). Successful support of the pathogen's life cycle is probably mediated in part by the stable maintenance of quiescent, polarized, differentiated hepatocytes that remain coupled by tight junctions. In addition, it is particularly desirable if the selected model system offers the capacity for miniaturization, as most human pathogens are difficult to source (for example, HBV, HCV, P. falciparum and P. vivax), and therefore test inoculum is typically a biomass limitation in the case of scaled experiments. To date, the in vitro study of hepatotropic pathogens most commonly uses cell lines.
The HCV community has typically relied on the Huh-7 and Huh-7.5 hepatoma cell lines, along with other subclones, to study infection. HBV work has relied upon the HepG2 hepatoblastoma cell line and variants containing integrated viral sequences, and DMSO-differentiated cultures of the hepatoma line HepaRG (ref. 38). These lines may exhibit numerous morphologic and functional abnormalities, meaning that findings obtained using these models, especially those relating to host-virus interactions39, often deviate from those observed in vivo. Other laboratories have developed primary human hepatocyte models that permit hepatropic pathogen infection, notably the Guguen-Guillouzo group for HBV infection40, and, more recently, other groups have established models of HCV infection41,42,43. Nonetheless, it remains to be determined whether these models offer the stability in hepatic phenotype, reproducibility and robustness that is routinely observed with MPCCs.
As is the case with HBV and HCV, our current understanding of the liver stages of P. falciparum and P. vivax, the major species of human malaria parasites, is also based in large part on the infection of human hepatoma cell lines44,45,46,47,48. In addition to the drawbacks of cell lines discussed above, the observation of parasite or virus development in liver cell lines is challenging after 6 d in culture, as the infected cells continue to proliferate and detach from culture49. Notably, Mazier and colleagues have combined the sandwich culture method with the use of Matrigel and HepaRG cells, and it has been shown to be permissive to P. falciparum infection and can maintain Plasmodium cynomolgi infection for up to 40 d (ref. 50). This prolonged culture period opens the door for the system to be suitable for P. vivax studies, and it offers a complementary platform for experiments aimed at achieving reactivation of hypnozoites. Nonetheless, given the abnormal, transformed nature of the HepaRG cells, the cultures established via this method may produce a random coculture of hepatocytes and cholangiocytes. This heterogeneous culture environment may well prove to be useful for certain malaria infection applications, yet it is also likely to be limited by experiment-to-experiment variability.
As early as 1984, the Mazier laboratory established the full life cycle of both liver forms of human malaria in cultured, primary human hepatocytes in two landmark studies51,52. Mazier and her collaborators have continued to leverage access to fresh liver tissue, infected mosquitos and culture expertise to apply their platform to the study of P. falciparum and P. vivax51,52,53,54. However, the use of primary hepatocyte systems by the broad liver-stage malaria community has been minimal, probably in part because of the difficulty in translating these cells into reproducible formats for screening, limited cell availability, as well as challenges in maintaining phenotypic function over extended periods in vitro55. Thus, for longitudinal studies of hepatotropic pathogens such as HBV, HCV and Plasmodium, the aforementioned models have offered limited progress in the field56,57.
Development of the MPCC protocol
Given the drawbacks of the above models, we sought to develop an engineered system that builds on existing advances to create reliable, robust miniaturized primary systems that replicate the authentic host biology of the primary human hepatocyte. In this paper, we discuss the development and use of an in vitro culture technology called MPCC, which we have developed, optimized and applied to problems in human health over the past 10 years28,55,56,57. This coculture system of primary human hepatocytes and 3T3-J2 mouse embryonic fibroblasts is robust, reproducible and miniaturized in a standard multiwell plate format compatible with automated workflow environments, and it sustains hepatocytes for 4–6 weeks in culture. Primary human hepatocytes can be sourced either fresh or cryopreserved from many human donors, and selected lots are then qualified for use in downstream applications. We have successfully used MPCCs to study the infection and drug response for HBV, HCV and malaria56,57,58. Our aim in sharing the historical derivation and our insight into the evolution of the MPCC is to provide a roadmap for the community to replicate, test, improve and hopefully adopt the model in order for it to expedite progress in achieving clinical impact for patients suffering from disease.
Before our foray into adapting in vitro models of human hepatocytes for the purposes of modeling liver infections, the original steps toward the development of MPCCs were inspired by early work focused on the role of physical homotypic (hepatocyte-hepatocyte) and heterotypic (hepatocyte-stromal) cell-cell interactions modulating hepatocyte functions in vitro. In particular, pioneering studies by Guillouzo and colleagues demonstrated transient induction of some functions in primary human hepatocytes cocultivated with an epithelial cell type, also derived from the liver59. However, in these early studies, the two cell types were randomly distributed onto planar surfaces, which did not allow exploration of the role of controlled cell-cell interactions on the hepatic phenotype without the confounding variable of cell seeding density. To circumvent this limitation, Toner and Bhatia adapted photolithography methods used by the semiconductor industry to physically position defined, 2D islands of primary rat hepatocytes on adsorbed collagen surrounded by supportive 3T3-J2 mouse embryonic fibroblasts55. Such micropatterning allowed tuning of the relative levels of homotypic and heterotypic interactions while keeping cell numbers and ratios between the two cell types constant across the various patterned configurations. The first iterations of these experiments revealed that defining various levels of homotypic cell-cell interactions alone had a crucial role in modulating hepatocyte functions by several fold. Functions of micropatterned hepatocyte colonies were then significantly augmented in both magnitude and longevity by contact coculture with stromal cells55. In particular, a functional screen revealed that the J2 subclone of 3T3 mouse embryonic fibroblasts (3T3-J2, isolated by Green and colleagues60) induced optimal functions in hepatocytes from multiple species (rat, human) as compared with other available 3T3 clones (i.e., NIH-3T3, Swiss-3T3, L1-3T3) (ref. 61).
Given significant differences between animals and humans in liver functions62,63,64, the aforementioned micropatterning configurations were applied to freshly isolated primary human hepatocytes. We noted that although an interplay between the ratio of homotypic and heterotypic interactions was still needed to induce optimal functions in human hepatocytes cocultivated with 3T3-J2 fibroblasts, the optimal configuration was distinct from that required by their rat-derived liver counterparts28. Another notable advance was achieved by Khetani and Bhatia when we developed soft-lithographic techniques that allowed for rapid creation of MPCCs in miniaturized formats for higher-throughput screening than that afforded by the serial process of photolithography for direct seeding of cells28. Furthermore, MPCC-creation protocols were optimized by investigators at Hepregen Corporation (Medford, Massachusetts, USA) to leverage advances in the production of cryopreserved primary human hepatocytes by multiple vendors (e.g., Life Technologies, BioreclamationIVT). The use of cryopreserved hepatocytes affords several advantages, which include the following: convenient on-demand experimentation as opposed to the unpredictability in procurement of fresh cells; longitudinal studies in the same donor when required, as opposed to significant inter-experimental variability observed with the use of fresh hepatocytes from different donors; and comparisons across responses in multiple donors for specific downstream applications. All of the aforementioned advances have culminated in modern-day MPCCs, which have been optimized for human hepatocyte functions in industry-standard 24- and 96-well formats (Figs. 1 and 2). Current 'best practice' cultures consist of 500-μm islands (∼200 hepatocytes per island), spaced 700–1,200 μm apart center-to-center, with supportive mouse 3T3-J2 fibroblasts in the remaining spaces. Such architectures remain intact in both fidelity of patterning and hepatic morphology and functions for several weeks.

Bright-field images of MPCCs constructed inside individual wells of a 96-well plate to facilitate medium-throughput screening. From left to right, progressively higher-magnification images of hepatocyte islands (H) surrounded by fibroblasts (J). These MPCCs were formed by patterning 40 islands of 500 μm in diameter per well. Note the maintenance of hepatocyte morphology with small, bright circular nuclei, dark cytoplasm and numerous bile canaliculi presenting as thin, white branches. Images are representative of MPCCs cultured for 18 d after fibroblast seeding. Reproduced with permission from ref. 56. Additional images can be seen in Khetani et al.13.
Applications of MPCC to hepatotropic infections
The earliest studies of MPCCs focused on the elucidation of basic mechanisms underlying hepatocyte-stromal cocultures and drug metabolism, as well as for preclinical evaluation of drug toxicity28,62,65,66,67,68,69. For instance, MPCCs have been shown to be ∼70–75% predictive of clinical outcomes for drug metabolite and drug-induced liver toxicity profiling, as opposed to <50% sensitivity in standard culture systems65. These and other characterizations of the MPCCs led us to assay whether the platform would satisfy the requirements of an in vitro model of human hepatropic pathogens, including host factor expression and facility to adapt the protocol to permit infection and development of candidate pathogens (Fig. 3). By collaborating with the Rice, Mota and Sanaria research groups, we have succeeded in applying MPCCs to study the life cycle of HCV57, HBV58 and human Plasmodium56,70 pathogens. In one example, we performed head-to-head comparisons and found that other conventional primary human hepatocyte culture models—such as hepatocytes on rigid collagen, collagen gel sandwiches, Matrigel spheroids and even randomly distributed co-cultures of the same two cell types used in MPCCs—do not sustain HBV or HCV infection as robustly as MPCCs57,58. On the basis of these successes, we have focused this report on the use of MPCCs with this collection of hepatropic infections (Figs. 4,5,6), but our findings suggest that our coculture platform can also be used to study hepatotropic pathogens broadly. Indeed, in our ongoing studies, we have confirmed that it is possible to achieve dengue virus infection of MPCCs, and although to date we have not used the platform to study infection with HAV and HEV (hepatitis A virus and hepatitis E virus, respectively) these are interesting areas for future study. After the demonstration that MPCCs support a pathogen's life cycle, a natural extension of this in vitro system is its use in candidate drug and vaccine screening to determine efficacy against the target pathogen. In this case, data derived from the study of these drugs in MPCCs are likely to be more representative of the human population when using primary human hepatocytes from multiple donors (Fig. 3) rather than single transformed donors. In addition, because of stable activities of drug metabolism enzymes (that is, cytochrome P450s) and drug transporters, MPCCs allow simultaneous evaluation of drug metabolism and/or toxicity, and the efficacy of candidate compounds in the same hepatocytes57.

(a) Schematic of sequential criteria filters used to select optimal human cryopreserved hepatocyte donors (lots) for infection with hepatotropic pathogens. (b) Functional characterization of plateable hepatocyte lots to assess their liver phenotype. Left, plots of albumin production, urea synthesis and cytochrome P450 activity (top to bottom), all performed on day 14 post seeding. Error bars represent the s.d. of triplicate wells. Right, representative images of optimal donor lots depicting hepatocyte morphology, CD81 expression (reproduced with permission from ref. 56) and SR-BI expression (top to bottom; staining performed on day 4 after seeding); CD81 and SR-BI are entry receptors for malaria and HCV. (c) Assessment of hepatocytes with robust liver phenotype for permissiveness to infection by malaria using fresh sporozoites and assessing infection at day 3 after infection (left; see Step 35D), or day 7 after infection for HCV (middle; see Step 35E) and HBV (right; adapted with permission from ref. 58; see Step 35G). Red boxes indicate preferred donor lots in this example screen, based on albumin secretion (b) or pathogen infection (c). Note that the most permissive human hepatocytes are not always the same for each pathogen. All error bars represent the s.d. of triplicate wells.

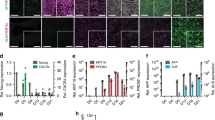
(a) Schematic adapted with permission from Portugal et al.95 depicting the obligate, initial liver-stage expansion before egress to the blood. Briefly, a Plasmodium-infected mosquito injects sporozoites into a human host, which travel via the bloodstream to the liver. After traversing through multiple hepatocytes, the sporozoites undergo asexual reproduction to form an EEF that eventually releases merozoites into the bloodstream, which leads to symptomatic pathology of Malaria because of host red blood cell lysis. (b) Left, representative images of (top) P. falciparum at days 3 and 5 after infection (parasites are identified by anti-HSP70 and anti-MSP-1 staining) and (bottom) P. vivax at days 5, 8 and 21 (parasites are identified by anti-PvHSP70 staining) in human MPCCs. The scale bars, antibody used for immunohistochemistry (blue, DAPI), and day of infection are indicated on each image. Middle, size distribution of P. falciparum (top) and P. vivax (bottom) parasites in MPCCs over time. Right, representative images of P. falciparum (top) and P. vivax (bottom) in human MPCCs stained on the indicated day of culture, and with the indicated fluorescent antigen (blue, DAPI). (c) Merozoites released from P. falciparum-infected MPCCs infect red blood cells; freshly prepared human type 0+ red blood cells were added to P. falciparum-infected MPCCs at a 2% hematocrit on day 6 after infection. Representative Giemsa stain reveals ring stage of the parasite life cycle. (d–g) Suitability of MPCCs for drug screening. (d) 96-well MPCCs were infected with cryopreserved P. falciparum sporozoites and tested for sensitivity to primaquine drug treatment56. Error bars represent the s.d. of triplicate wells. (e) Expression of drug metabolizing genes in MPCC relative to unpatterned cultures of the HC04 cell line were measured using LMA-Luminex analysis for 83 human-specific genes56. Each column represents one of three triplicate samples of RNA extracted from MPCC or HC04. (f) MPCCs have the capacity to distinguish live versus attentuated sporozoites in a candidate vaccine method, based on size distribution of EEFs in infected MPCCs after 5 d of culture56. (g) MPCCs are amenable to image analysis automation for medium-throughput screening. Top, image of candidate parasites that are detected via automated imaging platform are manually classified using Cell Profiler. This input data are used to establish a scoring algorithm that is applied to score all images in a test data set. Bottom, P. falciparum infection in MPCCs after two doses of fresh sporozoites is determined by manual counts (indicated by M) or by image analysis automation (indicated by A)56. Error bars represent s.e.m. Portions of this figure (b,d,e,f and part of g) are adapted with permission from ref. 56; other portions (c, part of g) are reproduced with permission from ref. 56. For more details, see March et al.56.

