
Abstract
Tobacco addiction is a global public health problem. Addiction to tobacco is thought to involve the effects of nicotine on the dopaminergic system. Only one study has previously investigated dopamine synthesis capacity in cigarette smokers. This study, exclusively in male volunteers, reported increased dopamine synthesis capacity in heavy smokers compared with non-smokers. We sought to determine whether dopamine synthesis capacity was elevated in a larger sample of cigarette smokers that included females. Dopamine synthesis capacity was measured in 15 daily moderate smokers with 15 sex- and age-matched control subjects who had never smoked tobacco. Dopamine synthesis capacity (indexed as the influx rate constant Kicer) was measured with positron emission tomography and 3,4-dihydroxy-6-[18F]-fluoro-l-phenylalanine. There was no significant group difference in dopamine synthesis capacity between smokers and non-smoker controls in the whole striatum (t28=0.64, p=0.53) or any of its functional subdivisions. In smokers, there were no significant relationships between the number of cigarettes smoked per day and dopamine synthesis capacity in the whole striatum (r=−0.23, p=0.41) or any striatal subdivision. These findings indicate that moderate smoking is not associated with altered striatal dopamine synthesis capacity.
Similar content being viewed by others
INTRODUCTION
Tobacco addiction is a major global public health problem (Ezzati and Lopez, 2003). Only 3–5% of self-quitters achieve prolonged abstinence for 6–12 months after quitting (Hughes et al, 2004). Treatment of tobacco addiction is successful in less than 19% of cases (West et al, 2000), indicating that there is a pressing need to develop improved treatments (Menossi et al, 2013). The development of better therapies for tobacco addiction is likely to need greater understanding of the neurobiological changes associated with tobacco use. Addiction to tobacco is thought to involve the effects of nicotine, its main addictive component (Stolerman et al, 1995), on the dopaminergic system (Balfour et al, 2000; Pidoplichko et al, 1997) as nicotinic receptors have been identified on nigrostriatal and mesolimbic dopaminergic neurons (Clarke and Pert, 1985). Supporting this, studies in rodents and non-human primates show that tobacco or nicotine increase dopamine neuron firing (Grenhoff et al, 1986; Zhang and Sulzer, 2004), increase dopamine release (Dewey et al, 1999; Gallezot et al, 2013; Marenco et al, 2004; Pontieri et al, 1996), and increase dopamine synthesis (Tsukada et al, 2005) in the striatum.
In humans, tobacco use has been associated with both increased striatal dopamine synthesis capacity (Salokangas et al, 2000) and dopamine release in response to acute cigarette use in smokers (Brody et al, 2004; Le Foll et al, 2013). However, two studies found that acute nicotine use did not elicit a significant dopamine release in smokers (Barrett et al, 2004; Montgomery et al, 2007), although these did find that the subjective hedonic response to acute nicotine was related to dopamine release. Smoking-induced dopamine release has been associated with a reduction in craving and the severity of tobacco dependence (Brody et al, 2004). Yet, unlike other drugs of addiction, drug-related (ie, smoking-related) cues did not result in significant dopamine release in smokers when compared with neutral images (Chiuccariello et al, 2013). Interestingly, one study (Busto et al, 2009) found that tobacco dependence was associated with reduced amphetamine-induced striatal dopamine release, although this is likely exacerbated by comorbid depression. These findings may reflect differences in the study design (Gallezot et al, 2013), the influence of other factors, such as sex effects, co-morbidity, or genetic variants (Dierker et al, 2002; Kendler et al, 1993; Lerman et al, 1998; Zhang et al, 2006), or the difficulty of imaging dopamine changes that are comparatively small (Egerton et al, 2010). In terms of other aspects of dopaminergic function, a large study on the dopamine transporter did not find an association between smoking and dopamine transporter availability (Thomsen et al, 2013).
As discussed above, studies indicate that striatal dopamine synthesis may be altered by nicotine exposure. Dopamine synthesis capacity can be indexed in humans using a radiolabeled dopamine precursor, L-3,4-dihydroxyphenylalanine (L-DOPA) with positron emission tomography (PET; Kumakura and Cumming, 2009). Our primary hypothesis was, therefore, that cigarette smokers would have increased dopamine synthesis capacity compared with non-smoker controls. To our knowledge, only one study has investigated dopamine synthesis capacity in cigarette smokers. This study, exclusively in male volunteers, found that striatal uptake of [18F]-DOPA was 16–29% higher in smokers than non-smokers (Salokangas et al, 2000). However, as this sample was exclusively of males and there is evidence of sex differences in the release of dopamine in response to nicotine (Dluzen and Anderson, 1997), we sought to determine whether dopamine synthesis capacity was elevated in a larger sample of cigarette smokers that included females and did not have a history of psychiatric co-morbidity including depression and alcohol use disorders.
