
Abstract
Exposure to stress elicits excitoxicity and neuroinflammation in the brain, contributing to cell death and damage in stress-related neurological and neuropsychiatric diseases. The endocannabinoid system is present in stress-responsive neural circuits and has been proposed as an endogenous neuroprotective system activated in some neuropathological scenarios to restore homeostasis. To elucidate the possible regulatory role of cannabinoid receptor 1 (CB1) in stress-induced excitotoxicity and neuroinflammation, both genetic and pharmacological approaches were used alternatively: (1) wild-type (WT) and CB1 knockout mice (CB1-KO) were exposed to immobilization/acoustic stress (2 h/day for 4 days) and (2) to specifically activate CB1, the selective CB1 agonist Arachidonyl-2′-chloroethylamide (ACEA) (2.5 mg/kg) was intraperitoneally administered daily to some groups of animals. Stress exposure increased CB1 mRNA and protein expression in the prefrontal cortex of WT mice in a mechanism related to N-methyl-D-aspartate glutamate receptor activation. Daily ACEA pretreatment prevented stress-induced: (1) upregulation of CB1 mRNA and protein, (2) decrease in glutamate uptake and glutamate astroglial transporter excitatory amino acid transporter 2 expression, (3) increase in consecutive proinflammatory molecules, such as cytokines (tumor necrosis factor-α and MCP-1), nuclear factor kappa B, and enzymatic sources, such as inducible nitric oxide synthase (NOS-2) and cyclooxygenase-2 (COX-2), (4) increase in lipid peroxidation; although having no effect on plasma corticosterone. Interestingly, a possible related mechanism could be the positive ACEA modulation of the antiinflammatory pathway deoxyprostaglandin/peroxisome proliferator-activated receptor γ (15d-PGJ2/PPARγ). Conversely, KO animal experiments indicated that a lack of CB1 produces hypothalamic/pituitary/adrenal (HPA) axis dysregulation and exacerbates stress-induced excitotoxic/neuroinflammatory responses. These multifaceted neuroprotective effects suggest that CB1 activation could be a new therapeutic strategy against neurological/neuropsychiatric pathologies with HPA axis dysregulation and an excitotoxic/neuroinflammatory component in their pathophysiology.
Similar content being viewed by others


INTRODUCTION
Uncontrolled excitotoxicity and neuroinflammation contribute to cell death and damage in neurological and neuropsychiatric diseases, including some that are related to stress exposure (neurodegenerative diseases, depression, post-traumatic stress disorder, and schizophrenia) (McLeod et al, 2001; Madrigal et al, 2006). Indeed, previous studies indicate that long-lasting stress affects synaptic plasticity, dendritic morphology, and neurogenesis in animals (reviewed in Kim and Yoon, 1998), and induces both clinical and anatomical features of neurotoxic damage in humans (Bremner et al, 1995). Moreover, the relationship between stressful events and the onset, evolution, and resolution of inflammatory-related diseases (cardiovascular, neurological, digestive, or immunological) is well documented (Baum and Posluszny, 1999; Slimmer et al, 2001). The precise molecular and cellular events induced by stress that are responsible for the brain damage found in these conditions are still a matter of debate. In this way, previous studies using stress models (physical, psychological, or mixed) reported a consistent brain response that is mainly characterized by excitotoxicity and a resulting activation of several consecutive inflammatory mediators (García-Bueno et al, 2008a). Briefly, stress exposure elicits excitotoxicity due to the massive release of the excitatory amino acid, glutamate, in some brain areas (eg, the prefrontal cortex) (Moghaddam, 1993), inducing the release of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) or interleukin 1β (Madrigal et al, 2002). Stress also activates the nuclear transcription factor-kappa B (NF-κB) pathway in a TNF-α-dependent mechanism (Bierhaus et al, 2003; Madrigal et al, 2002). NF-κB activation elicits the expression and activity of proinflammatory enzymatic sources, such as inducible nitric oxide (NO) synthase (NOS-2), and cyclooxygenase-2 (COX-2), among others (Madrigal et al, 2001a, 2003). The result of this sequence of events is the accumulation of oxidative and nitrosative mediators, which can attack membrane phospholipids and cause cell damage in a process known as lipid peroxidation (McEwen, 1998; Madrigal et al, 2001b) (see Figure 9 for a schematic view of this stress-activated proinflammatory pathway). However, there remain unresolved issues concerning the mechanisms, regulation, and functional significance of the excitotoxic/inflammatory processes induced by stress in the brain.