(a) A schematic depiction of the HCV life cycle in primary hepatocytes. The virus enters the cell through entry receptors including CD81, SR-BI, CLDN and OCLN, it unpacks in the cytosol and it uses its viral polymerase to undergo RNA-dependent RNA replication in both the reverse direction (generating negative-sense viral transcripts) and forward direction (generating positive-sense transcripts from the negative templates). Viral transcripts and structural proteins are packaged into a membrane-derived envelope during assembly and released. (b) Time course of infection of MPCCs with Jc1-Gluc strain of HCV, which expresses a secreted luciferase during viral translation. Viral translation is quantified using luciferase assay in relative light units (RLU), and it can be seen to be sensitive to inhibition with three separate drugs ITMN191 (a protease inhibitor), 2′CMA (a polymerase inhibitor) and IFN (an innate immune activator). At 10 d after infection, supernatant from DMSO (vehicle)-treated, infected cells was used to infect naïve Huh7.5 cells to perform a TCID50 assay (Step 35E). Error bars are s.e.m. of triplicate wells57. (c) MPCCs were transduced with the RFP-NLS-IPS reporter to allow for live-cell imaging of HCV infection. Upon successful infection of hepatocytes, reporter nuclear translocation can be seen (arrowheads). In this experiment, ∼0.8 translocations were seen per island, corresponding to an infection rate of 0.32%. Scale bar, 25 μm. (d) The responsiveness of HCV infection to antiviral drugs is tested using several doses of each drug, described in b. Multiple antiviral drugs with unique mechanisms of action show dose responsiveness with IC50 values between <0.01 μM and 0.13 μM. Error bars represent s.e.m. of triplicate wells. (e) Dose-dependent inhibition of HCV replication in infected MPCCs treated with antibodies against HCV glycoproteins (AP33, 3/11, CBH5, AR3A) or cellular CD81 (JS-81). Antibody concentrations are 0.1 (light gray), 1 (dark gray) and 10 (black) μg/ml. Error bars represent s.e.m. of triplicate wells. Portions of this figure (b,d) are adapted with permission from ref. 57; e is reproduced with permission from ref. 57.

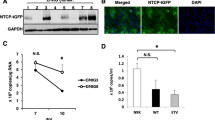
(a) Schematic of the HBV life cycle, including the viral parameters that are assayed in this protocol. (b) Immunofluorescence images of NTCP staining in MPCCs versus random cocultures (RCCs); NTCP is the entry receptor for HBV. White circle marks the approximate hepatocyte island boundary, Scale bars, 100 μm. (c) HBV infection of MPCCs versus RCCs shows that micropatterning is required for robust infection. Left, ELISA for HBsAg, secreted into the supernatant between 14 and 16 d after infection; right, HBV 3.5-kb mRNA expression at 16 d after infection. (d) Robust infection based on multiple measures of viral life cycle is enhanced by inhibition of innate antiviral signaling. Left, cumulative HBsAg release between 7 and 16 d after infection; right, cumulative copies of cccDNA per well extracted between 7 and 16 d after infection. (e) Prophylactic drug treatment regimen prevents viral expansion. MPCCs treated with DMSO or JAKi, with or without entecavir or IFN-β, were incubated with HBV infectious serum for 24 h, followed by continued drug treatment every 2 d. Collected supernatants were analyzed for HBV DNA after 3 weeks. Error bars are s.e.m. of triplicate wells. (f) Therapeutic drug treatment regimen abrogates established infection. HBV-infected MPCCs treated with JAKi were treated with either IFN-β or entecavir from 7 to 16 d after infection, when cell pellets were analyzed for 3.5-kb mRNA expression relative to nonantiviral-treated cells (left, 1 pellet per condition, consistent across multiple experiments) and for cccDNA (right, error bars represent range of duplicate pellets from one experiment, consistent across multiple experiments). Also at 16 d after infection, the medium collected over 48 h was analyzed for secreted HBV DNA (center, error bars are s.e.m. of triplicate wells, consistent across multiple experiments). Panel d is adapted with permission from ref. 58; (a–c,e,f) are reproduced with permission from ref. 58.
By virtue of their support of hepatropic pathogen life cycles, MPCCs can also be used for basic biological explorations of host-pathogen interactions that are not easily recapitulated in transformed and abnormal hepatic cell lines used in the field. As an example, we have recently used MPCCs to study the interplay between HBV/HCV/Plasmodium and the interferon axis present in hepatocytes, an inquiry that was previously challenging given the defective innate immune signaling exhibited by cell lines58. Another advantage of MPCCs is that hepatocytes exhibit polarized morphology over several weeks with appropriate localization of entry receptors and bile canaliculi, which in turn permit studies of cell-cell direct spread of HCV, for example ref. 28. In addition, beyond rodents and humans, MPCCs have been established using hepatocytes from other species, including nonhuman primates71,72. In some pathogens, such as Plasmodium, the human-tropic species can be challenging to source. One approach that has been taken in some laboratories is to evaluate the biology of another malaria species, P. cynomolgi, as a surrogate for P. vivax50,73.
Liver research programs over the past few decades have been stymied by poor access to in vitro models with stable liver functional phenotype and appropriate polarization. Thus, beyond infection, MPCCs can also be used to study more broad questions of liver physiology and pathophysiology (e.g., cholesterol trafficking, carbohydrate regulation, production of serum components)74. Further, as exemplified by studying infection and other assessments of drug toxicity (e.g., acetaminophen)66, MPCCs allow the study of primary human hepatocytes under backgrounds of specific disease states. This capacity makes it likely that MPCCs will assist in studies of fatty liver, fibrosis, alpha-1 antitrypsin deficiency (particularly by collecting hepatocytes from genetic mutant backgrounds) and aspects of hepatocellular carcinoma.
Experimental design
MPCC infection experiments consist of three main phases: fabricating the model (Steps 1–31), characterizing the components (Steps 32–33) and using MPCCs to interrogate infection (Steps 34–35; Fig. 1).
Fabricating the model. As primary hepatocytes need adsorbed ECM (i.e., collagen) to adhere to culture surfaces, the first protocol stage details the precise culture specifications that achieve seeding of the cultures. MPCCs have been adapted successfully to both industry-standard 24- and 96-well plate formats, to suit varying experimental requirements; 96-well layouts are typically used when greater throughput is sought, such as screening drug candidates, or when some aspect of the infection protocol is limiting (e.g., novel drugs, pathogens). The other primary specification to consider is the selection of MPCC island diameter and spacing; these parameters can influence essential experimental criteria, such as hepatocyte function, infection efficiency and the total number of target cells available for infection. In addition, we have recently found that the density of hepatocyte islands can also influence infection rates70.
Once these culture specifications are determined, a mold using elastomeric polymers (usually polydimethylsiloxane (PDMS)) can be fabricated using standard photo- and soft-lithographic techniques75. The mold then serves as the reusable instrument with which to micropattern collagen simultaneously into each well of multiwell plates. Briefly, well plates are coated with collagen; a PDMS mold is then placed on the well plate to protect collagen-coated islands to which hepatocytes will selectively attach (Fig. 1). At this point, oxygen plasma gas is used to ablate collagen-coated surfaces that are not protected by the mask, leaving only the array of collagen islands in prespecified architecture. The micropatterned plates can immediately be used for cell seeding, or they can be refrigerated for a few months in a desiccator without noticeable loss of cell attachment capacity. Each well of the plate is seeded with primary human hepatocytes (from fresh or cryopreserved sources) and shaken three or four times per hour to ensure uniform seeding of hepatocytes onto all ECM islands in the well. Once the islands are >85% filled with hepatocytes over 3–5 h, the cells that have not adhered are removed by washing the wells with culture medium to prevent nonspecific attachment of hepatocytes to bare plastic areas because of adsorption of serum proteins from culture medium. Adhered hepatocytes are then allowed 18–24 h to fully spread onto the islands to make fully confluent clusters. Stromal cells (i.e., mouse 3T3-J2 fibroblasts) are seeded onto the hepatocyte cultures the following day in the presence of serum proteins that mediate their attachment to the intervening regions (Fig. 2). If desired for the particular application, additional cell types can be seeded after this second stage. For example, Kupffer macrophages have been selected as an immune environment-modifying population; however, by themselves, these macrophages, liver sinusoidal endothelial cells76 and other liver-derived stromal cell types do not stabilize hepatic functions, and thus the 3T3-J2 cells remain an important component of the current MPCC platform. The controlled nature of the cultures achieves reproducibility between batches, especially with the use of prequalified cryopreserved human hepatocyte lots, and it allows for the testing of hypotheses related to the impact of specific cell types.
Characterizing the components of the model. Biomarkers for characterizing MPCCs can be used to optimize selection of a candidate hepatocyte cell source (i.e., commercially available, numbered lots of cells from specific donors), to confirm that cultures are highly functional while maintaining longevity of functions for the time-period required and to subsequently assess the impact of pathogen infection and/or therapeutic intervention (Fig. 3). Secretion of hepatocyte biosynthetic products (albumin, urea), CYP450 activity and morphology (including appropriate polarization with appearance of bile canaliculi) are biomarkers used at various time points during the culture life span. Typically, we screen a collection of hepatocyte lots from several human liver donors in parallel using the aforementioned biomarkers to select those donors that display stable phenotypic functions for several weeks in MPCCs. Cryopreserved hepatocyte lots that are sold under the label of 'plateable' by various vendors (i.e., BioreclamationIVT, Life Technologies, Triangle Research Labs) typically attach to collagen islands in MPCCs and display stable phenotype for 4–6 weeks in vitro. Hepatocytes from younger donors (<10 years of age) have been found to function for 6 weeks and longer in limited instances28.
Using MPCCs to interrogate infection. MPCCs that successfully maintain longevity can be used for infection experiments. Such experiments can be tweaked as per the needs of the researcher. The typical approach is to infect with the pathogen of interest and then use assays that are specific to that pathogen (Figs. 4,5,6). Below, we will discuss details of this process for HCV, HBV and Plasmodium infection. Assays can be used to monitor infection, for example, in the context of drug screening or other perturbations in basic biology studies.
Experimental controls. During the course of developing MPCCs as infection models for Plasmodium, HBV and HCV, several controls were performed to compare the rates of infection of MPCCs with randomly organized cocultures with the same cell types, as well as other culture models (e.g., cell lines, monolayer culture, sandwich culture). In a given MPCC experiment, to control for any unknown variables that are present in the assays that we use to quantify infection, it is advisable to include drug treatment controls in each experiment, in which known antiviral or antiparasite drugs are dosed during and after infection to provide a negative control.
Limitations
Compared with conventional seeding of a cell line on well plates, establishment of MPCCs is more involved when the process for patterning the plates and seeding of multiple cell types are taken into account. However, the ability to store collagen-patterned plates for several months at 4 °C decouples the batch patterning process in multiwell plates from the seeding of the cell types on two separate days. Regardless, the added effort to create MPCCs affords the researcher the ability to evaluate prolonged drug dosing regimens on stable primary hepatocytes, which otherwise may require time-consuming and far more challenging investigations in vivo.
In certain experimental contexts, the current MPCC format can present a problem in that it requires the use of mouse fibroblasts as the supportive cell type. For instance, mouse fibroblasts may represent a confounding detail in certain immunologic studies that require cells with an exclusively human background. In addition, any time a non-hepatocyte-specific biomarker is assessed in MPCCs, fibroblast-only controls typically need to be carried out to ascertain hepatocyte-specific responses. However, as the MPCCs are a 2D monolayer of cells, high-content imaging has been successfully used to determine the effects of perturbations on hepatocytes and fibroblasts, allowing assessment of both hepatocyte-specific and non-hepatocyte-specific effects in the same well, a key advantage of MPCCs36,68. Ultimately, it would be ideal to replace the fibroblasts altogether with stromal cell–derived molecules that stabilize the hepatic phenotype. Considerable efforts are being made to achieve fibroblast replacement using defined microenvironment cues such as small molecules, proteoglycans, cadherins and biomaterials, yet the complete temporal and spatial combinatorial sequence of fibroblast-derived molecules has not yet been determined36,61,77,78.
MPCCs are also currently limited by their finite life span, although these cultures do persist far longer than other hepatocyte systems (that is, hours to days versus 4–6 weeks). As such, infection experiments cannot be monitored over the course of many months (e.g., in the study of fibrosis related to HCV or HBV infection, or for the purposes of assaying for hypnozoite reactivation in long latency P. vivax strains). Currently, MPCCs are better suited for the study of initial infection and acute disease progression, whereas it is challenging to apply this model for the study of true 'chronic' HCV or HBV infection. We do see a decline in viral protein production coupled to maintenance of covalently closed circular DNA (cccDNA) level at time points 2 weeks after HBV infection58, which may be indicative of a switch from an acute to a chronic phase of infection, although there are other explanations for this phenomenon—such as innate immune stimulation limiting host and viral translation. Overall, the inability to maintain MPCC function indefinitely is likely to be due to some component of in vivo physiology (e.g., certain nonparenchymal cell types) and/or aspects of 3D architecture that are missing in vitro. Related to this limitation is the finding that, as in other in vitro systems used in the community, infection is not as robust in MPCCs as it is in vivo. Recent preliminary data suggest that innate immune signaling is relatively more active in the in vitro cultures than had been anticipated on the basis of in vivo findings, and this difference may contribute to the dampened HCV infection efficiency observed in this platform (data not shown). Nonetheless, as MPCCs are built 'bottom-up' from individual components, they can be used as a base platform on which to engineer additional liver-specific microenvironmental cues in order to better mimic liver physiology and disease states13.