MATERIALS AND METHODS
A case–control design was used to compare striatal [18F]-DOPA uptake in smokers to that in non-smokers. The study was conducted in accordance with the Declaration of Helsinki and followed National Research Ethics Service and the Administration of Radioactive Substances Advisory Committee approval. All participants received full information about the study and gave informed written consent to take part.
Participants
Participants were recruited through advertisements in the press. The non-smokers were matched to the smokers on the basis of age (within 5 years) and sex. Inclusion criteria for all subjects were: minimum age 18 years, good physical health with no history of major medical condition, and capacity to give written informed consent. The exclusion criteria for all participants were: presence of any significant current medical disorder or treatment including history of head injury resulting in loss of consciousness and any neurological disorder; contraindications to PET including pregnancy or breast-feeding; a diagnosis of past or current psychiatric disorders including personality disorder using the Structured Clinical Interview for DSM-IV (SCID; First et al, 1996) including alcohol or any other substance dependence or abuse (apart from Nicotine Use Disorders in cigarette smokers); evidence of an At Risk Mental State for Psychosis; drug use other than alcohol or cigarettes in the 3 months before PET scanning; a family history of any psychotic disorder in first- or second-degree relatives. All participants provided urine samples to screen for drug use (Monitect HC12, Branan Medical Corporation, Irvine, California), and in women, for pregnancy test. Participants were excluded if either sample came back with positive result on the day of the scan. No subject was taking psychotropic medication at the time of study participation.
Smoking Data
Smoking data were collected via a semi-structured questionnaire for assessing exposure to cigarettes and alcohol (from the Cannabis Experiences Questionnaire interview; Barkus et al, 2006). The non-smoker group was defined as those with no lifetime use of tobacco.
PET
All participants were asked to not to eat or drink (except water), and refrain from alcohol for 12 h before the scan. The smokers were allowed tobacco 3 h before the scan. This time period was selected so that nicotine levels were at steady state (Benowitz et al, 1982). In addition, this is a similar time period to the only other study that has investigated dopamine synthesis capacity in smokers (Salokangas et al, 2000). Less than 2 h would coincide with peak plasma nicotine levels and longer durations would likely be measuring the dopamine system in a state of nicotine withdrawal. Imaging data from the PET scans were obtained on a Siemens CTI ECAT HR+962 PET scanner (Siemens, Erlanger, Germany) in three-dimensional mode with an axial field of view of 15.5 cm. One hour before the scan, participants received 400 mg entacapone, a peripheral catechol-0-methyltransferase inhibitor, and 150 mg carbidopa, a peripheral aromatic acid decarboxylase inhibitor, in order to increase specific signal detection, as these compounds decrease the formation of radiolabeled metabolites that may cross the blood–brain barrier (Cumming et al, 1993; Guttman et al, 1993). A 10 min transmission scan was conducted before the radiotracer injection using a 150-MBq cesium-137 rotating point source to correct for scatter and attenuation. Participants were positioned in the scanner with the orbitomeatal line parallel to the transaxial plane of the tomograph. Head position was marked and monitored via laser crosshairs and a camera, and movement was minimized using a head strap.
A 17 MeV GE PET-trace cyclotron was used for radionuclide production. The gas target was filled with 18O2 and bombarded at 40 μA for 30 min followed by a passivation bombardment of 0.1% F2 in argon at 20 μA for 20 min. This produced [18F]-F by the 18O(p,n) 18F reaction. An electrophilic fluorination procedure was then used to synthesize 6-[18F]fluoro-L-DOPA. In brief, [18F]F2 was bubbled through a solution of 6-trimethylstannyl-L-DOPA (60 mg) stirring in Deutero-chloroform (5 ml) over 20 min at 5 °C. 6 M HCl (2 ml) was added and the chloroform evaporated at 70 °C. The resulting aqueous mixture was heated at reflux for 10 min before allowing to cool. The cooled crude mixture was purified by semi-prep high-pressure liquid chromatography polymer X column eluting with ammonium acetate buffer. The peak corresponding to [18F]-L-DOPA eluted at 15 min was stabilized with 1 mg ascorbic acid and sodium phosphate dibasic. Typical yields were 2.96–3.33 GBq. For quality assurance purposes, a sample was taken from each synthesis and analyzed by reverse phase high-pressure liquid chromatography to confirm identity and purity. To be able to proceed with the injection, a radiochemical purity of 95.0% or higher was required. Approximately 180 MBq of [18F]-DOPA was administered as a bolus intravenous injection 30 s after the start of the emission scan. Emission data were acquired as 26 frames of increasing duration over the 90 min scan. This comprised a 30-s background frame, 4 60-s frames, 3 120-s frames, 3 180-s frames, and finally 15 300-s frames.