Schematic view of the regulatory role of CB1 on stress-induced excitotoxicity and neuroinflammatory balance. NMDA, N-methyl-D-asparte receptor; EAAT-2, excitatory aminoacid transporter-2; CB1, cannabinoid receptor 1; NF-κB, nuclear factor kappa B; NOS-2, nitric oxide synthase type II; COX-2, cyclooxygenase type II; L-PGDS, lipocalin prostaglandin D synthase; 15d-PGJ2, 15d-prostaglandin J2; PPARγ, peroxisome proliferators-activated receptor γ; (−), inhibition; (+), stimulation; ?, alternative mechanisms. Black, proinflammatory mediators. Grey, antiinflammatory mediators.
In spite of stress-induced production and accumulation of potentially cytotoxic and/or proinflammatory mediators like glutamate, NO, peroxynitrite anion (ONOO−), or prostaglandin E2 (PGE2), various authors have discussed the possibility that some of the many changes caused by stress response effectors are not damaging to the neurons, but in fact predominantly beneficial to their structure and function (McEwen, 1998). One possible mechanism involved as a possible neuroprotective pathway that has received considerable experimental attention is led by some COX-2-derived lipid mediators, such as prostaglandin 15d-PGJ2 and its peroxisome proliferator-activated nuclear receptor γ (PPARγ). This COX-derived sub-pathway is activated in the brain in response to stress and other neuroinflammatory stimuli, constituting a possible endogenous antiinflammatory defense mechanism against excessive excitotoxicity and neuroinflammation (García-Bueno et al, 2008a) (see Figure 8 for a schematic view of this stress-activated antiinflammatory pathway).

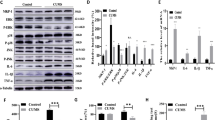
Validation of CB1-mediated ACEA effects. (a) Western blot and densitometric analysis of the astroglial glutamate transporter EATT-2 in prefrontal cortex homogenates of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice and stressed + daliy ACEA (SKO + ACEA) CB1 KO mice. Data are normalized by β-actin (lower band), and are representative of three experiments. *p<0.05 vs CWT; #p<0.05 vs SWT; $p<0.05 vs SWT + ACEA (one-way ANOVA followed by Newman–Keuls post hoc test). AU, arbitrary units. (b) NOS-2 protein expression analysis by western blot and densitometric analysis of the band of interest in prefrontal cortex samples of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice and stressed + daliy ACEA (SKO + ACEA) CB1 KO mice. Data are normalized by β-actin (lower band), and are representative of three experiments. *p<0.05 vs CWT; #p<0.05 vs SWT; $p<0.05 vs SWT + ACEA (one-way ANOVA followed by Newman–Keuls post hoc test). AU, arbitrary units. (c) MDA levels (lipid peroxidation index) in prefrontal cortex homogenates of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice and stressed + daily ACEA (SKO + ACEA) CB1 KO mice. Data represents the mean ± SEM of six mice.
The study of alternative/related antiinflammatory pathways deserves further attention to understand pathophysiological inflammatory alterations as well as to develop possible new therapeutic approaches for the treatment of stress-related neuropathologies. Currently, the endocannabinoid system (ECS) is considered as an endogenous homeostatic system activated by different immune challenges, restoring brain balance at different levels (Mechoulam and Shohami, 2007) in different experimental settings (Galve-Roperh et al, 2008; Wolf et al, 2008; Bambico et al, 2009; Cabral and Griffin-Thomas, 2009). ECS refers to a group of endogenous arachidonate-based lipids (ie, anandamide and 2-arachidonoylglycerol), widely known as ‘endocannabinoids’; their cannabinoid G protein-coupled receptors, namely cannabinoid receptor 1 (CB1) and CB2, and the enzymes fatty acid amide hydrolase and monoacylglycerol lipase that are responsible for their degradation and/or reuptake.