With regard to Plasmodium parasite growth, the sizes reported in our system are similar to what has been previously observed in other in vitro systems at day 5 (refs. 51,73,79), but they are smaller than those observed in vivo at day 5 (refs. 80,81) or in humanized mouse models82. It is possible that conformational cues may be important for parasite growth (for example, 2D versus 3D contexts), or that EEF size is affected by hepatocyte packing density. Notably, we recently reported that in vitro parasite growth can be increased in MPCCs by modulating the cell surface oxygen tension experienced by the hepatocytes70.
Materials
REAGENTS
Culturing fibroblasts
-
3T3-J2 fibroblasts (courtesy of H. Green, Harvard Medical School)60 or NIH-3T3 fibroblasts (ATCC)83
Critical
Caution
The cell lines used in your research should be regularly checked to ensure that they are authentic and that they are not infected with mycoplasma.
-
High-glucose DMEM (DMEM with L-glutamine; Corning, Cellgro, cat. no. 10-017-CV)
-
BS (bovine serum; Life Technologies, Gibco, cat. no. 16170-078)
-
Penicillin-streptomycin (Corning, Cellgro, cat. no. 300-002-Cl)
-
Trypsin-EDTA (0.05-0.25% (wt/vol); Corning, Cellgro, cat. no. 25-053-Cl)
-
Fibroblast medium: DMEM, 10% (vol/vol) BS, 1% (vol/vol) penicillin-streptomycin
Seeding and culturing human primary hepatocytes in MPCCs
-
Cryopreserved primary human hepatocytes (we have used BioreclamationIVT, Lot NON; Invitrogen, Lot Hu4151 and Lot Hu8085)
Caution
Request vials of cryopreserved primary human hepatocytes of different donors from vendors permitted to sell products derived from human organs in your home country. For example, we procure in the United States from federally designated Organ Procurement Organizations that include Life Technologies, Lonza, BioreclamationIVT, Corning Life Sciences and Triangle Research Labs. Ensure that human hepatocytes are tested by these vendors to be negative for HIV, HCV and HBV.
-
ITS+ (insulin/human transferrin/selenous acid and linoleic acid) premix (BD Biosciences, cat. no. 354352)
-
Dexamethasone (Sigma, cat. no. D8893)
-
Absolute ethanol (Sigma, cat. no. E7023)
-
DMEM with L-glutamine (Corning, Cellgro, cat. no. 10-017-CV)
-
FBS (Gibco, cat. no. 1600-044)
-
HEPES, 1 M, pH 7.6 (Gibco, cat. no. 15630-080)
-
Penicillin-streptomycin (Corning, Cellgro, cat. no. 300-002-Cl)
-
Glucagon (Sigma, cat. no. G2044)
-
Trypan blue (Sigma, cat. no. T8154)
-
JAK inhibitor (EMD Millipore, cat. no. 420097)
Pathogen sources
-
Fresh or cryopreserved P. falciparum and P. vivax sporozoites (Sanaria)
Caution
It is highly recommended that persons using this product ensure that they are fully trained in all aspects of safety related to the product.
-
Generate HCV stocks for infection by electroporation of HCV RNA into Huh7.5.1 cells, followed by collection, purification and concentration of secreted infectious virions85 (Box 1)
Caution
It is highly recommended that persons using this product are fully trained on all aspects of safety related to the product, and that they adhere to Biosafety Level 2 or higher practices during the use of this product.
-
De-identified HBV+ human plasma (tested negative for HIV, HCV; Red Cross, see Box 2) or HBV from infected from HepG2.2.15 cells86 (Box 3)
Caution
Plasma derived from humans must be derived from sources that obtain patient consent. It is highly recommended that persons using this product are fully trained on all aspects of safety related to the product, and that they adhere to Biosafety Level 2 practices during use of this product.
Making MPCC masters
-
PDMS (Sylgard 184 kit, Dow Corning)
-
Arch punch (13 mm for a 24-well plate, 5 mm for a 96-well plate; McMaster Carr)
-
Silicon master; micro-domain of 500-μm-diameter islands. 1,200 μm center-to-center island spacing in a 24-well format or 700–900 μm spacing in a 96-well format
-
Trichloro(1H,1H,2H,2H-perfluorooctyl)silane (Sigma, cat. no. 448931)
-
Glass Petri dish (150 mm diameter; Fisher, cat. no. S00056)
-
Plastic Petri dishes (150 mm diameter; Sigma, cat. no CLS430597)
-
Hexane, mixture of isomers (Sigma, cat. no 650544)
Preparing micropatterned cocultured plates
-
Tissue culture plates, 96 well or 24 well (For high-magnification fluorescence microscopy, use glass-bottom plates.)
-
Glass-bottom plates, 96 well (Greiner Bio-One, cat. no. 655892)
-
Glass-bottom plates, 24 well (Greiner Bio-One, cat. no. 662892)
-
Coverslips, 12 mm circle (VWR, cat. no. 48366-251)
-
PDMS (Sylgard 184 kit, Dow Corning) master. Micro-domain of 500-μm-diameter islands. 1,200 μm center-to-center island spacing in a 24-well format or 700–900 μm spacing in a 96-well format. Production is described in the protocols; the PDMS MPCC master must be made before patterning MPCC plates
-
Rat tail collagen solution type I (BD Biosciences, cat. no. 354236)
-
Sterile ddH2O
Functional characterization of hepatocytes
-
Human albumin ELISA quantitation set (Bethyl, cat. no. E80-129)
-
Urea nitrogen BUN test (Stanbio, cat. no. 0580-250)
-
CYP3A4 activity assay (Promega, cat. no. V9002)
-
Bile canaliculi live stain (Life Technologies, cat. no. C1165)
Immunofluorescence staining of malaria-infected MPCCs
-
Primary antibodies: anti-CD81 (BD Pharmingen, cat. no. 555675), anti-SRB1 (BD Pharmingen, cat. no. NB400-104); for P. falciparum: anti-PfHSP70 clone 4C9, anti-PfCSP clone 2A10, anti-PfEBA-175 and anti-PfMSP1; for P. vivax: anti-PvCSP (subtype VK210, anti-PvCSP clone 2F2; subtype VK247 requires a distinct clone). In our experiments, we used P. vivax subtype VK210
-
Secondary antibodies: Alexa Fluor 594 donkey anti mouse IgG (Invitrogen, cat. no. A21203) or Alexa Fluor 488 goat anti-mouse IgG (Invitrogen, cat. no. A11029)
-
Methanol (MeOH; Sigma, cat. no. 179337)
-
Paraformaldehyde (PFA; Electron Microscopy Sciences, cat. no. 15714)
-
Hoechst 33342 (Life Technologies, cat. no. H3570)
-
Aqua Mount fluorescent mounting medium (Lerner Laboratories, cat. no. 13800)
-
Fluoromount G (Southern Biotech, cat. no. 17984-25)
-
Dulbecco's PBS with CaCl2 and MgCl2 (Life Technologies, cat. no. 14040)
-
BSA (Sigma-Aldrich, cat. no. A7906)
HCV RNA quantification
-
EraGen MultiCode RTx HCV kit (Luminex Corp.)
HCV luciferase reporter assay
-
Promega luciferase assay system (Promega, cat. no. E1501)
-
White assay plates, 96 well (e.g., Corning, cat. no. 3600)
HCV NS5A staining
-
Poly-L-lysine (Sigma, cat. no. P8920)
-
Tissue culture–treated plate, 96 well (Corning)
-
HCV NS5A antibody 9E10 (ref. 87)
-
Goat anti-mouse horseradish peroxidase (HRP) secondary antibody (ImmPress kit, Vector Labs)
-
DAB+ substrate (Dako)
HCV stock generation
-
Huh-7.5 or Huh-7.5.1 cells
-
HCV RNA for electroporation
-
2-mm-Gap cuvettes
-
100 kDa MWCO Millipore Amicon ultracentifugal filters (Millipore, cat. no. EW-29968-28)
-
Cryovials (e.g., Corning)
HBV stock preparation or stock generation
-
HBV-positive patient plasma (tested HIV-, HCV-) from the Red Cross
-
1.25 M CaCl2 solution
-
Sterile 1.5-ml Eppendorf tubes
-
HepG2.2.15 cells
-
L-Glutamine solution (Sigma, cat. no. G7513)
-
Sodium pyruvate (Sigma, cat. no. P8574)
-
MEM Nonessential amino acid solution (Sigma, cat. no. M7145)
-
Williams E medium (Life Technologies, cat. no. 12551-032)
-
Hydrocortisone, water soluble (Sigma, cat. no. H0396)
-
Inosine (Sigma, cat. no. I4125)
-
DMSO (Sigma, cat. no. D8418)
-
Centricon Plus-70 centrifugal filter units (100 kDa; EMD Millipore, cat. no. UFC710008)
IPS-1 reporter lentivirus preparation and transduction
-
pTRIP-RFP-NLS-IPS reporter plasmid DNA88
-
VSV-G and HIV gag-pol plasmid DNA88
-
HEK 293T cells (e.g., ATCC)
-
Poly-L-lysine (Sigma, cat. no. P8920)
-
Tissue culture–treated well plates or flasks
-
Transfection reagent (XtremeGene9, Lipofectamine, etc.)
-
HEPES (Life Technologies, cat. no. 15630-80)
-
Polybrene (Sigma, cat. no. 107689)
Drug controls
-
DMSO (Sigma, cat. no. D8418)
-
2′-C-Methyladenosine (Santa Cruz, cat. no. 15397-12-3)
HBV infection ELISA
-
HBsAg ELISA: GS HBsAg EIA 3.0 kit (Bio-Rad, cat. no. 32591)
-
HBeAg ELISA: HBeAg Ab ELISA kit (AbNova, cat. no. KA0290)
-
3,3,5,5-Tetramethyl-benzidine substrate (Pierce, cat. no. 34029)
HBV 3.5 kb RNA and total RNA quantification
-
RNeasy Plus mini kit (Qiagen, cat. no. 74134)
-
RNase-free DNase kit (Qiagen, cat. no. 79254)
-
SuperScript III first-strand synthesis system (Invitrogen, cat. no. 18080)
-
Primers (see PROCEDURE Step 35J)
-
SYBR Premix Ex Taq kit (TaKaRa, cat. no. RR820)
HBV DNA and cccDNA quantification
-
QIAamp DNA blood mini kit (Qiagen, cat. no. 51104)
-
QIAamp Minelute virus spin kit (Qiagen, cat. no. 57704)
-
TaqMan universal PCR master mix (Applied Biosystems, cat. no. 4391128)
-
Primers (see PROCEDURE Step 35K)
-
2× HBV plasmid (known quantities for standard curve)
Southern blotting for HBV DNA forms
-
Proteinase K (Qiagen, cat. no. 19131)
-
LE-agarose (Ambion, cat. no. AM9040)
-
TAE buffer (for example, Thermo, cat. no. B49)
-
Hybond-N+ (GE Healthcare Life Science, cat. no. RPN119B)
-
Prime-It II random primer labeling kit (Agilent, cat. no. 300385)
Immunofluorescence staining of HBV core protein
-
32% (wt/vol) PFA (Electron Microscopy Sciences, cat. no. 15714)
-
Senso-Plate glass-bottom tissue culture plates (Greiner Bio-one, cat. no. 662892)
-
Triton-X 100 (Sigma, cat. no. X100)
-
Rabbit polyclonal anti-HBV core antibody (Dako, cat. no. B0586)
-
Donkey anti-rabbit DyLight 594–Alexa Fluor 594 conjugate (Jackson Immunoresearch, cat. no. 712-585-153)
-
Hoechst 33342 dye (Thermo, cat. no. 66249)



EQUIPMENT
Culturing fibroblasts
-
Tissue culture flasks
-
Tissue culture centrifuge
-
Microscope
-
Fibroblast medium
Seeding and culturing human primary hepatocytes
-
Collagen micropatterned plates
-
Tissue culture centrifuge
Preparing micropatterned cocultures plates
-
PDMS masks
-
Collagen type I
-
Plasma chamber (for example, SPI Plasma Prep III, Plasma-Etch PE-50 or PE-75)
-
Tissue culture (TC) hood with UV light
HCV stock generation
-
Electroporator
-
Ultracentrifuge for virus concentration
HBV stock preparation
-
Benchtop centrifuge
HCV infection analysis
-
Luminometer (machine capable of reading Gaussia luciferase luminescence)
-
Light microscope (for HRP+ quantification of NS5A staining)
HBV infection analysis
-
Luminometer
-
Plate reader
-
ABI 7500 or Light Cycler 480 (for RT-PCR)
-
Gel box
-
Phosphorimager
-
Epifluorescence microscope
REAGENT SETUP
ITS stock
-
Combine 20 ml of ITS, 30 ml of 1 M HEPES and 20 ml of penicillin-streptomycin. ITS stock should be used within 3 weeks when it is stored at 4 °C.
Dexamethasone stock
-
Prepare a 20 μg/ml stock by adding 1 ml of absolute ethanol to 1 mg of dexamethasone, gently swirl it to dissolve and add 49 ml of sterile DMEM medium. Aliquots can be stored at −20 °C for up to 6 months.
Glucagon stock
-
Prepare the stock at a concentration of 0.1 mg/ml in ddH2O. Aliquots can be stored at −80 °C for up to 6 months.
Hepatocyte medium
-
Prepare 200 ml of hepatocyte medium by combining and subsequently filter-sterilizing the following quantities: 7.2 ml of ITS stock, 172.8 ml of DMEM, 20 ml of FBS, 400 μl of dexamethasone and 14 μl of glucagon stock.
Critical
Hepatocyte full medium should be used within 2 weeks when it is stored at 4 °C. Do not warm and rewarm the entire volume of medium for use; remove aliquots as needed for the experiment.
Sporozoites
-
Feed mosquitoes on P. falciparum– and P. vivax–infected blood using membrane feeding. For the P. vivax Chesson strain, feed mosquitoes directly on infected monkeys44,45,89. Extract sporozoites from infected mosquitoes by dissection of their salivary glands and by passing the glands back and forth through a 26G needle fitted to a 1-ml syringe. Further purification and cryopreservation are optional45,90.