Image Analysis
To correct for head movement in the scanner, non-attenuation-corrected dynamic images were denoized using a level 2, order 64 Battle-Lemarie wavelet filter. We used nonattenuation-corrected images used for the realignment algorithm as they include greater scalp signal, improving re-alignment compared with attenuated-corrected images (Turkheimer et al, 1999). Frames were realigned to a single ‘reference’ frame, acquired 10 min post-injection, employing a mutual information algorithm (Studholme et al, 1996). The transformation parameters were then applied to the corresponding attenuated-corrected dynamic images. The realigned frames were then summated, creating a movement-corrected dynamic image, which was used in the analysis. The cerebellar reference region (Kumakura and Cumming, 2009) was defined using a probabilistic atlas (Martinez et al, 2003), and as previously described, regions of interest (ROI) in the whole striatum and its functional sub-divisions were delineated to create an ROI map (Egerton et al, 2010). The functional subdivisions of the striatum reflect the topographical arrangement of corticostriatal projections. Projections to the sensorimotor striatum come from the motor cortex and related areas for instance the premotor cortex, primary motor cortex, and supplementary motor cortex; projections to the associative striatum start in associative regions such as dorsolateral prefrontal cortex; and projections to the limbic striatum are from limbic areas such as the amgydala and hippocampus (Haber, 2003). SPM5 (http://www.fil.ion.ucl.ac.uk/spm) was then used to normalize the ROI map together with the tracer-specific ([18F]-DOPA) template (Egerton et al, 2010, Howes et al, 2009) to each individual PET summation image. This nonlinear transformation procedure allowed ROIs to be automatically placed on individual [18F]-DOPA PET dynamic images. The influx constant (Kicer, written as Ki in some previous publications (Howes et al, 2009)) for the entire striatal ROI and the functional subdivisions bilaterally were calculated compared with uptake in the reference region using a graphical approach adapted for a reference tissue input function (Egerton et al, 2010).
Statistical Analysis
Normality of distribution and homogeneity of variance were assessed using Kolmogorov–Smirnov and Levene’s tests, respectively. Group (smoker vs non-smoker) differences in demographic and imaging variables were determined using independent samples t-tests for normally distributed data, Mann–Witney U-tests for non-normally distributed data, and the χ2 test for dichotomous variables. The influence of sex on group differences in Kicer was determined using a two-way analysis of variance. Within the smoker group, the relationship between Kicer and level of cigarette consumption was tested using Pearson’s product-moment correlation coefficient. A two-tailed significance level of p=0.05 was employed throughout. A power calculation determined that we needed a sample size of 15 per group to have 80% or greater power to detect the effect size reported in the Salokangas et al, study (Cohen’s d=1.1) at this significance level.
RESULTS
Subject Characteristics and Scan Parameters
Fifteen smokers were recruited into the study. All smokers consumed tobacco as a cigarette. Mean (SD) cigarette consumption was 8.1 (4.1) per day (range: 1–17). Twelve smokers met DSM-IV(TR) criteria for Nicotine Dependence. Mean (SD) cigarette consumption was 9.6 (2.9) per day in smokers who met Dependency criteria (range: 5–17). Mean (SD) cigarette consumption was 2.0 (1.0) per day in smokers who did not meet criteria.
Fifteen non-smoker control subjects were matched to the smoker group for age (±5 years) and sex. Group demographics are reported in Table 1. Urine drug screens were negative for all substances (cannabis, amphetamine, opiates, cocaine, methamphetamine, benzodiazepines) in every subject.
Subjects were well matched for age and sex. There was no significant group difference in the amount of radioactivity or specific activity of [18F]-DOPA administered (Table 1). No subjects had a history of alcohol dependence or abuse according to DSM-IV(TR) criteria and subjects were well matched for alcohol use. There was no relationship between age and Kicer in the whole striatum (r=−0.10, p=0.62) or its subdivisions in the whole sample or in either group (data available on request).
Striatal Dopaminergic Function
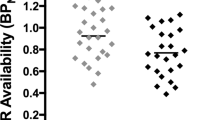
There was no significant group difference in Kicer in the whole striatum (Figure 1), or any striatal subdivision (Table 2). No significant differences in Kicer were detected after removing the three smokers who did not meet DSM-IV(TR) diagnostic criteria for nicotine dependency from the analysis (t25=0.85, p=0.40).

Whole striatal dopamine synthesis capacity (indexed as the influx rate constant Kicer) in smokers compared to non-smokers. Error bars indicate standard deviations.
The Relationship between Striatal Dopamine Synthesis Capacity and Tobacco Use
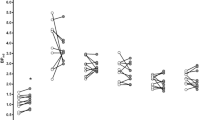
In smokers, there were no significant relationships between the number of cigarettes smoked per day and Kicer in the whole striatum (r=−0.23, p=0.41; Figure 2), or any striatal subdivision (associative: r=−0.16, p=0.57; sensorimotor: r=−0.33, p=0.23; limbic: r=−0.22, p=0.44).