Interestingly, several epidemiological studies have identified that cannabinoids affect anxiety and stress responsivity (Ledent et al, 1999), and the presence of the ECS in stress-responsive neural circuits suggests that it may have a critical role in regulating hypothalamic/pituitary/adrenal (HPA) axis activation and the resulting excitotoxic/neuroinflammatory response. Although it is widely assumed that this response is mainly mediated by the CB2 receptor, which is expressed by a wide range of immune cells (Arévalo-Martín et al, 2003), recent evidence suggests that pharmacological enhancement of CB1 protects neurons from acute brain injury, demonstrating a potentially attractive alternative therapeutic strategy against CNS pathologies that deserves further investigation (Cabral and Griffin-Thomas, 2009).
Taking into account all this background, the aim of the present study was to explore the effect of sub-chronic stress exposure on CB1 expression in the prefrontal cortex in mice, and the possible regulatory role of CB1 in conditions of stress-induced excitotoxicity and neuroinflammation, using alternatively pharmacological (activation of CB1 with the selective agonist arachidonyl-2′-chloroethylamide (ACEA)) and genetic (CB1 receptor-deficient mice) strategies.
MATERIALS AND METHODS
Animals
Adult male CB1 double mutant CB1 (−/−) mice and wild-type CB1 (WT) littermates (ICR (Swiss) strain) were used in all the experiments. The generation of mice lacking the CB1 receptor has been described previously (Ledent et al, 1999). WT and CB1 (−/−) mice used in a given experiment, originated from the same breeding series and were matched for age and weight (age=2–3 months; weight=25–30 g). All experimental protocols adhered to the guidelines of the animal welfare committee of the Universidad Complutense in accordance with European legislation (DC 86/609/CEE). The mice were housed individually with standard temperature and humidity conditions and in a 12-h light/dark cycle (lights on at 0800 h) with free access to food and water. All the animals were maintained under constant conditions for 4 days before the exposure to stress.
Stress Protocol
Mice were exposed to sub-chronic immobilization and acoustic stress using an ultrasound bath (2 h from 1300 h to 1500h for 4 days) as previously described (Kiank et al, 2006; Depke et al, 2008; Olfe et al, 2010). The restraint/acoustic stress model is classified as both physical and psychological, predictable, and inescapable, suitable for studying the neurochemical effects of stress on brain function. Some authors have evaluated restraint and acoustic stress, both separately and in combination, and found that larger and more predictable responses were obtained with the combined stressors (Qiu et al, 1999). Stressed animals were put down immediately after the restraint (while still in the restrainer) using sodium pentobarbital (320 mg/kg i.p.). Control animals were not submitted to stress but were handled at 1300 h for a few seconds, and food and water were removed. They were put down at the same time as the stressed animals (1500 h). Blood for plasma determinations was collected by cardiac puncture and anticoagulated in the presence of trisodium citrate (3.15% (wt/vol), 1 vol citrate per 9 vol blood). After decapitation, the brain was removed from the skull, and after careful removal of the meninges and blood vessels, the prefrontal cortical areas from both brain hemispheres were excised and frozen at −80°C until assayed. Mouse brain prefrontal cortex was chosen because of its high levels of CB1 receptor, proinflammatory (NOS-2, prostaglandin E2), and antiinflammatory (PPARγ, lipocalin prostaglandin D synthase) mediators, its susceptibility to excitotoxic/neuroinflammatory process elicited by inflammatory/immune stimulus (García-Bueno et al, 2008b), and finally because this brain area is an important neural substrate for the regulation of the HPA axis response to stress (Radley et al, 2006).