Procedure
Preparation of the PDMS etch mask
Timing 3–5 d
Critical Step
Steps 1–14 only need to be completed once.
-
1
Machine Teflon pieces that contain base and pillars for 24- or 96-well plate formats.
-
2
Screw two Teflon layers together tightly to prevent leakage of PDMS between Teflon layers.
-
3
Mix the PDMS base and curing agent at a 10:1 ratio, and place the mixture in a vacuum desiccator under vacuum to degas the PDMS mixture.
-
4
Pour degassed PDMS into Teflon molds, making sure to fill in entirety the 'pillars' that extend into the well plates and to maintain a flat rectangular base of PDMS on top of pillars (this section needs to be at least ∼2 mm thick to ensure flatness along the PDMS).
-
5
Cure the PDMS base piece in a Teflon mold in the oven overnight at 65 °C.
-
6
Carefully pull PDMS off the Teflon piece after casting, being sure to peel slowly to avoid tearing the PDMS. If large defects arise during this process, the PDMS should be recast and cured.
Pause point
PDMS is stable, and the cured bases can be stored at room temperature (RT; 20–25 °C) for years.
-
7
Design and produce high-resolution transparencies containing the island patterns for MPCCs. Standard island sizes are circles with a 500-μm diameter, with 700–1,200 μm center-to-center spacing between islands.
-
8
Order patterned silicon masters using standard foundry services (e.g., Trianja technologies, SimTech, FlowJem), ensuring that island regions are recessed in the master with a feature height of 150–250 μm.
-
9
Upon receiving the silicon master, glue the wafer into a glass Petri dish of appropriate size by smearing a few drops of PDMS (10:1 base to curing agent) onto a Petri dish and then pressing the wafer onto these drops. Ensure a tight seal such that the wafer remains flat on the Petri dish surface (that is, by using a weight), and cure it overnight at 65 °C.
-
10
Silanize the master by dropping a few drops of trichloro(1H,1H,2H,2H-perfluorooctyl)silane into a plastic weighing dish and suspending the master upside down, directly above this weighing dish in a vacuum desiccator for 2 h. Replace the trichloro(1H,1H,2H,2H-perfluorooctyl)silane and repeat the 2-h treatment.
-
11
Slowly pour the degassed PDMS mixture (10:1 base to curing agent) over the silicon master until a uniform layer of PDMS coats the glass dish into which the master is glued. Degas PDMS in vacuum desiccator again after it is poured, and note the exact volume of PDMS used to fill the dish so that future casts are performed with the same volume. After degassing the mixture, cure PDMS overnight at 65 °C.
-
12
Carefully cut the cured PDMS from the outside edge of the glass dish using a scalpel. Next, peel the cured PDMS layer off the silicon master by gently loosening the PDMS from the outside edge of the glass dish and then by slowly peeling the entire PDMS layer off. Be careful not to bend PDMS too far, or else PDMS may tear. The PDMS layer that was on top of the silicon master now has the negative replica of the features present on the master.
-
13
Use metal punches (McMaster Carr) to core out individual 'buttons' (for each well of the multiwell plate) from the PDMS with the relevant features. In particular, use a 14-mm punch for a 24-well plate and a 6-mm punch for a 96-well plate format.
-
14
Attach the PDMS buttons to the large PDMS base layer (cast on the Teflon molds) to create the final two-part PDMS etch mask. First, mix a 10:1 ratio of PDMS base to the curing agent, and add 1:1 vol/vol hexanes to this mixture to create 'PDMS glue.' Then, flip the PDMS base layer such that the pillars for each well are facing up, cover each pillar surface with PDMS glue using a spatula or brush and firmly place one cored-out PDMS button containing raised islands onto each pillar. Ensure that each button is firmly attached to the pillar below, and then cure the entire assembly overnight at 65 °C to complete fabrication of the PDMS etch mask. Place a weight (i.e., a flat glass surface with a metal weight on top) on the entire assembly to ensure that all the buttons glued onto the base structure are at the same height and leveled.
Pause point
The PDMS etch mask can be stored at RT indefinitely. Store it with clear tape covering the buttons.
Preparation of micropatterning plates
Timing 2 h
-
15
Prepare 25–50 μg/ml collagen solution. Dilute stock collagen with ddH2O. PBS can be used; however, because it leaves a salt residue in each well, the number of washes (Step 18) needs to be increased to a total of five to remove the salt, which can affect patterning.
-
16
Fill each well of a 24- or 96-well plate with 250–400 or 50–100 μl, respectively, of collagen solution. Up to 8–10 plates can be prepared at once with relative simplicity, although more can be made for large experiments.
-
17
Incubate the plates for 60–120 min at RT or for 30–60 min at 37 °C.
-
18
Rinse them three times with ddH2O.
-
19
Dry the plates thoroughly—bang the plates onto paper towels and let them dry in the back of culture hoods near the air vent for at least 2 h, although overnight drying is also acceptable.
Pause point
The plates can be stored for up to 3 months sealed in a plastic bag with desiccant pack at 4 °C.
-
20
Clamp the etch mask to the plate. Tighten it such that the pattern is evenly visible on the bottom of the plate.
Critical Step
Do not overtighten; columns may bend, thus exposing those regions that are to be shielded from plasma ablation.
-
21
Insert it into the plasma chamber and close it.
-
22
Expose it to 50–100 W oxygen plasma for 30–120 s (this may need to be optimized to obtain optimal protein patterning, depending on the plasma system used).
-
23
Release the vacuum, open the plasma chamber and remove the device. Unclamp the mask from the plate.
-
24
Sterilize the plate in a tissue culture hood under UV light for 15 min. If UV light is used, it is important to use an appropriate meter to ensure that the UV intensity in the bulb of the tissue culture hood has not diminished significantly. If UV is not available, wells can be incubated with 70% ethanol in ddH2O (300 μl per well) for 20 min followed by 3× rinses with sterile ddH2O.
Pause point
The plates can be stored for up to 3 months sealed in a plastic bag with a desiccant pack at 4 °C.
Handling of primary human hepatocytes
-
25
If cryopreserved hepatocytes are used, follow option A; if fresh hepatocytes suspended in preservation buffer are used, follow option B.
-
A
Thawing of cryopreserved primary human hepatocytes • TIMING 30 min
-
i
If a cryopreserved vial is taken out of a liquid nitrogen dewar, loosen the cap very slightly (half a turn on a thread should be sufficient) to let any nitrogen gas out and prevent the vial from bursting open upon thawing in the next step. Then, tighten the cap before thawing.
-
ii
Thaw the vial in the water bath at 37 °C. Typically, 2 min is sufficient to thaw the vial such that a small ice crystal is still visible. However, the thawing time should be optimized depending on the volume in the vial and vendor instructions.
-
iii
Spray the vial with 70% (vol/vol) ethanol and wipe it clean quickly to ensure that the potentially contaminated water from the water bath is removed before Step 25A(v).
-
iv
While the vial is still tightly closed, invert it a few times in the tissue culture hood to mix the contents, as hepatocytes often settle to the bottom of the vial.
-
v
Uncap the vial in the tissue culture hood and immediately add the contents of the vial on top of 40 ml of prewarmed (in 37 °C water bath for 15–30 min) culture medium (DMEM with 1% (vol/vol) penicillin-streptomycin without serum) in a 50-ml conical tube.
Critical Step
It is important not to use serum in the medium in order to achieve specific patterning on collagen islands in subsequent steps without nonspecific attachment to the plastic regions because of adsorption of serum proteins.
-
vi
Spin down the cells at 100g for 6 min at RT.
-
vii
Carefully remove the supernatant with an aspirator, ensuring not to disturb the hepatocyte pellet. It is acceptable to leave ∼0.25–0.5 ml of medium in the tube if the hepatocyte pellet begins to move.
-
viii
Resuspend the cells in DMEM with 1% (vol/vol) penicillin-streptomycin (without serum).
-
ix
Count the cells. The trypan blue exclusion method and manual counting using a hand tally counter works well. Hepatocytes are much larger than nonparenchymal cells and red blood cells, which may be present in very small quantities. Thus, it is relatively straightforward to identify hepatocytes under a light microscope.
Critical Step
It is also important to execute Step 25A(iii–v) as quickly as possible (<1 min) to avoid injury to hepatocytes because of higher temperatures and the presence of concentrated cryoprotectant in the vial. Thus, thawing no more than one or two vials at once is recommended.
-
i
-
B
Fresh isolated primary human hepatocytes • TIMING 15 min
-
i
Execute Step 25A(vi–viii) to remove the preservation buffer in which fresh cells are typically suspended. Then, add warm culture medium.
-
ii
Execute Step 25A(ix) to obtain a cell suspension of known cell density for seeding.
-
i
-
A
Seeding cells in ECM micropatterned plates
Timing 2 d (2–4 h active time)
-
26
Plate the cells in each well. For a 24-well plate, seed 250,000 hepatocytes in a final volume of 300 μl per well; for a 96-well plate, seed 70,000 hepatocytes in a final volume of 70 μl per well. These seeding densities can be optimized depending on the attachment efficiency of the hepatocyte lot. We have used as low as 150,000 hepatocytes and 30,000 hepatocytes in 24- and 96-well formats, respectively.
-
27
Put the cultures in the incubator. Then, three or four times each hour, take the plate out of the incubator and shake it three times by hand in both the horizontal and then the vertical perpendicular planar axes (not circular) to homogenize the cells in each well, as hepatocytes settle to the edges of the wells given their size distribution. The cells start seeding onto the islands in 15–20 min, and they should be visible from the bottom of the plate macroscopically under appropriate lighting.
Critical Step
The plate should be out of the incubator for no more than 30 s each time. In addition, ensure that shaking is not done so vigorously so as to splash the cell suspension out of the well and into the space between wells.
-
28
Every 2 h after seeding, inspect island-filling density under the microscope. When 85–90% of the islands are covered with hepatocytes, typically after 2–4 h, proceed to the next step.
-
29
Rinse unattached cells away by gentle aspiration from each well. Next, rinse 2–3 times with culture medium (DMEM with 1% vol/vol penicillin-streptomycin without serum) in immediate succession until a clear pattern is observed and minimal cells are observed under the microscope between the hepatocytes islands.
Critical Step
Approximately 1–5% of the cells will still be in suspension in each well even after the washings. These cells typically do not attach well onto the plastic, and they are washed away the next day upon fibroblast seeding.
-
30
24 h after seeding hepatocytes, remove the medium from each well with gentle aspiration, and seed 3T3-J2 mouse embryonic fibroblasts in hepatocyte culture medium. Use 40,000 total cells in a volume of 300–400 μl volume per well of a 24-well plate and 7,000 total cells in 50–70 μl per well of a 96-well plate.
-
31
Culture the plates, which can now be described as MPCCs, for 4–6 weeks or until desired for infection and/or analysis. Collect the supernatant from MPCCs every 24–48 h, and replace it with fresh hepatocyte medium. Supernatant samples can be stored at −20 or −80 °C until further use, typically for up to 12 months at −80 °C.
Hepatocyte selection and evaluation (functional assays and infectibility)
Timing multiple 30 min–3 h assay points over 3 weeks
-
32
As a first step toward qualifying candidate hepatocyte donor lots for use in long-term infection experiments (e.g., HCV persistence and Plasmodium vivax hypnozoite biology), keep MPCC cultures for at least 3 weeks and perform the observations described in each of the following options (Fig. 3). For option A, perform microscopic observations every 2–4 d. For option B, assess human albumin and urea secretion at the end of the 3-week period.
-
A
Microscopic observations • TIMING 30 min
-
i
Monitor the maintenance of morphological traits associated with differentiated primary adult human hepatocytes, such as a polygonal shape, distinct nuclei and nucleoli, the presence of bile canaliculi (bright borders between cells with rough edges) and the absence of dark granularity in the hepatocyte cytoplasm. These features are admittedly qualitative and take some practice to detect, yet hepatocytes have a very distinct prototypical morphology that, with experience, is clearly distinguishable from the surrounding fibroblasts and when compared with spread out de-differentiated hepatocytes cultured for a few days in the absence of stromal support cells.
-
i
-
B
Human albumin and urea secretion • TIMING 3 h to overnight
-
i
Collect MPCC supernatant and keep it at −20 °C.
Pause point
The supernatant can be stored at −20 °C for 6 months.
-
ii
Assay the levels of human albumin and urea secretion using appropriate kits (specific kits that can be used for this purpose are listed in the reagents section).
-
iii
Select a hepatocyte lot to use for malaria, HBV or HCV infection that exhibits stable (within 25%) albumin secretion and urea production for at least 3 weeks in culture (Fig. 3).
-
i
Critical Step
Screen 5–10 hepatocyte lots from distinct donors for suitability in each application, and bank 1–3 qualified lots for continued and on-demand use in infection studies.
-
A
-
33
Maintain hepatocytes that show both suitable morphological traits and consistent human albumin and urea secretion over a period of at least 3 weeks. Functional maintenance of a particular lot of hepatocytes is necessary but not sufficient to ensure infectibility by either malaria parasites, HBV or HCV. As such, it is recommended that hepatocyte lots that pass the functional screening described above be further assayed for their capacity to support infection by either pathogen (Step 34A–C; Fig. 3).
Infection of MPCCs with hepatotropic pathogens
Critical Step
If the analysis method planned requires microscopic analysis, ensure that MPCC cultures are established on glass-bottomed 96-well plates or 12-mm glass coverslips in the wells of a plastic 24-well plate. Use 50 or 300 μl of medium per well of a 96- or 24-well plate, respectively. For some analysis methods, the procedure will need to be modified while performing the following steps; check this before starting this section.
-
34
Infection conditions have been optimized for each pathogen. If you are infecting MPCCs with Plasmodium sporozoites, follow Step 34A. If you are infecting them with HCV or HBV, follow Step 34B or C, respectively (Fig. 3c).