The relationship between tobacco use and dopamine synthesis capacity (indexed as the influx rate constant Kicer) in smokers.
To examine whether nicotine dependency was specifically associated with elevated Kicer, we divided the tobacco user group into subjects who met DSM-IV(TR) diagnostic criteria for nicotine dependency (n=12), and those who did not meet criteria (n=3). Mean (SD) Kicer was 0.0127 (0.0010) min−1 in smokers meeting Nicotine Dependence criteria and 0.0131 (0.0019) min−1 in smokers who did not meet criteria. t-Tests showed no significant differences between the nicotine dependency sub-group and non-smoker controls (t25=0.85, p=0.40). There was no significant relationship between tobacco use and dopamine synthesis capacity in the whole striatum (r=0.18, p=0.57) or any of its functional subdivisions (data available on request) within the nicotine-dependent sub-group.
Sex
We performed a further explorative analysis to examine for possible sex effects. Mean (SD) Kicer was 0.0127 (0.0008) min−1 in females and 0.0130 (0.0012) min−1 in males. Among males, mean (SD) Kicer was 0.0130 (0.0013) min−1 in smokers and 0.0131 (0.0010) min−1 in non-smokers. Among females, mean (SD) Kicer was 0.0125 (0.0010) min−1 in smokers and 0.0130 (0.0006) min−1 in non-smokers. Two-way analysis of variance did not reveal a significant interaction between smoking status and sex on dopamine synthesis capacity in the whole striatum (F1,26=0.23, p=0.64) or any functional subdivision (associative striatum: F1,26=0.08, p=0.79; sensorimotor striatum: F1,26=0.34, p=0.54; limbic striatum: F1,26=0.73, p=0.40).
DISCUSSION
This study found no evidence for altered striatal dopamine synthesis in tobacco smokers compared with non-smokers, or relationship between the levels of daily cigarette smoking and dopamine synthesis capacity. Furthermore, we did not find an effect of nicotine dependence on dopamine synthesis capacity. Our findings are therefore not consistent with our hypothesis that dopamine synthesis capacity would be elevated in smokers compared with non-smokers.
These negative findings are in contrast to a previous report of elevated dopamine synthesis capacity in 9 male smokers compared with 10 non-smokers (Salokangas et al, 2000). Although striatal dopamine synthesis capacity may be higher in females (Laakso et al, 2002), this was not evident in our sample. The subjects in the study by Salokangas et al were heavy smokers (at least 15 cigarettes/day, mean 19.8 cigarettes/day compared with mean 8.1 cigarettes/day in our study), which could explain the difference with our findings. Although we found no evidence of a relationship between Kicer and the level of daily cigarette consumption, this may indicate that elevations in presynaptic dopamine synthesis capacity are only apparent in heavy smokers.
The study by Salokangas et al (2000) used the same methodology as a previous study by Hietala et al (1999), ie, Carbidopa 100 mg only was administered 90 min before PET scan (Personal Communication from Professor Salokangas), followed by measurement of radiolabeled metabolites in the arterial input function. This is in comparison to the carbidopa 150 mg and entacapone 400 mg administered 1 h before PET scan followed by a cerebellar reference region approach in the present study. To our knowledge, data on the effects of smoking on the pharmacodynamics of entacapone are lacking. Entacapone undergoes rapid hepatic metabolism via the uridine 5'-diphospho-glucuronosyltransferase pathway (Lautala et al, 2000). Data from studies in humans (Bock and Köhle, 2004) and mice models (Villard et al, 1998) indicate that cigarette smoke is a potent inducer of uridine 5'-diphospho-glucuronosyltransferase, which would thus lead to a faster elimination of entacapone and therefore a potential reduction in plasma [18F]-DOPA in smokers vs controls whom have had entacapone administered. As our reference region approach theoretically eliminates the plasma input function, we would predict the main effect of any potential reduction in plasma [18F]-DOPA to result in increased variability of Kicer without altering mean Kicer. However, there are limited data directly comparing Ki values obtained via a tissue reference region and arterial plasma input function method, as was the case in the study by Salokangas et al (2000). Sossi et al (2003) reported a comparison between a reference region and arterial input function approach. In that study, Sossi et al used the occipital lobe as the reference region, rather than the cerebellum used in our study. Therefore, it remains possible that a difference in entacapone metabolism in smokers may be underlying the disparity in results between our study and that of Salokangas et al (2000). Nevertheless, the entry into and exit from the brain of radiolabeled plasma metabolites may affect graphical analysis (Boyes et al, 1986; Cumming et al, 1987) and could bias results if metabolism is selectively altered in one group. Compared with non-smokers, smokers have reduced CSF levels of the dopamine metabolite homovanillic acid (Geracioti et al, 1999) and there is evidence of reduced MAO-A and MAO-B activity in smokers (Fowler et al, 1996a, 1996b). However, as these differences would, if anything, reduces the production of radiolabeled metabolites in smokers, they are unlikely to explain the failure to detect an elevation in smokers. In summary, even if entacapone clearance is higher in our smoker group, we remain unable to conclude that it would be sufficient to impact metabolite production within our experimental window. Furthermore, even if there was a sufficient impact on metabolite production, our use of a reference region approach would be expected to be sufficiently robust to overcome this problem.