Pharmacological Tools
Various groups of animals were injected with the potent and highly selective CB1 receptor agonist (Ki=1.4 nM; >1400-fold CB1 selectivity over CB2) ACEA, using a dose of 2.5 mg/kg administered intraperitoneally (i.p.) daily or its vehicle (a solution of 5% ethanol, 5% dextrose, and 5% Tween 80 in sterile saline). The dose of ACEA was used based on preceding studies in neuroinflammation-related models (Arévalo-Martín et al, 2003). To block N-methyl-D-aspartate (NMDA) glutamate receptor-dependent effects, two groups of animals were i.p. injected with dizocilpine [(+)-5-methyl-10,11-dihydroxy-5H-dibenzo (a, d) cyclohepten-5,10-imine; MK-801], a specific noncompetitive NMDA antagonist (MK; 0.1 mg/kg). This low dose was chosen to avoid possible neurotoxic, hallucinogenic, and HPA axis disturbing effects from this compound when it is used at higher doses, and was based on preceding studies from our laboratory and others (García-Bueno et al, 2008b; Jezova et al, 1995). ACEA and vehicle were injected at the onset of the stress (1300 h), and MK-801 and its vehicle (saline) were respectively i.p. injected 1 h before the onset of the stress (1200 h). None of the parameters studied were modified in the two different vehicle-treated groups of mice when compared with the noninjected animals. To simplify figures, both groups were unified into one vehicle-injected group in control (CWT) and stress (SWT) conditions. Each experimental group included at least six animals.
Preparation of Cytosolic and Nuclear Extracts
A modified procedure based on the method of Schreiber et al (1989) was used. Tissues (cerebral cortex) were homogenized with 300 μl of buffer (10 mmol/l N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.9; 1 mmol/l ethylenediamine tetraacetic acid (EDTA), 1 mmol/l EGTA, 10 mmol/l KCl, 1 mmol/l DTT), 0.5 mmol/l phenylmethylsulfonyl fluoride, 0.1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml Nα-p-tosyl-L-lysine-chloromethyl ketone, 5 mmol/l NaF, 1 mmol/l NaVO4, 0.5 mol/l sucrose, and 10 mmol/l Na2MoO4). After 15 min, Nonidet P-40 (Roche) was added to a 0.5% concentration level. The tubes were gently vortexed for 15 s, and nuclei were collected by centrifugation at 8000 g for 5 min. Supernatants were taken as a cytosolic fraction. The pellets were resuspended in 100 μl of buffer supplemented with 20% glycerol and 0.4 mol/l KCl, and gently shaken for 30 min at 4 °C. Nuclear protein extracts were obtained by centrifugation at 13 000 g for 5 min, and aliquots of the supernatant were stored at −80°C. All steps of the fractionation were carried out at 4°C.
Western Blot Analysis
After determining and adjusting protein levels, homogenates of prefrontal cortex tissue, once centrifuged (12000 g, 20 min at 4°C) were mixed with Laemmli sample buffer (Bio-Rad, Hercules, CA) (SDS 10%, distilled H2O, 50% glycerol, 1M Tris HCl, pH 6.8, dithiothreitol, and blue bromophenol) with beta mercaptoethanol (50 μl/ml of Laemmli) and 20 μl (2 μg/μl) were loaded into an electrophoresis gel. Once separated on the basis of molecular weight, proteins from the gels were blotted onto a polyvinylidene difluoride membrane (Millipore, Bedford, MA) or nitrocellulose membrane (Amersham Ibérica, Spain) with a semi-dry transfer system (Bio-Rad) and were incubated with specific antibodies: (1) rabbit polyclonal CB1, raised against the synthetic peptide MSVSTDTSAEAL, corresponding to C-terminal amino acids 461–472 of human CB1 in a dilution of 1 : 1000 in 5% bovine serum albumin (BSA) in TBS-Tween (Abcam, Cambridge, UK); (2) rabbit polyclonal excitatory amino acid transporter 2 (EAAT-2) raised against an epitope corresponding to amino acids 1–85 mapping at the N-terminus of EAAT-2 of human origin in a dilution of 1 : 1000 in 5% BSA in TBS-Tween (sc-15317) (Santa