-
A
Infection of MPCCs with Plasmodium sporozoites • TIMING 4 h
-
i
Ensure that you have a suspension of sporozoites at a ratio of 1:5 to 1:10 (attached hepatocytes:infectious sporozoites) using hepatocyte culture medium, plus hepatocytes cultured as described up to the end of Step 29, which have been confirmed by a previous study to have optimal function (as demonstrated by following Steps 32 and 33 on a previous culture).
Critical Step
Optimal infection rates are achieved if infections are conducted 1 d after hepatocyte seeding (Step 29). However, infections remain possible, albeit diminished, for at least 3 weeks after establishing MPCCs (Steps 30 and 31), offering flexibility with the timing of sporozoite seeding and experimental schedules.
Critical Step
The number of infective sporozoites is indirectly defined using a sporozoite gliding motility assay (performed as described in Step 35A). For cryopreserved sporozoites, sporozoite motility depends on the source material and freezing protocol used. Typically, ∼30% of cryopreserved sporozoites demonstrate gliding motility after thawing, whereas 80–90% of sporozoites freshly obtained via dissection of mosquito salivary glands demonstrate gliding motility. We typically infect MPCCs according to a ratio of 1:5 hepatocytes:gliding sporozoites for cryopreserved parasites, and a ratio of 1:6 for fresh. Example cell and parasite numbers used in a typical infection experiment are shown in the table below.
Table 2 -
ii
Remove the culture medium and add the suspension of sporozoites to the relevant wells of the plate. Shake the plates in a perpendicular axis within the same plane, two times in each direction.
-
iii
Centrifuge the plate at 600g for 5 min at RT.
-
iv
Incubate the infected plates at 37 °C and 5% CO2 for 3 h.
-
v
After 3 h, wash the wells three times with hepatocyte culture medium (50 and 300 μl per well of a 96- or 24-well plate, respectively).
-
vi
Seed 3T3-J2 mouse embryonic fibroblasts in hepatocyte culture medium, as described in Step 30. Replace the medium 24 h after the fibroblast seeding and subsequently daily.
Critical Step
If nonaseptic mosquitoes are used, increase the concentration of penicillin-streptomycin to 3% during and after the 3-h infection period. Supplement the hepatocyte medium with amphotericin B, which is also called Fungizone (1:1,000), only after the 3-h infection period—i.e., with the addition of the 3T3-J2 fibroblasts.
-
i
-
B
Infection of MPCCs with HCV • TIMING 1 h
-
i
Prepare hepatocytes that have been confirmed by a previous study to have optimal function (as demonstrated by following Steps 31–32 on a previous culture) by culturing as described up to the end of Step 29.
-
ii
Seed 3T3-J2 mouse embryonic fibroblasts (40,000 cells per well of a 24-well plate) in fibroblast medium onto the hepatocyte culture, as described in Step 30, 24 h after seeding the hepatocytes.
-
iii
Incubate the cells for 24 h after MPCCs are established. Ensure that you have HCV stock prepared as described in Box 1.
-
iv
Inoculate the cells with HCV JFH-1-based stocks (typically final concentration TCID50 per ml >106 as titered on permissive Huh-7.5 hepatoma cells is ideal, diluted in hepatocyte medium). Infection is typically done by incubating MPCCs with virus for 24 h.
Caution
Materials in contact with HCV should be decontaminated after use according to the appropriate safety protocols followed in your research environment.
Critical Step
It is also possible to infect cells with HCV before 3T3-J2 seeding, as described for malaria in Step 34A. However, we typically infect soon after fibroblast seeding. Waiting to infect for too long post fibroblast seeding typically reduces subsequent infection rates.
-
v
Wash the cultures five times with DMEM + 10% FBS and replace with fresh medium.
-
vi
Every 48 h, remove the supernatants and wash the cultures three times with medium to remove leftover luciferase. If desired (Step 35E), retain the supernatants to measure viral titer. Collection and washing frequency can be increased if daily samples are desired.
-
i
-
C
Infection of MPCCs with HBV • TIMING 1–3 h
-
i
Prepare hepatocytes that have been confirmed by a previous study to have optimal function (as demonstrated by following Steps 31 and 32 on a previous culture, by culturing as described up to the end of Step 29).
-
ii
Seed 3T3-J2 mouse embryonic fibroblasts (40,000 cells per well of a 24-well plate) in fibroblast medium onto the hepatocyte culture, 24 h after seeding of the hepatocytes, and incubate them for a further 24 h.
-
iii
Obtain HBV as described in Boxes 2 and 3.
Caution
Materials in contact with HBV should be decontaminated after use according to the appropriate safety protocols followed in your research environment.
Critical Step
It is possible to infect cells with HBV before or after 3T3-J2 seeding; however, we typically infect soon after fibroblast seeding. Infection can be performed up to at least 5 d after 3T3-J2 seeding, depending on the application, although infection rates may decrease. We recommend infecting 48 h after hepatocyte seeding.
-
iv
If desired, preincubate MPCCs for 24 h with 10 μM JAKi before infection; in our hands, this step boosts HBV infection.
-
v
Dilute HBV stocks 1:10–1:20 in culture medium, and infect MPCCs for 24 h.
-
vi
After 24 h, collect the initial inoculum supernatant (to determine the initial viral titer), wash MPCCs five times with DMEM and replace the standard culture medium.
-
vii
Collect and replace the medium every 2 d, and store it at −80 °C until the collected medium is used for analysis of HBsAg, HBeAg and secreted viral DNA.
Critical Step
Even with rigorous washing, HBsAg, HBeAg and viral DNA from initial viral stocks may remain for up to 5 d after infection; thus, measurements taken before this time are subject to false positives.
Pause point
Supernatant from infection experiments can be stored in standard polystyrene multiwell plates with a plate sealer cover at −80 °C for up to 1 month.
-
viii
For intracellular DNA and RNA endpoint measurements, collect cells from MPCCs by washing them twice with PBS, and then by mechanically disrupting cocultures and collecting the cells from each well into separate Eppendorf tubes. Centrifuge the tubes in a microcentrifuge at 1,000–2,000g for 5 min, and remove the supernatant, leaving the cell pellet at the bottom of each tube, and then flash-freeze the tubes on dry ice.
Pause point
Cell pellets from infection experiments can be stored in Eppendorf tubes at −80 °C for up to 1 month.
-
i
-
A
Infection analysis
-
35
Perform infection analysis, by following the appropriate method(s) for each pathogen. If MPCCs are infected with Plasmodium sporozoites, follow option A for a sporozoite gliding assay, option B for a cell membrane wounding and repair assay, option C for a double-staining assay for sporozoite entry and option D for immunofluorescence staining of Plasmodium-infected MPCCs. If MPCCs are infected with HCV, follow option E for a Gaussia luciferase-HCV assay, option F for a viral (NS5B) polymerase inhibitor usage assay and option G for a viral production assay. Because HBV has a complex life cycle, it can be important to measure several markers of viral persistence and replication. If MPCCs are infected with HBV, the HBsAg ELISA (option G) and HBeAg ELISA (option H), each reflect measures of viral protein production, with HBeAg being detected reliably only in high-level infection. Viral 3.5-kb RNA, which represents both the pregenomic RNA and the longest viral mRNA coding for the core protein, is used as a measure of viral transcription, and its quantification is covered in option I. Viral DNA can be quantified both intracellularly and from the supernatant (option J), and it is a measure of virion production; cccDNA is the long-lived genomic form of HBV that is considered a hallmark of true, productive infection (option K). Information about when to use particular assays is given at the start of each option.
-
A
Sporozoite gliding assay • TIMING 4 h
-
i
Prepare a 10 μg/ml solution of monoclonal antibody against Plasmodium circumsporozoite protein (anti-CSP) in PBS.
-
ii
Coat 12-mm glass coverslips or glass-bottomed multiwell plates with the anti-CSP solution for 30 min at 37 °C.
-
iii
Wash the coverslips or wells twice with PBS.
-
iv
Add 20,000 sporozoites, either freshly dissected or cryopreserved (see REAGENTS), in 300 μl of DMEM to each coverslip, centrifuge them at 600g for 5 min at RT, and then incubate them at 37 °C for 40 min to allow motile sporozoites to glide.
-
v
Wash the coverslips or wells twice with PBS, add 4% (wt/vol) PFA and incubate them for 10–15 min at RT.
-
vi
Wash the samples three times with PBS.
Pause point
Samples can be stored at 4 °C in PBS for 2 weeks.
-
vii
Block coverslips or wells with blocking solution (PBS + BSA 2% (wt/vol)) for 30 min at RT.
-
viii
Add anti-CSP at 10 μg/ml in blocking solution and incubate the samples for 1 h at RT.
-
ix
Wash the samples three times with PBS.
-
x
Add an anti-mouse IgG conjugated to Alexa Fluor 594 at a 5 μg/ml concentration in blocking solution, and then incubate the samples for 1 h at RT.
-
xi
Wash three times with PBS.
-
xii
Place one drop of Aqua Mount into each well of the 96-well plate. For MPCCs fabricated on 12-mm glass coverslips in the wells of plastic 24-well plates, the glass coverslips should be mounted with Fluoromount G (∼20 μl per coverslip) on glass slides for immunofluorescence imaging.
-
xiii
Place the mounted coverslips or the 96-well plates at 4 °C overnight.
Pause point
Seal the 96-well plates with plate sealers to allow for a longer-term storage (∼3 weeks) of the samples at 4 °C.
-
xiv
Image the coverslips or the plates using an inverted fluorescence microscope, and quantify the percentage of gliding sporozoites. A gliding sporozoite is defined as one that leaves a CSP trail of at least one circle, and we typically score at least 200 CSP+ sporozoites (10 fields with an average of 20 sporozoites per field) per sample for their gliding activity.
Critical Step
Both fresh and cryopreserved Plasmodium sporozoites may demonstrate significant batch-to-batch variability in gliding motility56. Therefore, it may be helpful to quantify the percentage of gliding sporozoites as an indicator of sporozoite infectivity in parallel with the actual MPCC infection so as to allow the comparison of data from independent experiments, if such comparisons are desired. This sporozoite gliding assay is conceptually similar to a sandwich ELISA for sporozoites.
-
i
-
B
Cell membrane wounding and repair assay • TIMING 3.5 h
-
i
Infect MPCCs (24 h after hepatocyte seeding) with sporozoites in the presence of 1 mg/ml 10,000-Da tetramethylrhodamine-dextran (lysine-fixable) (Sigma).
-
ii
At 3 h after infection, wash the wells twice with PBS, add 1% (wt/vol) PFA and incubate the samples for 20 min at RT.
-
iii
Wash them three times with PBS, and mount the samples as described above (Step 35A(xii and xiii)).
-
iv
Image the coverslips or the plates using an inverted fluorescence microscope. Migration of sporozoites through cells is quantified by the number of dextran-positive hepatocytes per island.
Critical Step
This is an alternative sporozoite quality control assay that measures sporozoite traversal of hepatocytes, which involves the use of a cell-impermeable fluorescent dextran in a cell membrane wounding and repair assay.
-
i
-
C
Double-staining assay for sporozoite entry • TIMING 5 h
-
i
At 3 h after infection, wash the wells twice with PBS, add 4% (wt/vol) PFA and incubate the samples for 10–15 min at RT.
-
ii
Wash the samples three times with PBS.
Pause point
Samples can be stored at 4 °C in PBS for 2 weeks.
-
iii
Block the samples with blocking solution (PBS + BSA 2% (wt/vol)) for 30 min at RT.
-
iv
Add anti-CSP at a concentration of 10 μg/ml in blocking solution, and incubate the samples for 1 h at RT.
-
v
Wash the samples three times with PBS.
-
vi
Add the secondary antibody anti-mouse IgG conjugated to Alexa Fluor 488 at a concentration of 5 μg/ml in blocking solution and incubate for 1 h at RT.
-
vii
Wash the samples twice with PBS.
-
viii
Permeabilize the samples with −20 °C methanol for 10 min at 4 °C. This allows the subsequent steps to label intracellular sporozoites.
-
ix
Wash the samples twice with PBS.
-
x
Add anti-CSP at a concentration of 10 μg/ml in blocking solution, and incubate the samples for 1 h at RT.
-
xi
Wash them three times with PBS.
-
xii
Add secondary antibody anti-mouse IgG conjugated to Alexa Fluor 594 at a concentration of 5 μg/ml in blocking solution and incubate the samples for 1 h at RT.
-
xiii
Wash the samples three times with PBS, counterstain them with Hoechst and mount them as described in Step 32A(xii).
-
xiv
Image the coverslips or the plates using an inverted fluorescence microscope. Sporozoites that successfully entered hepatocytes should stain only with Alexa Fluor 594 (red), whereas sporozoites that remain extracellular stain with both Alexa Fluor 594 and Alexa Fluor 488 (green) and hence appear yellow. Similar protocols have already been described91,92, and they can be referred to for more information.
Critical Step
Depending on the goal of the experiment (e.g., testing the efficacy of a Plasmodium-specific blocking antibody), it may also be important to determine the efficiency of sporozoite invasion of hepatocytes at a very early time point after infection. This can be done using this double staining assay for sporozoite entry at 3 h after infection.
-
i
-
D
Immunofluorescence staining of Plasmodium -infected MPCCs • TIMING 3 h
-
i
At the desired terminal time point, aspirate the medium completely and wash the wells two times with PBS. We recommend fixing the cultures at days 3 and 6 after infection.
-
ii
Add cold methanol (stored at −20 °C) to each well and incubate the wells at 4 °C for 10 min.
-
iii
Completely aspirate methanol and wash the wells three times with PBS.
Pause point
Samples can be stored at 4 °C in PBS for 2 weeks.
-
iv
Block each well with blocking solution (PBS + BSA 2% (wt/vol)) for 30 min at RT.
-
v
Add the desired primary antibody diluted in blocking solution and incubate the samples for 1 h at RT.
-
vi
Aspirate the primary antibody solution and wash the samples twice with PBS.