Survey data in Great Britain indicate that over the last few decades there has been a gradual decline in the number of cigarettes consumed per day among smokers, such that the average number of cigarettes smoked per day is now 12 for men and 11 for women (Office for National Statistics, 2013). Our sample of moderate smokers is therefore in the same range as that of the general population. Overall our findings and those of Salokangas et al thus indicate that there is no markedly altered dopamine function in moderate smokers, but alterations are apparent in heavy smokers. A further contributing factor for the discrepancy in findings from those of the study by Salokangas et al and the present study is that the nicotine content of cigarettes has decreased since the former. However, we did not collect data on the type of cigarette consumed by each tobacco-smoking subject, which would have enabled an estimation of the amount of nicotine consumed. Despite proposals for nicotine reduction policies (Benowitz and Henningfield, 1994), which have intended for the nicotine content of cigarettes to be reduced, there is some evidence that the nicotine yield of cigarettes in American brands may, in fact, have increased since the Salokangas study (Connolly et al, 2007). Future studies assessing both the effects of heavy vs moderate smoking and nicotine dose of cigarettes on dopaminergic function are therefore warranted.
Study Limitations
Although our sample size was larger than the only other study to investigate dopamine synthesis capacity in smokers, it is important to consider the possibility of a type II error in our findings. We had 84% power to detect the effect size (Cohen’s d=1.12) seen in the Salokangas et al study, which is above the 80% power threshold conventionally considered adequate. Nevertheless, even at this power, there is a 16% chance that a true effect of this magnitude has been missed. As we only included moderate smokers, a limitation of our study would be that we did not include a group of heavy smokers for comparison. A potential limitation of the correlation between cigarette use and dopamine synthesis capacity is that seven of the smokers in our study consumed 10 cigarettes per day, which likely reflects the fact that tobacco in the United Kingdom is sold in packets of 10 and 20 cigarettes, but limits the power of the correlational analysis. Our sample contained five female subjects per group and so was underpowered to detect sex–group interactions, indicating that these analyses should only be considered exploratory. Gonadal hormones may influence dopaminergic function. However, we did not assess this or phase of the menstrual cycle in the females.
A further limitation of our study is that we did not include the Fagerström Test for Nicotine Dependence, which would have enabled the exploration of the relationships between dopamine synthesis capacity and subjective craving. Likewise, we did not measure plasma nicotine, cotinine, or carbon dioxide levels. Future studies would therefore benefit from including these measures in larger sample sizes.
Implications for Understanding Tobacco Addiction
The role of the dopamine system in drug reinforcement has long been accepted from animal studies (eg, Koob, 1992) and there is mounting evidence that dysregulated dopamine function is central to addiction behaviors in humans (as reviewed by Volkow et al, 2011). There is growing evidence that chronic drug abuse is associated with abnormal striatal dopaminergic functioning in humans, as has been found with alcohol (Heinz et al, 2005), cannabis (Bloomfield et al, 2013), cocaine (Wu et al, 1997), methamphetamine (Wang et al, 2012), and ecstasy (Tai et al, 2011). However, our study suggests that dopamine dysregulation may only become apparent at higher levels of use, either because it is below the level of detection with more moderate use, or because it is a cumulative consequence of heavy use.
CONCLUSIONS
This study found that moderate smoking was not associated with marked effects on striatal dopamine synthesis capacity, in contrast to a previous finding of elevated dopamine synthesis capacity in heavy smokers. Further studies in smokers of presynaptic dopaminergic function using heavy and moderate smokers are warranted to determine whether dopaminergic effects only become evident with heavier use.
FUNDING AND DISCLOSURE
Dr Bloomfield and Dr Mouchlianitis receive funding from the Medical Research Council (UK). Dr Howes has received research funding from the Medical Research Council (UK) (Grant: MC-A656-5QD30) and a National Institute of Health Research Biomedical Research Council grant to King’s College London for this study. He has received investigator-led charitable research funding or has been on the speaker bureaux for Astra-Zeneca, BMS, Eli Lilly, and Jansenn Cilag. The remaining authors declared no conflict of interest.
References
Balfour DJ, Wright AE, Benwell ME, Birrell CE (2000). The putative role of extra-synaptic mesolimbic dopamine in the neurobiology of nicotine dependence. Behav Brain Res 113: 73–83.