Cruz Biotechnology, CA); (3) specific rabbit polyclonal PPARγ (epitope corresponding to amino acids 8–106 mapping at the N-terminus of PPARγ1 of human origin) in a dilution of 1 : 1000 in 5% BSA in TBS-Tween (sc-7196) (Santa Cruz Biotechnology); (4) rabbit polyclonal IκBα (epitope mapping at the C-terminus of IκB-α of human origin) in a dilution of 1 : 1000 in 5% skimmed milk in BSA (Santa Cruz Biotechnology) (5) rabbit polyclonal NF-κB p65 (epitope mapping within the N-terminus of NFκB p65 of human origin) in a dilution of 1 : 500 in BSA 2% (sc-109) (Santa Cruz Biotechnology); (6) NOS-2 rabbit polyclonal antibody from Santa Cruz Biotechnology raised against a peptide mapping at the amino terminus of NOS-2 of human origin in a dilution of 1 : 1000 in TBS-Tween (sc-651); (7) COX-2 goat polyclonal antibody from Santa Cruz Biotechnology raised against a peptide mapping at the C-terminus of COX-2 of human origin in a dilution of 1 : 750 in 5% BSA in TBS-Tween (sc-1745); (8) L-PGDS rabbit polyclonal antibody, which recognizes human L-PGDS amino acids 30–41 (VQPNFQPDKFLG) in a dilution of 1 : 500 in 5% BSA in TBS-Tween (Cayman Chemical Europe, Tallinn, Estonia). Proteins recognized by the respective horseradish peroxidase-linked secondary antibodies (in a dilution of 1 : 1000 in 2–5% BSA in TBS-Tween) and visualized on X-ray film by chemiluminescence following manufacturer's instructions (Amersham). Autoradiographs were quantified by densitometry (program Image J, NIH), and several time expositions were analyzed to ensure the linearity of the band intensities. All densitometries are expressed in arbitrary units (AU). In all the WB analyses, the housekeeping gene β-actin was used as a loading control (the blots are shown in the respective figures).
mRNA Analysis
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, Barcelona, Spain); aliquots were converted to cDNA using random hexamer primers. Quantitative changes in mRNA levels were estimated by real-time PCR (Q-PCR) using the following cycling conditions: 35 cycles of denaturation at 95°C for 10 s, annealing at 58–61°C for 15 s, depending on the specific set of primers, and extension at 72°C for 20 s. Reactions were carried out in the presence of SYBR green (1 : 10 000 dilution of stock solution from Molecular Probes, Eugene, OR), carried out in a 20-μl reaction in a Corbett Rotor-Gene (Corbett Research, Mortlake, NSW, Australia).
The primers used for CB1 were: forward 5′-AACTGTGCCAGGCAATCTGTTGTG-3′ and reverse 5′-GGCCTTCCCATTGAAGCACAAACT-3′; for TNF-α: forward ACCACTCTCCCTTTGCAGAACTCA and reverse TCTCATGCACCACCATCAAGGACT; and for tubulin were: forward 5′-CCCTCGCCATGGTAAATACAT-3′ and reverse 5′-ACTGGATGGTACGCTTGGTCT-3′. Relative mRNA concentrations were calculated from the take-off point of reactions using the included software, and tubulin levels were used to normalize data.
Preparation of Synaptosomes
After decapitation, half of the forebrain was dissected on ice. All subsequent steps were performed at 4°C. Cortical tissue was immediately homogenized in 25 vols of 0.32 M sucrose in a glass homogenizer fitted with a Teflon pestle. The homogenate was centrifuged at 200 g for 10 min, and the supernatant was then collected and centrifuged at 20 000 g for 20 min. The pellet was resuspended in 0.32 M sucrose and centrifuged at 20 000 g for 20 min. The crude synaptosomal pellet was finally resuspended in 1 ml of 0.32 M sucrose. Synaptosomal preparations, consisting of pre- and post-synaptic elements of neurons and associated astrocytic endfeet, have provided insights into the regulation of a variety of synaptic functions, including neurotransmitter release and reuptake, energy metabolism, and ion transport systems in physiological, neurological, and neurodegenerative disorders (Begley et al, 1998).