-
vii
Add the desired secondary antibody conjugated to a suitable fluorophore in blocking solution (Invitrogen; diluted 1:400 or at 5 μg/ml), and incubate the samples for 50 min at RT.
-
viii
Without washing, add the desired nuclear stain (DAPI or Hoechst 33342) at a final dilution of 2 μg/ml to the secondary antibody solution, and then incubate the samples for an additional 10 min at RT.
-
ix
Aspirate the secondary antibody and nuclear stain, and then wash each well three times with PBS.
-
x
Place one drop of Aqua Mount into each well of the 96- or 24-well plate. For MPCCs fabricated on 12-mm glass coverslips in the wells of plastic 24-well plates, the glass coverslips should be mounted with Fluoromount G (∼20 μl per coverslip) on glass slides for immunofluorescence imaging.
-
xi
Place the mounted coverslips or the 96-well plates at 4 °C overnight. Seal the 96-well plates with plate sealers to allow for a longer-term storage of the samples at 4 °C.
Pause point
Samples can be stored at 4 °C for 2–6 months.
-
xii
Image the coverslips or the plates using an inverted fluorescence microscope. The identification of liver-stage malaria parasites, also known as EEFs, is discussed in the ANTICIPATED RESULTS.
Critical Step
Routine analysis of productive liver-stage malaria infection in MPCCs at various time points after infection is also carried out using a standard immunofluorescence assay to detect various Plasmodium antigens expressed during the liver stage, including heat-shock protein 70 (HSP70, pan-liver stage), merozoites surface protein-1 (MSP-1, mid-late liver stage) and erythrocyte-binding antigen-175 (EBA-175, late liver-stage).
-
i
-
E
HCV Infection analysis by Gaussia luciferase-HCV assay • TIMING 2 h
-
i
Collect the supernatants after infection with Gluc-expressing HCV. Perform the assay as described in the kit.
Critical Step
Lysis buffer is essential to inactivate virus and to stabilize luciferase activity.
Critical Step
To ensure that luciferase detected in MPCC supernatants after infection with a Gluc-based reporter virus is indicative of active viral replication and not residual inoculum, we use a viral polymerase inhibitor control (2′-C-methyladenosine (2′CMA)). 2′CMA is used at ∼50× IC50 (2.16 mM), and it should be applied after the HCV inoculum is removed. DMSO (0.1% (vol/vol)) acts as a vehicle control, and both 2′CMA and DMSO are added fresh with medium changes over the course of the experiment.
Critical Step
RT-PCR kits are available for the assessment of viral content in supernatants or cell lysates (e.g., EraGen); however, we typically use a sensitive Gaussia luciferase (Gluc)–based viral reporter prepared as previously described93. This reporter expresses luciferase that is secreted into the culture medium, permitting viral quantification of supernatant samples.
-
i
-
F
HCV Infection analysis by fluorescent reporter • TIMING 4 d (Step 35F(i–vii)) and 4 d (Step 35F(viii–x)) and 24 h (Step 35F(xi))
-
i
Generation of IPS-1 reporter-expressing lentivirus. Coat six-well plates with poly-L-lysine (100 μg/ml), and then aspirate and wash the plates once with sterile water. This protocol can be scaled up to larger dishes if desired. Allow the plates to dry while preparing cells.
-
ii
Plate 4–5 × 105 293T cells per well of a six-well plate and incubate the cells overnight at 37 °C.
Critical Step
Propagate 293T cells in the absence of antibiotics that can adversely affect transfection efficiency.
-
iii
Before the addition of DNA/XtremeGene9/OptiMEM mix to cells, replace the medium on 293T cells with DMEM/3% (vol/vol) FBS (1 ml/well in a six-well plate) to slow cell growth.
-
iv
Transfect cells using XtremeGene9 (Roche) according to the manufacturer's protocol, by selecting a ratio of 6 μl of transfection reagent to 100 μl of OptiMEM and using the following amounts of plasmid DNA: 0.2 μg of VSV-G, 1 μg of pTRIP and 0.8 μg of HIV gag-pol per well.
-
v
Replace the medium 6 h after transfection with 1.5 ml of DMEM/3% (vol/vol) FBS per well. Collect the supernatant at 24, 48 and 72 h after transfection, pass it through a 0.45-μm filter, and store it at 4 °C protected from light. Replace the medium at each collection with DMEM/3% (vol/vol) FBS (1.5 ml/well).
-
vi
Pool the collected supernatants, and add HEPES (20 mM final concentration) and polybrene (4 μg/ml final concentration). Divide it into aliquots into cryovials and freeze them at −80 °C.
Pause point
Samples can be stored at −80 °C for up to 12 months. Avoid repeat freeze-thaw of stocks.
-
vii
Titrate lentiviral stocks by testing for infection of 293T via a limiting dilution assay using several test concentrations of lentivirus, as in Box 4 (limiting dilution assay to quantify infectious virus production).
Critical Step
293T cells from different sources can differ in their efficiency of lentivirus production; if titers are low, consider comparing different lines or using lower passages.
-
viii
Transduction with IPS-1 reporter. In this modification, transduce cells with lentivirus encoding the IPS-1 reporter 24 h after 3T3-J2 fibroblast seeding. To transduce cells, treat cultures for ∼20 s with 0.025% (wt/vol) EDTA/trypsin before washing.
-
ix
Add lentivirus diluted in polybrene (4 μg/ml)-containing hepatocyte medium if necessary, and then incubate overnight. After 24 h, wash the cultures to remove excess luciferase, and maintain them for 3 d to give cultures a chance to stabilize immunologically and for IPS-1 reporter signals to become active.
-
x
Perform HCV infection as described in Step 34B 72 h after transduction initiation, a total of 5 d after hepatocyte seeding.
-
xi
Monitor cultures via fluorescence microscopy regularly to observe relocalization of IPS-1 to the nucleus, the earliest traces of which are typically seen 24 h after infection. This can be monitored with time to watch the spread of viral infection and ultimately clearance in the culture.
Critical Step
Our alternative tool for HCV infection analysis is the use of a fluorescent reporter that permits real-time evaluation of infection in single cells88. The RFP-NLS-IPS-reporter88 uses HCV NS3/4a-mediated cleavage of IPS-1 to liberate RFP from the mitochondrial membrane. The incorporation of a nuclear localization sequence (NLS) results in RFP translocation to the nucleus in cells with replicating HCV. The following describes the generation of RFP-NLS-IPS-expressing lentivirus by co-transfection of 293T cells with the pTRIP provirus expressing the reporter, retroviral gag-pol genes and VSV glycoprotein coding sequence.
-
i
-
G
HBsAg ELISA • TIMING 3 h
-
i
Thaw the medium collected from Step 34B(vi).
-
ii
Transfer 100 μl per sample onto GS HBsAg EIA 3.0 ELISA plate coated with mouse monoclonal anti-HBsAg antibodies (Bio-Rad), and follow the manufacturer's instructions for performing the assay.
-
iii
Read luminescence from plates using standard luminometer (e.g., FLUOstar Omega; BMG Labtech)
-
iv
Calculate the cutoff for positive HBsAg results as the average of three negative control samples + 0.07 (this value has been optimized from our experimentation).
-
i
-
H
HBeAg ELISA • TIMING 3 h
-
i
Thaw the medium collected from Step 34B(vi).
-
ii
Transfer 100 μl/sample onto an ELISA plate coated with mouse monoclonal anti-HBeAg antibodies (AbNova) and follow the manufacturer's instructions for performing the assay.
-
iii
Perform assay readout using HRP detection and 3,3′,5,5′- tetramethyl-benzidine substrate (Thermo), and read out on a standard plate reader.
Critical Step
Ideally, this assay will be performed side by side with the HBsAg ELISA to minimize freeze-thaw cycles, if needed.
-
i
-
I
HBV 3.5-kb RNA and total RNA quantification • TIMING 4 h
-
i
Isolate total RNA from MPCC cell pellets (Step 34C(viii)) using the Qiagen RNeasy Plus mini kit according to the manufacturer's instructions. To rule out DNA contamination, treat total RNA with DNase I (this can be done on-column using Qiagen instructions).
-
ii
Quantify RNA using a NanoDrop, and use 200–500 ng of total RNA for first-strand cDNA synthesis.
Pause point
cDNA can be stored for up to 12 months at −20 °C.
-
iii
Use the SuperScript III RT kit (Invitrogen) to synthesize cDNA.
Pause point
cDNA can be stored for up to 12 months at −20 C.
-
iv
Perform qPCR on HBV 3.5-kb RNA and total RNA using the following primers and the SYBR Premix Ex Taq kit, according to the manufacturer's instructions on a real-time PCR instrument (e.g., ABI 7,500 or LightCycler 480) using the following primers (F: 5-GAGTGTGGATTCGCACTCC-3; R: 5-GAGGCGAGGGAGTTCTTCT-3 for HBV 3.5-kb transcripts (123-bp fragment); F: 5-TCACCAGCACCATGCAAC-3; R: 5-AAGCCACCCAAGGCACAG-3 for total HBV-specific transcripts (92-bp fragment)) and the following cycling conditions:
Table 3 -
v
Normalize results to qRT-PCR for the human RPS11 gene or other housekeeper, and verify with an additional housekeeping gene.
-
i
-
J
HBV DNA or cccDNA quantification • TIMING 4 h
-
i
Isolate viral DNA from MPCC cell pellets (Step 34C(viii)) using either the QIAamp DNA blood mini kit (Qiagen) for medium supernatants or the QIAamp Minelute Virus spin kit (Qiagen) for cell pellets, according to the manufacturer's instructions to extract DNA. Elute the final product in 60 μl of water and use 5 μl for each qPCR.
Pause point
Viral DNA can be stored for up to 12 months at −20 C.
-
ii
For HBV DNA, perform qPCR using the TaqMan Universal PCR master Mix (Applied Biosystems) using the following primers (5′-CCGTCTGTGCCTTCTCATCTG-3′ (sense); 5′-AGTCCAAGAGTCCTCTTATGTAAGACCTT-3′ (antisense); 5′-/56-FAM/CCG TGT GCA/ZEN/CTT CGCTTC ACCTCT GC/3IABkFQ/-3′ (probe)) and the following conditions on a LightCycler 480 (Roche):
Table 4 -
iii
Perform quantification using a standard curve composed of known quantities of a 2×HBV plasmid, with a concentration range from 101 to 109 copies.
-
iv
For HBV cccDNA quantification, first subject DNA extracted from cells to overnight digestion with a plasmid-safe DNase (Epicentre), at 5–10 units DNase in 50 μl volume, followed by DNase inactivation at 70 °C for 30 min.
-
v
Add 2 to 20 μl of PCR reaction mix, using the SYBR Premix Ex Taq kit (TaKaRa), and perform PCR with the following primers94 (F: 5′-TGCACTTCGCTTCACCT-3′; R: 5′-AGGGGCATTTGGTGGTC-3′) with the following conditions on a LightCycler 480 (Roche) to yield a 735-bp fragment. Perform quantification using a standard curve composed of known quantities of a 2×HBV plasmid, with a concentration range from 101 to 109 copies.
Table 5
-
i
-
K
Southern blotting for HBV DNA forms • TIMING 3 h
-
i
Extract total DNA from MPCC cell pellets (Step 34C(viii)) using the QIAamp DNA blood mini kit (Qiagen), including proteinase K treatment.
Pause point
Viral DNA can be stored for up to 12 months at −20 °C.
-
ii
Run total DNA on a 0.8% (wt/vol) agarose-TAE gel, and then denature and transfer it to a Hybond-N nylon membrane (Amersham).
-
iii
Detect the viral DNA by hybridization with a [α-32P] dCTP-labeled random primed HBV probe, using the Prime-It II random primer labeling kit (Agilent)
Caution
It is highly recommended that persons using radioactivity are fully trained on all aspects of related safety, and that they adhere to appropriate safety practices during use of this product.
-
iv
Wash the membrane once each with 2× SSC/0.1% (wt/vol) SDS, 1× SSC/0.1% (wt/vol) SDS and 0.5× SSC/0.1% (wt/vol) SDS at 67 °C for 20 min.
-
v
Detect using autoradiographic exposure.
-
i
-
L
Immunofluorescence staining of HBV core protein • TIMING 1 d
-
i
At the appropriate time during the experiment, fix MPCC cultures by washing twice with PBS, incubating for 15 min with 4% (wt/vol) PFA (Electron Microscopy Sciences) in PBS, and washing twice with PBS. We recommend performing immunofluorescence staining between 5 and 21 d after infection, depending on what time points are required for the experiment in question. (Note that for optimal results, MPCCs should be created on glass-bottom plates (Greiner Bio-One) for immunofluorescence staining).
Pause point
Plates can be stored with PBS at 4 °C for up to 2 weeks.
-
ii
Wash the cultures once with PBS, and then block and permeabilize them by incubation for 1 h in 0.1% donkey serum/0.1% (vol/vol) Triton X-100 in PBS
-
iii
Wash the plates twice with PBS, and then incubate them overnight at 4 °C with primary antibody (rabbit polyclonal anti-HBV core; we used an antibody provided by Shaul and colleagues58, but several commercial options exist; these must be tested for optimal signal-to-background and dilutions)
-
iv
Wash with PBS, and then incubate the plates for 1 h at RT with secondary antibody, donkey anti-rabbit DyLight 594 conjugate (Jackson Immunoresearch; dilution should be optimized in-house).
-
v
Wash the cultures twice with PBS, and then counterstain with Hoechst dye (Invitrogen) and wash them again.
-
vi
Image on a standard epifluorescence microscope.