Barkus EJ, Stirling J, Hopkins RS, Lewis S (2006). Cannabis-induced psychosis-like experiences are associated with high schizotypy. Psychopathology 39: 175–178.
Barrett SP, Boileau I, Okker J, Pihl RO, Dagher A (2004). The hedonic response to cigarette smoking is proportional to dopamine release in the human striatum as measured by positron emission tomography and [11C]raclopride. Synapse 54: 65–71.
Benowitz NL, Henningfield JE (1994). Establishing a nicotine threshold for addiction. The implications for tobacco regulation. N Engl J Med 331: 123–125.
Benowitz NL, Jacob P 3rd, Jones RT, Rosenberg J (1982). Interindividual variability in the metabolism and cardiovascular effects of nicotine in man. J Pharmacol Exp Ther 221: 368–372.
Bloomfield MA, Morgan CJ, Egerton A, Kapur S, Curran HV, Howes OD (2013). Dopaminergic Function in Cannabis Users and Its Relationship to Cannabis-Induced Psychotic Symptoms. Biol Psychiatry 75: 470–478.
Bock KW, Köhle C (2004). Coordinate regulation of drug metabolism by xenobiotic nuclear receptors: UGTs acting together with CYPs and glucuronide transporters. Drug Metab Rev 36: 595–615.
Boyes BE, Cumming P, Martin WR, McGeer EG (1986). Determination of plasma [18F]-6-fluorodopa during positron emission tomography: elimination and metabolism in carbidopa treated subjects. Life Sci 39: 2243–2252.
Brody AL, Olmstead RE, London ED, Farahi J, Meyer JH, Grossman P et al (2004). Smoking-induced ventral striatum dopamine release. Am J Psychiatry 161: 1211–1218.
Busto UE, Redden L, Mayberg H, Kapur S, Houle S, Zawertailo LA (2009). Dopaminergic activity in depressed smokers: a positron emission tomography study. Synapse 63: 681–689.
Chiuccariello L, Guranda M, Rusjan PM, Wilson AA, Zawertailo L, Houle S et al (2013). Presentation of smoking-associated cues does not elicit dopamine release after one-hour smoking abstinence: A [11C]-(+)-PHNO PET study. PLoS One 8: e60382.
Clarke PB, Pert A (1985). Autoradiographic evidence for nicotine receptors on nigrostriatal and mesolimbic dopaminergic neurons. Brain Res 348: 355–358.
Connolly GN, Alpert HR, Wayne GF, Koh H (2007). Trends in nicotine yield in smoke and its relationship with design characteristics among popular US cigarette brands, 1997-2005. Tob Control 16: e5.
Cumming P, Leger GC, Kuwabara H, Gjedde A (1993). Pharmacokinetics of plasma 6-[18F]fluoro-L-3,4-dihydroxyphenylalanine ([18F]Fdopa) in humans. J Cereb Blood Flow Metab 13: 668–675.
Cumming P, Boyes BE, Martin WR, Adam M, Grierson J, Ruth T et al (1987). The metabolism of [18F]6-fluoro-L-3,4-dihydroxyphenylalanine in the hooded rat. J Neurochem 48: 601–608.
Dewey SL, Brodie JD, Gerasimov M, Horan B, Gardner EL, Ashby CR Jr (1999). A pharmacologic strategy for the treatment of nicotine addiction. Synapse 31: 76–86.
Dierker LC, Avenevoli S, Stolar M, Merikangas KR (2002). Smoking and depression: an examination of mechanisms of comorbidity. Am J Psychiatry 159: 947–953.
Dluzen DE, Anderson LI (1997). Estrogen differentially modulates nicotine-evoked dopamine release from the striatum of male and female rats. Neurosci Lett 230: 140–142.
Egerton A, Demjaha A, McGuire P, Mehta MA, Howes OD (2010). The test-retest reliability of 18F-DOPA PET in assessing striatal and extrastriatal presynaptic dopaminergic function. Neuroimage 50: 524–531.
Ezzati M, Lopez AD (2003). Estimates of global mortality attributable to smoking in 2000. Lancet 362: 847–852.
First MB SR, Gibbon M, Williams JBW (1996) Structured Clinical Interview for DSM-IV Axis I Disorders, Clinician Version (SCID-CV).. American Psychiatric Press: Washington, DC.
Fowler JS, Volkow ND, Wang GJ, Pappas N, Logan J, MacGregor R et al (1996a). Inhibition of monoamine oxidase B in the brains of smokers. Nature 379: 733–736.
Fowler JS, Volkow ND, Wang GJ, Pappas N, Logan J, Shea C et al (1996b). Brain monoamine oxidase A inhibition in cigarette smokers. Proc Natl Acad Sci USA 93: 14065–14069.
Gallezot JD, Kloczynski T, Weinzimmer D, Labaree D, Zheng MQ, Lim K et al (2013). Imaging nicotine- and amphetamine-induced dopamine release in Rhesus monkeys with [11C]PHNO vs [11C]raclopride PET. Neuropsychopharmacology 39: 866–874.