[3H]Glutamate Uptake by Synaptosomes
Sodium-dependent glutamate uptake in synaptosomes was measured according to the procedure previously described by Robinson et al (1991) with some modifications. In brief, 25-ml aliquots of synaptosomes were added to 250 ml of incubation buffer (5 mM Tris, 10 mM HEPES, 2.5 mM KCl, 1.4 M NaCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 1.2 mM KH2PO4, and 10 mM dextrose, pH 7.4) containing L-[3H]glutamic acid 0.125 mM (1 mCi/ml; Amersham Biosciences Europe GmbH, Freiburg, Germany) and incubated for 3 min at 37°C in a shaking waterbath. The reaction was terminated using 1 ml of ice-cold choline buffer (an incubation buffer in which an equimolar concentration of choline chloride was substituted for NaCl), and the samples were centrifuged at 10 000 g for 2 min to recover the synaptosomes. The 3H-bound radioactivity was measured using a liquid scintillation counter.
Plasma Corticosterone Levels
Plasma was obtained from blood samples by centrifuging them at 1000 g for 15 min immediately after the stress exposure. All plasma samples were stored at −20°C before assay with a commercially available radioimmunoassay kit (131I-labeled corticosterone; DPC, Los Angeles, CA). A gamma counter was used to measure radioactivity of the samples. The values obtained in the control animals (100±34.22 ng/ml) matched the kit manufacturer's expected values in adult mice at the time of blood extraction (approximately 1500 h).
MCP-1 Measurement
MCP-1 levels in the prefrontal cortex homogenates were detected using a specific ELISA kit for MCP-1, according to manufacturer's instructions (R&D systems, Abingdon, UK). The assay detection limits were 31.3–2000 pg/ml. To determine tissue levels of MCP-1, brain prefrontal cortices were removed and homogenized by sonication for 10 s at 4°C in PBS containing complete protease inhibitor cocktail (Roche Diagnostics). The homogenates were centrifuged (13 000 g, 10 min, 4°C), and supernatants were used for determinations using the ELISA kit. Protein content was determined by Bradford's method using BSA as standard.
Lipid Peroxidation
Lipid peroxidation was measured by the thiobarbituric acid test for malondialdehyde (MDA) following the method described by Das and Ratty (1987) with some modifications. Cerebral cortex was sonicated in 10 vol of 50 mmol/l phosphate buffer and deproteinized with 40% trichloroacetic acid and 5 mol/l HCl, followed by the addition of 2% (wt/vol) thiobarbituric acid in 0.5 mol/l NaOH. The reaction mixture was heated in a waterbath at 90°C for 15 min and centrifuged at 12 000 g for 10 min. The pink chromogen was measured at 532 nm in a Beckman DU-7500 spectrophotometer (Beckman). The results were expressed as nmol/mg of protein.
Brain PGE2 and 15d-PGJ2 Levels
PGE2 and 15d-PGJ2 brain levels were measured by enzyme immunoassay (EIA) using reagents in kit form (Prostaglandin E2 EIA Kit-Monoclonal; (Cayman Chemical) and 15-deoxy-Δ12,14-prostaglandin J2 ELISA Kit (DRG Diagnostics, Marburg, Germany), respectively. Samples were sonicated in 300 μl homogenization buffer (0.1 M phosphate buffer, pH, 7.4, 1 mM EDTA, and 10 μM indomethacin) and purified by incubation in ethanol at 4 × sample volume for 5 min at 4°C, and then centrifugation at 3000 g for 10 min. They were acidified with glacial acetic acid to pH 3.5, and PGE2 was extracted using SPE (C-18) columns (Amersham Biosciences, Buckinghamshire, UK) rinsed with methanol and water. After the application of samples, columns were washed with water and hexane, and PGE2 and 15d-PGJ2 were eluted with ethyl acetate. Samples were then evaporated to dryness under nitrogen and resuspended in EIA buffer. Levels of PGE2 and 15d-PGJ2 were measured in a 96-well plate and read at 405 nm following manufacturer's instructions. The sensitivity of the assay for PGE2 was 15 pg/ml; intra- and interassay coefficients of variation were 6.6 and 15.5%, respectively, at 62.5 pg/ml, and for 15d-PGJ2 the sensitivity was 36.8 pg/ml; intra- and interassay coefficient of variations were 7.4 and 13%, respectively, at 4.4 pg/ml.
Protein Assay
Protein levels were measured using Bradford method based on the principle of protein-dye binding (Bradford, 1976).