-
i
-
A

Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
Steps 1–14, preparation of PDMS etch mask: 3–5 d (only needs to be completed once)
Steps 15–24, preparation of micropattering plates: 2 h
Step 25, handling of primary human hepatocytes: 15–30 min
Steps 26–31, seeding cells in ECM micropatterned plates: 2 d (2–4 h total active time, depending on the hepatocyte lot)
Steps 32 and 33, hepatocyte selection/evaluation: multiple 30 min–3 h assay points over a period of 3 weeks (only needs to be completed once per qualified donor lot; several lots can be assayed in parallel)
Step 34A, infection of MPCC with Plasmodium sporozoites: 4 h
Step 34B, infection of MPCC with HCV: 1 h
Step 34C, infection of MPCC with HBV: <1 h (for cell culture–derived virus) to 3 h (for patient-derived virus)
Plasmodium infection analysis
Step 35A, sporozoite gliding assay: 4 h
Step 35B, cell membrane wounding and repair assay: 3.5 h
Step 35C, double-staining assay for sporozoite entry: 5 h
Step 35D, immunofluorescence staining of infected MPCCs: 3 h
HCV infection analysis
Step 35E, Gaussia luciferase-HCV assay: 2 h
Step 35F, transduction with fluorescent IPS-1 reporter: 5 d
HBV infection analysis
Step 35G, HBsAg ELISA: 3 h
Step 35H, HBeAg ELISA: 3 h (in parallel with 35G)
Step 35I, HBV 3.5kb RNA and total RNA quantification: 4 h
Step 35J, HBV intracellular DNA or cccDNA quantification: 4 h
Step 35K, Southern blotting for HBV DNA: 3 h
Step 35L, immunofluorescence staining of HBV protein: 1 d
Box 1, generation of HCV stocks: 5 d
Box 2, preparation and characterization of patient-derived HBV stocks: 6 h
Box 3, preparation and characterization of HBV from infected HepG2.2.15 cells: 6 d
Box 4, limiting dilution assay to quantify infectious virus production: 4 d
Anticipated results
After seeding the hepatocytes on the micropatterned surfaces and establishing a coculture with the fibroblasts, the functional hepatocyte phenotype can be maintained for up to 4–6 weeks. Levels of infection with malaria, HBV or HCV vary across pathogen and hepatocyte donors, and to date no direct correlation has been established with respect to the levels of hepatocyte function or expression of host factors that forecast the extent of Plasmodium sporozoite or hepatitis infection. That is, these factors have been shown to be necessary but not sufficient for infection (data not shown). MPCCs remain susceptible to Plasmodium or HCV infection for many weeks after the patterning process; however, infection rates are optimal at day 1 or day 2 after seeding for Plasmodium, and at day 5 for HCV.
For malaria
If high-quality fresh sporozoites are used to infect MPCCs at 1–2 d after seeding, an infection rate between 0.5 and 1.5% can be expected at day 3 after infection. If MPCCs are infected at later time points, the infection rate at day 3 after infection decreases at different rates, depending mainly on the hepatocyte donor and secondarily on the motility of the sporozoite batch. If cryopreserved sporozoites are used, a reduction of ∼7–10 fold is expected. The difference between the number of EEFs observed at day 3.5 versus the number at day 5.5 is defined as the 'progression rate', and this metric varies between hepatocyte donors. At day 3.5, EEFs in MPCCs exhibit an average size of 5 μm in diameter and a granular appearance (Fig. 4). These two features allow for users to easily differentiate mature EEFs from undeveloped or axenic EEFs (those that initiate development extracellularly but that do not progress), and they are important measures to collect in order to avoid false positives.
At days 5 and 7 after infection, MPCCs infected with P. falciparum parasites can range in size, in part because of differences in sporozoite quality and/or the hepatocyte donor cells used, but they typically exhibit an average size of 10–15 μm and 15–20 μm, respectively, and they are similar in size to those that have been previously reported in other in vitro systems at day 5 (refs. 51,73,79). However, larger EEFs have been observed in vivo at day 5 (30–50 μm)80,81 or in humanized mouse models (18–22 μm at day 5 and 54–69 μm at day 7) (ref. 82). Nonetheless, we consistently observe that P. falciparum EEFs continue to grow in MPCCs for at least 7 d after infection56.
Infection of MPCCs with P. vivax leads to the presence of big forms (schizonts) with an average of 25 μm on day 8, and small forms (<10 μm) that persist as long as the MPCCs are maintained56. The sizes of the MPCC-derived schizonts are similar to those that have been previously reported in vitro52. By using cryopreserved P. vivax sporozoites, the infection efficiency, based on the percentage of hepatocytes containing large and small PvCSP+ EEF forms at day 6 after infection, is ∼0.013% and 2%, respectively. These numbers depend mainly on the P. vivax strain used in the infections. Similar numbers of small forms are observed for up to 21 d. Importantly, 85% of the small forms observed were shown to be intracellular (thus ruling out possible axenic infection)56. In contrast, cultures infected with P. falciparum contained very few small forms after 2 weeks in culture, and those few observed were shown to be predominantly extracellular. Reactivation of potentially dormant, small P. vivax small forms has not been observed yet using this in vitro system, but it is an active area of investigation.
For HCV
If high-titer Gluc-expressing HCV is used to infect a highly permissive hepatocyte donor, infection generally results in a 10-fold increase in luminescent signal (RLU) over baseline at 5 d after infection (ideal stocks have TCID50 per ml ∼107 and are diluted 1:10 for infection, yielding a final multiplicity of infection (MOI) of ∼5 for hepatocytes; MOI ∼1 if 3T3-J2 cells are included in calculation). This signal decays over time, but it typically can be resolved over background for ∼2 weeks (Fig. 5). If the hepatocytes are transduced with the RFP-NLS-IPS reporter before infection, nuclear translocation of the RFP reporter (representing successful viral infection) is observed in ∼1.5% of hepatocytes. It is sometimes possible to see small clusters of hepatocytes within certain islands exhibiting nuclear translocation, which may be a result of direct cell-cell transmission of HCV. In addition, infectious virions can be collected from the supernatant of infected MPCC cultures, typically maintaining a titer of 101–102 TCID50 per ml at 10–14 d after infection.
The HCV luciferase signal does decay over 2 weeks, yet we do find that both the luciferase readout and RFP-NLS-IPS reporter are sufficiently sensitive to perform testing of antiviral compounds. By using the luciferase reporter, treatment with a protease inhibitor (ITMN191), polymerase inhibitor (2′CMA) or immune activator (interferon; IFN) results in rapid decay of the signal to background levels, as early as 5–6 d after infection. By RFP-NLS-IPS reporter, untreated 'islands' contain about four hepatocytes exhibiting nuclear translocation of the reporter as early as 2 d after infection, whereas 2′CMA-treated islands rarely contain infected hepatocytes (∼0.07 hepatocytes/island).
For HBV
If high-titer patient-derived virus (starting titer of ∼108–109 GE per ml before clot removal; yield after clot removal is about tenfold lower, and then diluted 1:10–1:20 in culture medium) is used to infect a highly permissive hepatocyte donor, HBV infection results in sustained production of HBsAg, robust transcription of viral RNAs and establishment of a cccDNA pool (Fig. 6). As discussed more thoroughly in a recent publication58, inhibition of the hepatocyte interferon-inducible innate immune response, by treating cultures with a pan-JAK inhibitor (JAKi), results in even more robust infection, including sustained production of HBV E antigen (a clinical marker of active viral replication), and growth in the cccDNA pool over time. Upon JAKi treatment, ∼20–25% of hepatocytes stain positive for HBV Core protein, which generally exhibits nuclear localization as expected, given its incorporation into the HBV cccDNA 'minichromosome'.
HBV-infected MPCCs respond to antiviral drug treatment in ways that match clinical outcomes, providing a useful platform for testing new antivirals. Upon pretreatment of MPCCs with either entecavir (a reverse transcriptase inhibitor) or IFN, HBV titers are markedly reduced, along with all other viral readouts including cccDNA. However, when drug treatment is started after an infection is established (e.g. at 7 d after infection), responses to the two drugs differ. IFN, a multifunctional innate immune stimulator that can generate cures in a small minority of prolonged HBV infections, effectively decreases the amount of viral transcription and the levels of cccDNA in infected cells. Entecavir, in contrast, is very effective at reducing the viral titer by preventing reverse transcription, but it does not affect forward transcription or cccDNA levels. This mimics the clinical scenario, in which entecavir prevents viral rebound but cannot cure chronically infected patients.
References
Lavanchy, D. The global burden of hepatitis C. Liver Int. 29 (suppl. 1), 74–81 (2009).
Ganem, D. & Prince, A.M. Hepatitis B virus infection—natural history and clinical consequences. N. Engl. J. Med. 350, 1118–1129 (2004).
Razavi, H. et al. Chronic hepatitis C virus (HCV) disease burden and cost in the United States. Hepatology 57, 2164–2170 (2013).
Alonso, P.L. et al. A research agenda for malaria eradication: drugs. PLoS Med. 8, e1000402 (2011b).
Wells, T.N., Alonso, P.L. & Gutteridge, W.E. New medicines to improve control and contribute to the eradication of malaria. Nat. Rev. Drug Discov. 8, 879–891 (2009).
Wells, T.N., Burrows, J.N. & Baird, J.K. Targeting the hypnozoite reservoir of Plasmodium vivax: the hidden obstacle to malaria elimination. Trends Parasitol. 26, 145–151 (2010).
Billerbeck, E., de Jong, Y., Dorner, M., de la Fuente, C. & Ploss, A. Animal models for hepatitis C. Curr. Top. Microbiol. Immunol. 369, 49–86 (2013).
Kaushansky, A., Mikolajczak, S.A., Vignali, M. & Kappe, S.H. Of men in mice: the success and promise of humanized mouse models for human malaria parasite infections. Cell Microbiol. 16, 602–611 (2014).
Lohmann, V. & Bartenschlager, R. On the history of hepatitis C virus cell culture systems. J. Med. Chem. 57, 1627–1642 (2014).
Farkas, D. & Tannenbaum, S.R. In vitro methods to study chemically-induced hepatotoxicity: a literature review. Curr. Drug Metab. 6, 111–125 (2005).
Godoy, P. et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 87, 1315–1530 (2013).
Guillouzo, A. Liver cell models in in vitro toxicology. Environ. Health Perspect. 106 (suppl. 2), 511–532 (1998).
Khetani, S.R. et al. Microengineered liver tissues for drug testing. J. Lab. Autom. 20, 216–250 (2015).
Guillouzo, A. & Guguen-Guillouzo, C. Evolving concepts in liver tissue modeling and implications for in vitro toxicology. Expert Opin. Drug Metab. Toxicol. 4, 1279–1294 (2008).
LeCluyse, E.L. Human hepatocyte culture systems for the in vitro evaluation of cytochrome P450 expression and regulation. Eur. J. Pharm. Sci. 13, 343–368 (2001).
Lagaye, S. et al. Efficient replication of primary or culture hepatitis C virus isolates in human liver slices: a relevant ex vivo model of liver infection. Hepatology 56, 861–872 (2012).
van Midwoud, P.M., Merema, M.T., Verweij, N., Groothuis, G.M. & Verpoorte, E. Hydrogel embedding of precision-cut liver slices in a microfluidic device improves drug metabolic activity. Biotechnol. Bioeng. 108, 1404–1412 (2011).
Gerets, H.H. et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biol. Toxicol. 28, 69–87 (2012).
Wilkening, S., Stahl, F. & Bader, A. Comparison of primary human hepatocytes and hepatoma cell line Hepg2 with regard to their biotransformation properties. Drug Metab. Dispos. 31, 1035–1042 (2003).
Andersson, T.B., Kanebratt, K.P. & Kenna, J.G. The HepaRG cell line: a unique in vitro tool for understanding drug metabolism and toxicology in human. Expert Opin. Drug Metab. Toxicol. 8, 909–920 (2012).
Le Vee, M. et al. Functional expression of sinusoidal and canalicular hepatic drug transporters in the differentiated human hepatoma HepaRG cell line. Eur. J. Pharm. Sci. 28, 109–117 (2006).
Le Vee, M., Noel, G., Jouan, E., Stieger, B. & Fardel, O. Polarized expression of drug transporters in differentiated human hepatoma HepaRG cells. Toxicol. In Vitro 27, 1979–1986 (2013).
Lubberstedt, M. et al. HepaRG human hepatic cell line utility as a surrogate for primary human hepatocytes in drug metabolism assessment in vitro. J. Pharmacol. Toxicol. Methods 63, 59–68 (2011).
Szabo, M., Veres, Z., Baranyai, Z., Jakab, F. & Jemnitz, K. Comparison of human hepatoma HepaRG cells with human and rat hepatocytes in uptake transport assays in order to predict a risk of drug-induced hepatotoxicity. PLoS ONE 8, e59432 (2013).
Xu, J.J. et al. Cellular imaging predictions of clinical drug-induced liver injury. Toxicol. Sci. 105, 97–105 (2008).
Bi, Y.A., Kazolias, D. & Duignan, D.B. Use of cryopreserved human hepatocytes in sandwich culture to measure hepatobiliary transport. Drug Metab. Dispos. 34, 1658–1665 (2006).
Dunn, J.C., Yarmush, M.L., Koebe, H.G. & Tompkins, R.G. Hepatocyte function and extracellular matrix geometry: long-term culture in a sandwich configuration. FASEB J. 3, 174–177 (1989).
Khetani, S.R. & Bhatia, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 26, 120–126 (2008).
Sivaraman, A. et al. A microscale in vitro physiological model of the liver: predictive screens for drug metabolism and enzyme induction. Curr. Drug Metab. 6, 569–591 (2005).
LeCluyse, E.L., Witek, R.P., Andersen, M.E. & Powers, M.J. Organotypic liver culture models: meeting current challenges in toxicity testing. Crit. Rev. Toxicol. 42, 501–548 (2012).
Ramsden, D., Zhou, J. & Tweedie, D.J. Determination of a degradation constant for CYP3A4 by direct suppression of mRNA in a novel human hepatocyte model, HepatoPac. Drug Metab. Dispos. 43, 1307–1315 (2015).
Si-Tayeb, K. et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 51, 297–305 (2010).