Geracioti TD Jr, West SA, Baker DG, Hill KK, Ekhator NN, Wortman MD et al (1999). Low CSF concentration of a dopamine metabolite in tobacco smokers. Am J Psychiatry 156: 130–132.
Guttman M, Leger G, Reches A, Evans A, Kuwabara H, Cedarbaum JM et al (1993). Administration of the new COMT inhibitor OR-611 increases striatal uptake of fluorodopa. Mov Disord 8: 298–304.
Grenhoff J, Aston-Jones G, Svensson TH (1986). Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta Physiol Scand 128: 351–358.
Haber SN (2003). The primate basal ganglia: parallel and integrative networks. J Chem Neuroanat 26: 317–330.
Heinz A, Siessmeier T, Wrase J, Buchholz HG, Gründer G, Kumakura Y et al (2005). Correlation of alcohol craving with striatal dopamine synthesis capacity and D2/3 receptor availability: a combined [18F]DOPA and [18F]DMFP PET study in detoxified alcoholic patients. Am J Psychiatry 162: 1515–1520.
Hietala J, Syvälahti E, Vilkman H, Vuorio K, Räkköläinen V, Bergman J et al (1999). Depressive symptoms and presynaptic dopamine function in neuroleptic-naive schizophrenia. Schizophr Res. 35: 41–50.
Howes OD, Montgomery AJ, Asselin MC, Murray RM, Valli I, Tabraham P et al (2009). Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry 66: 13–20.
Hughes JR, Keely J, Naud S (2004). Shape of the relapse curve and long-term abstinence among untreated smokers. Addiction 99: 29–38.
Kendler KS, Neale MC, MacLean CJ, Heath AC, Eaves LJ, Kessler RC (1993). Smoking and major depression. A causal analysis. Arch Gen Psychiatry 50: 36–43.
Koob GF (1992). Neural mechanisms of drug reinforcement. Ann N Y Acad Sci 654: 171–191.
Kumakura Y, Cumming P (2009). PET studies of cerebral levodopa metabolism: a review of clinical findings and modeling approaches. Neuroscientist 15: 635–650.
Laakso A, Vilkman H, Bergman J, Haaparanta M, Solin O, Syvälahti E et al (2002). Sex differences in striatal presynaptic dopamine synthesis capacity in healthy subjects. Biol Psychiatry 52: 753–763.
Lautala P, Ethell BT, Taskinen J, Burchell B (2000). The specificity of glucuronidation of entacapone and tolcapone by recombinant human UDP-glucuronosyltransferases. Drug Metab Dispos 28: 1385–1389.
Le Foll B, Guranda M, Wilson AA, Houle S, Rusjan PM, Wing VC et al (2013). Elevation of Dopamine Induced by Cigarette Smoking: Novel Insights from a [11C]-(+)-PHNO PET Study in Humans. Neuropsychopharmacology 39: 415–424.
Lerman C, Caporaso N, Main D, Audrain J, Boyd NR, Bowman ED et al (1998). Depression and self-medication with nicotine: the modifying influence of the dopamine D4 receptor gene. Health Psychol 17: 56–62.
Marenco S, Carson RE, Berman KF, Herscovitch P, Weinberger DR (2004). Nicotine-induced dopamine release in primates measured with [11C]raclopride PET. Neuropsychopharmacology 29: 257–268.
Martinez D, Slifstein M, Broft A, Mawlawi T, Hwang DR, Huang YY et al (2003). Imaging human mesolimbic dopamine transmission with positron emission tomography. Part II: amphetamine-induced dopamine release in the functional subdivisions of the striatum. J Cerebr Blood F Met 23: 285–300.
Menossi HS, Goudriaan AE, de Azevedo-Marques Périco C, Nicastri S, de Andrade AG, D’Elia G et al (2013). Neural bases of pharmacological treatment of nicotine dependence—insights from functional brain imaging: a systematic review. CNS Drugs 27: 921–941.
Montgomery AJ, Lingford-Hughes AR, Egerton A, Nutt DJ, Grasby PM (2007). The effect of nicotine on striatal dopamine release in man: A [11C]raclopride PET study. Synapse 61: 637–645.
Office for National Statistics (2013) Opinions and Lifestyle Survey, Smoking Habits Amongst Adults, 2012. Office for National Statistics: London, United Kingdom.
Pidoplichko VI, DeBiasi M, Williams JT, Dani JA (1997). Nicotine activates and desensitizes midbrain dopamine neurons. Nature 390: 401–404.
Pontieri FE, Tanda G, Orzi F, Di Chiara G (1996). Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature 382: 255–257.
Salokangas RK, Vilkman H, Ilonen T, Taiminen T, Bergman J, Haaparanta M et al (2000). High levels of dopamine activity in the basal ganglia of cigarette smokers. Am J Psychiatry 157: 632–634.
Sossi V, Holden JE, de la Fuente-Fernandez R, Ruth TJ, Stoessl AJ. (2003). Effect of dopamine loss and the metabolite 3-O-methyl-[18F]fluoro-dopa on the relation between the 18F-fluorodopa tissue input uptake rate constant K occ and the [18F]fluorodopa plasma input uptake rate constant Ki. J Cereb Blood Flow Metab. 23: 301–309.
Stolerman IP, Mirza NR, Shoaib M (1995). Nicotine psychopharmacology: addiction, cognition and neuroadaptation. Med Res Rev 15: 47–72.
Studholme C, Hill DL, Hawkes DJ (1996). Automated 3-D registration of MR and CT images of the head. Med Image Anal 1: 163–175.
Tai YF, Hoshi R, Brignell CM, Cohen L, Brooks DJ, Curran HV et al (2011). Persistent nigrostriatal dopaminergic abnormalities in ex-users of MDMA (‘Ecstasy’): an 18F-dopa PET study. Neuropsychopharmacology 36: 735–743.
Thomsen G, Knudsen GM, Jensen PS, Ziebell M, Holst KK, Asenbaum S et al (2013). No difference in striatal dopamine transporter availability between active smokers, ex-smokers and non-smokers using [123I]FP-CIT (DaTSCAN) and SPECT. EJNMMI Res 3: 39.
Tsukada H, Miyasato K, Harada N, Nishiyama S, Fukumoto D, Kakiuchi T (2005). Nicotine modulates dopamine synthesis rate as determined by L-[beta-11C]DOPA: PET studies compared with [11C]raclopride binding in the conscious monkey brain. Synapse 57: 120–122.
Turkheimer FE, Brett M, Visvikis D, Cunningham VJ (1999). Multiresolution analysis of emission tomography images in the wavelet domain. J Cereb Blood Flow Metab 19: 1189–1208.
Villard PH, Herber R, Sérée EM, Attolini L, Magdalou J, Lacarelle B (1998). Effect of cigarette smoke on UDP-glucuronosyltransferase activity and cytochrome P450 content in liver, lung and kidney microsomes in mice. Pharmacol Toxicol 82: 74–79.
Volkow ND, Wang GJ, Fowler JS, Tomasi D, Telang F (2011). Addiction: beyond dopamine reward circuitry. Proc Natl Acad Sci USA 108: 15037–15042.
Wang GJ, Smith L, Volkow ND, Telang F, Logan J, Tomasi D et al (2012). Decreased dopamine activity predicts relapse in methamphetamine abusers. Mol Psychiatry 17: 918–925.
West R, McNeill A, Raw M (2000). Smoking cessation guidelines for health professionals: an update. Health Education Authority. Thorax 55: 987–999.
Wu JC, Bell K, Najafi A, Widmark C, Keator D, Tang C et al (1997). Decreasing striatal 6-FDOPA uptake with increasing duration of cocaine withdrawal. Neuropsychopharmacology 17: 402–409.
Zhang H, Sulzer D (2004). Frequency-dependent modulation of dopamine release by nicotine. Nat Neurosci 7: 581–582.
Zhang L, Kendler KS, Chen X (2006). The mu-opioid receptor gene and smoking initiation and nicotine dependence. Behav Brain Funct 2: 28.
Acknowledgements
We thank our subjects and the staff of GE Imanet for their assistance with the positron emission tomography scans, in particular Ms Hope McDevitt, Mr Andrew Blyth, Ms Andreanna Williams, and Ms Stephanie McKnight.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article
Cite this article
Bloomfield, M., Pepper, F., Egerton, A. et al. Dopamine Function in Cigarette Smokers: An [18F]-DOPA PET Study. Neuropsychopharmacol 39, 2397–2404 (2014). https://doi.org/10.1038/npp.2014.87
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2014.87
This article is cited by
-
Striatal dopaminergic alterations in individuals with copy number variants at the 22q11.2 genetic locus and their implications for psychosis risk: a [18F]-DOPA PET study
Molecular Psychiatry (2023)
-
Dopamine dysregulation in psychotic relapse after antipsychotic discontinuation: an [18F]DOPA and [11C]raclopride PET study in first-episode psychosis
Molecular Psychiatry (2021)
-
The relationship between grey matter volume and striatal dopamine function in psychosis: a multimodal 18F-DOPA PET and voxel-based morphometry study
Molecular Psychiatry (2021)
-
Glutamatergic and dopaminergic function and the relationship to outcome in people at clinical high risk of psychosis: a multi-modal PET-magnetic resonance brain imaging study
Neuropsychopharmacology (2020)
-
Amphetamine-induced striatal dopamine release in schizotypal personality disorder
Psychopharmacology (2020)