Schwartz, R.E., Fleming, H.E., Khetani, S.R. & Bhatia, S.N. Pluripotent stem cell-derived hepatocyte-like cells. Biotechnol. Adv. 32, 504–513 (2014).
Cai, J. et al. in StemBook 1.52.1 (The Stem Cell Research Community) (Harvard Stem Cell Institute, 2012).
Berger, D.R., Ware, B.R., Davidson, M.D., Allsup, S.R. & Khetani, S.R. Enhancing the functional maturity of induced pluripotent stem cell-derived human hepatocytes by controlled presentation of cell-cell interactions in vitro. Hepatology 61, 1370–1381 (2015).
Shan, J. et al. Identification of small molecules for human hepatocyte expansion and iPS differentiation. Nat. Chem. Biol. 9, 514–520 (2013).
Bhatia, S.N. & Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 32, 760–772 (2014).
Gripon, P. et al. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 99, 15655–15660 (2002).
Rehermann, B. & Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 5, 215–229 (2005).
Gripon, P. et al. Hepatitis B virus infection of adult human hepatocytes cultured in the presence of dimethyl sulfoxide. J. Virol. 62, 4136–4143 (1988).
Banaudha, K. et al. Primary hepatocyte culture supports hepatitis C virus replication: a model for infection-associated hepatocarcinogenesis. Hepatology 51, 1922–1932 (2010).
Buck, M. Direct infection and replication of naturally occurring hepatitis C virus genotypes 1, 2, 3 and 4 in normal human hepatocyte cultures. PLoS ONE 3, e2660 (2008).
Harding, M.J. et al. An implantable vascularized protein gel construct that supports human fetal hepatoblast survival and infection by hepatitis C virus in mice. PLoS ONE 5, e9987 (2010).
Chattopadhyay, R. et al. Establishment of an in vitro assay for assessing the effects of drugs on the liver stages of Plasmodium vivax malaria. PLoS ONE 5, e14275 (2010).
Epstein, J.E. et al. Live attenuated malaria vaccine designed to protect through hepatic CD8+ T cell immunity. Science 334, 475 (2011).
Hollingdale, M.R., Collins, W.E. & Campbell, C.C. In vitro culture of exoerythrocytic parasites of the North Korean strain of Plasmodium vivax in hepatoma cells. Am. J. Trop. Med. Hyg. 35, 275–276 (1986).
Karnasuta, C. et al. Complete development of the liver stage of Plasmodium falciparum in a human hepatoma cell line. Am. J. Trop. Med. Hyg. 53, 607–611 (1995).
Sattabongkot, J. et al. Establishment of a human hepatocyte line that supports in vitro development of the exo-erythrocytic stages of the malaria parasites Plasmodium falciparum and P. vivax. Am. J. Trop. Med. Hyg. 74, 708–715 (2006).
Yokoo, H. et al. Proteomic signature corresponding to α-fetoprotein expression in liver cancer cells. Hepatology 40, 609–617 (2004).
Dembele, L. et al. Persistence and activation of malaria hypnozoites in long-term primary hepatocyte cultures. Nat. Med. 20, 307–312 (2014).
Mazier, D. et al. Complete development of hepatic stages of Plasmodium falciparum in vitro. Science 227, 440–442 (1985).
Mazier, D. et al. Cultivation of the liver forms of Plasmodium vivax in human hepatocytes. Nature 307, 367–369 (1984).
van Schaijk, B.C. et al. Gene disruption of Plasmodium falciparum p52 results in attenuation of malaria liver stage development in cultured primary human hepatocytes. PLoS ONE 3, e3549 (2008).
Yalaoui, S. et al. Scavenger receptor BI boosts hepatocyte permissiveness to Plasmodium infection. Cell Host Microbe 4, 283–292 (2008).
Bhatia, S.N., Balis, U.J., Yarmush, M.L. & Toner, M. Effect of cell-cell interactions in preservation of cellular phenotype: cocultivation of hepatocytes and nonparenchymal cells. FASEB J. 13, 1883–1900 (1999).
March, S. et al. A microscale human liver platform that supports the hepatic stages of Plasmodium falciparum and vivax. Cell Host Microbe 14, 104–115 (2013).
Ploss, A. et al. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc. Natl. Acad. Sci. USA 107, 3141–3145 (2010).
Shlomai, A. et al. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc. Natl. Acad. Sci. USA 111, 12193–12198 (2014).
Guguen-Guillouzo, C. et al. Maintenance and reversibility of active albumin secretion by adult rat hepatocytes co-cultured with another liver epithelial cell type. Exp. Cell Res. 143, 47–54 (1983).
Rheinwald, J.G. & Green, H. Serial cultivation of strains of human epidermal keratinocytes: the formation of keratinizing colonies from single cells. Cell 6, 331–343 (1975).
Khetani, S.R., Szulgit, G., Del Rio, J.A., Barlow, C. & Bhatia, S.N. Exploring interactions between rat hepatocytes and nonparenchymal cells using gene expression profiling. Hepatology 40, 545–554 (2004).
Leclercq, L. et al. Which human metabolites have we MIST? Retrospective analysis, practical aspects, and perspectives for metabolite identification and quantification in pharmaceutical development. Chem. Res. Toxicol. 22, 280–293 (2009).
Olson, H. et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 32, 56–67 (2000).
Shih, H., Pickwell, G.V., Guenette, D.K., Bilir, B. & Quattrochi, L.C. Species differences in hepatocyte induction of CYP1A1 and CYP1A2 by omeprazole. Hum. Exp. Toxicol. 18, 95–105 (1999).
Wang, W.W., Khetani, S.R., Krzyzewski, S., Duignan, D.B. & Obach, R.S. Assessment of a micropatterned hepatocyte coculture system to generate major human excretory and circulating drug metabolites. Drug Metab. Dispos. 38, 1900–1905 (2010).
Khetani, S.R. et al. Use of micropatterned cocultures to detect compounds that cause drug-induced liver injury in humans. Toxicol. Sci. 132, 107–117 (2013).
Chan, T.S., Yu, H., Moore, A., Khetani, S.R. & Tweedie, D. Meeting the challenge of predicting hepatic clearance of compounds slowly metabolized by cytochrome P450 using a novel hepatocyte model, HepatoPac. Drug Metab. Dispos. 41, 2024–2032 (2013).
Trask, O.J. Jr., Moore, A. & LeCluyse, E.L. A micropatterned hepatocyte coculture model for assessment of liver toxicity using high-content imaging analysis. Assay Drug Dev. Technol. 12, 16–27 (2014).
Ramsden, D., Tweedie, D.J., Chan, T.S., Taub, M.E. & Li, Y. Bridging in vitro and in vivo metabolism and transport of faldaprevir in human using a novel cocultured human hepatocyte system, HepatoPac. Drug Metab. Dispos. 42, 394–406 (2014).
Ng, S. et al. Hypoxia promotes liver-stage malaria infection in primary human hepatocytes in vitro. Dis. Model Mech. 7, 215–224 (2014).
Ukairo, O. et al. Long-term stability of primary rat hepatocytes in micropatterned cocultures. J. Biochem. Mol. Toxicol. 27, 204–212 (2013).
Aoyama, S., Lambirth, S. & Khetani, S.R. A long-term culture model for primary hepatoyctes from Cynomolgus hepatocytes. Drug Metab. Rev. 43, 124 (2011).
Dembele, L. et al. Towards an in vitro model of Plasmodium hypnozoites suitable for drug discovery. PLoS ONE 6, e18162 (2011).
Davidson, M.D., Lehrer, M. & Khetani, S.R. Hormone and drug-mediated modulation of glucose metabolism in a microscale model of the human liver. Tissue Eng. Part C Methods 21, 716–725 (2015).
Qin, D., Xia, Y. & Whitesides, G.M. Soft lithography for micro- and nanoscale patterning. Nat. Protoc. 5, 491–502 (2010).
March, S., Hui, E.E., Underhill, G.H., Khetani, S. & Bhatia, S.N. Microenvironmental regulation of the sinusoidal endothelial cell phenotype in vitro. Hepatology 50, 920–928 (2009).
Chen, A.A., Khetani, S.R., Lee, S., Bhatia, S.N. & Van Vliet, K.J. Modulation of hepatocyte phenotype in vitro via chemomechanical tuning of polyelectrolyte multilayers. Biomaterials 30, 1113–1120 (2009).
Khetani, S.R., Chen, A.A., Ranscht, B. & Bhatia, S.N. T-cadherin modulates hepatocyte functions in vitro. FASEB J. 22, 3768–3775 (2008).
Meis, J.F. et al. Fine structure of the malaria parasite Plasmodium falciparum in human hepatocytes in vitro. Cell Tissue Res. 244, 345–350 (1986).
Shortt, H. History of recent researches on tissue phases of the malaria parasite at the London School of Hygiene and Tropical Medicine. Trans. R. Soc. Trop. Med. Hyg. 45, 175–188 (1951).
Shortt, H.E., Garnham, P.C. & Malamos, B. The pre-erythrocytic stage of mammalian malaria. Br. Med. J. 1, 192–194 (1948).
Vaughan, A.M. et al. Complete Plasmodium falciparum liver-stage development in liver-chimeric mice. J. Clin. Invest. 122, 3618–3628 (2012).
Todaro, G.J. & Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17, 299–313 (1963).
Bhatia, S.N., Yarmush, M.L. & Toner, M. Controlling cell interactions by micropatterning in co-cultures: hepatocytes and 3T3 fibroblasts. J. Biomed. Mater. Res. 34, 189–199 (1997).
Zhong, J. et al. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102, 9294–9299 (2005).
Sells, M.A., Chen, M.L. & Acs, G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA 84, 1005–1009 (1987).
Lindenbach, B.D. et al. Complete replication of hepatitis C virus in cell culture. Science 309, 623–626 (2005).
Jones, C.T. et al. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat. Biotechnol. 28, 167–171 (2010).
Albuquerque, S.S. et al. Host cell transcriptional profiling during malaria liver stage infection reveals a coordinated and sequential set of biological events. BMC Genomics 10, 270 (2009).
Hoffman, S.L. et al. Development of a metabolically active, non-replicating sporozoite vaccine to prevent Plasmodium falciparum malaria. Hum. Vaccin. 6, 97–106 (2010).
Renia, L. et al. Malaria sporozoite penetration. A new approach by double staining. J. Immunol. Methods 112, 201–205 (1988).
Silvie, O. et al. Hepatocyte CD81 is required for Plasmodium falciparum and Plasmodium yoelii sporozoite infectivity. Nat. Med. 9, 93–96 (2003).
Marukian, S. et al. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 48, 1843–1850 (2008).
Glebe, D. et al. Pre-s1 antigen-dependent infection of Tupaia hepatocyte cultures with human hepatitis B virus. J. Virol. 77, 9511–9521 (2003).
Portugal, S., Drakesmith, H. & Mota, M.M. Superinfection in malaria: Plasmodium shows its iron will. EMBO Rep. 12, 1233–1242 (2011).
Arzberger, S., Hosel, M. & Protzer, U. Apoptosis of hepatitis B virus-infected hepatocytes prevents release of infectious virus. J. Virol. 84, 11994–12001 (2010).
Reed, L.J. & Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27, 493–497 (1938).
Acknowledgements
We thank R. Wirtz (Centers for Disease Control and Prevention) and F. Zavala (Johns Hopkins University) for PvCSP and HSP70 monoclonal antibodies, respectively, and the Malaria Research and Reference Reagent Resource Center (MR4) for monoclonal antibodies recognizing PfCSP (clone 2A10, deposited by E. Nardin) and EBA175 (clone R217, deposited by the US National Institute for Allergy and Infectious Disease (NIAID)). We are grateful to S. Hoffman and the Sanaria manufacturing team for the production of P. falciparum sporozoites and P. vivax sporozoites. We also thank J. Sattabongkot Prachumsri and R. Patrapuvich (Mahidol University) for providing fresh P. vivax sporozoites, and H. Green (Harvard University) for providing 3T3-J2 fibroblasts. This work has been supported in part by funding from the NIH (RO1 DK85713), a Skolkovo Institute of Science and Technology Grant 022423-003 and the Bill and Melinda Gates Foundation (OPP1023607). S.N.B is a Howard Hughes Medical Institute Investigator. S.R.K. acknowledges funding from the National Science Foundation (CAREER CBET-1351909) and Colorado State University. This paper is dedicated to the memory of Dr. Howard Green, a visionary and inspiring scientist.
Author information
Authors and Affiliations
Contributions
S.M., V.R., K.T., M.M.M., S.R.K., C.M.R. and S.N.B. conceived the study; S.M., V.R., K.T., S.N., M.S. and A.S. designed the experiments; malaria studies were conducted by S.M., S.N., N.G. and A.G.; hepatitis experiments were conducted by V.R., K.T., M.S. and A.S.; and the manuscript was prepared by S.M., V.R., K.T., N.G., S.R.K., H.E.F. and S.N.B.
Corresponding author
Ethics declarations
Competing interests
S.N.B. and S.R.K. are the founders of Hepregen Corporation. The remaining authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
March, S., Ramanan, V., Trehan, K. et al. Micropatterned coculture of primary human hepatocytes and supportive cells for the study of hepatotropic pathogens. Nat Protoc 10, 2027–2053 (2015). https://doi.org/10.1038/nprot.2015.128
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2015.128
This article is cited by
-
Comparative analyses of functional antibody-mediated inhibition with anti-circumsporozoite monoclonal antibodies against transgenic Plasmodium berghei
Malaria Journal (2023)
-
Modelling host–microbiome interactions in organ-on-a-chip platforms
Nature Reviews Bioengineering (2023)
-
Generation of proliferating human adult hepatocytes using optimized 3D culture conditions
Scientific Reports (2021)
-
In vitro and in vivo inhibition of malaria parasite infection by monoclonal antibodies against Plasmodium falciparum circumsporozoite protein (CSP)
Scientific Reports (2021)
-
Two chemoattenuated PfSPZ malaria vaccines induce sterile hepatic immunity
Nature (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