Chemicals and Statistical Analyses
Unless otherwise stated, the chemicals were from Sigma Spain, Madrid. Data in text and figures are expressed as mean ± SEM. For multiple comparisons, a one-way ANOVA followed by the Newman–Keuls post hoc test to compare all pairs of means between groups was made. A p-value <0.05 was considered statistically significant.
RESULTS
CB1 Expression in the Prefrontal Cortex of Control and Stressed WT Mice. Glutamate-Related Mechanism
Our first study was aimed to elucidate the possible effect of stress exposure on CB1 expression in the prefrontal cortex in mice. Western blot studies and subsequent densitometric analysis of the band of interest (Figure 1a) revealed a significant increase in CB1 protein expression (≈60 kDa band) elicited by stress. As expected, this band is absent in CB1 knockout (KO) mice in control and stress conditions, validating the specificity of the antibody used. This observed increase in protein expression is in agreement with CB1 mRNA upregulation, quantified by Q-PCR (Figure 1b).

CB1 upregulation is elicited by stress exposure in mouse prefrontal cortex in a glutamate-related mechanism. (a) Western blot and densitometric analysis of CB1 band in the prefrontal cortex of control (CWT) and stressed during 4 days (SWT) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. The densitometric data are normalized by β-actin (lower band) and are representative of three experiments. *p<0.05 vs CWT (one-way ANOVA followed by Newman–Keuls post hoc test). AU: arbitrary units. (b) Q-PCR analysis of CB1 mRNA in the prefrontal cortex of control (CWT), stressed during 4 days (SWT), stressed + ACEA (SWT + ACEA), and stressed + MK-801 (SWT + MK) wild-type mice. Data are normalized by tubulin, and are representative of three experiments. *p<0.05 vs CWT; #p<0.05 vs SWT (one-way ANOVA followed by Newman–Keuls post hoc test. AU, arbitrary units. (c) Western blot and densitometric analysis of CB1 band in the prefrontal cortex of control (CWT), stressed during 4 days (SWT), stressed + ACEA (SWT + ACEA), and stressed + MK-801 (SWT + MK) wild-type mice. Data are normalized by β-actin and are representative of three experiments. *p<0.05 vs CWT; #p<0.05 vs SWT (one-way ANOVA followed by Newman–Keuls post hoc test). AU, arbitrary units.
Prestress daily ACEA administration blocked stress-induced CB1 mRNA upregulation (Figure 1b) and protein expression (Figure 1c) in the prefrontal cortex in mice. In control conditions, neither ACEA nor MK treatments were able to modify CB1 protein and mRNA expression (data not shown to simplify Figures 1b and c).
Stress exposure leads to the massive release of glutamate into the synaptic cleft, reaching excitotoxic levels in some brain areas (ie, hippocampus, prefrontal cortex) (Moghaddam, 1993). To elucidate whether stress-induced CB1 upregulation is related to this massive glutamate release, a group of animals was i.p. injected with the NMDA-selective blocker, MK-801, at 1 h before the onset of the stress exposure. Interestingly, daily administration of MK-801 partly inhibited stress-induced CB1 mRNA upregulation (Figure 1b) and protein overexpression (Figure 1c) in the prefrontal cortex of WT mice.
CB1-Dependent Effects on Stress-Induced Changes in Synaptosomal Glutamate Uptake and Expression of Glutamate Transporters
The ECS has been shown to confer neuroprotection inhibiting glutamatergic excitotoxic neurotransmission by mechanism/s still not completely understood. We decided to explore the possible effects of CB1 on glutamate transport in brain cortical synaptosomes from WT and CB1 KO mice submitted to stress.
As it has been previously described (García-Bueno et al, 2007), stress exposure leads to a marked decrease in glutamate uptake compared with what was seen in the control group (Figure 2a). Pretreatment with ACEA fully prevented this stress-induced impairment in glutamate uptake function (Figure 2a). Conversely, CB1 KO mice showed dramatically reduced glutamate transport in cortical synaptosomes in both control and stress conditions (Figure 2a).

CB1 effects on synaptosomal glutamate uptake and transporters. (a) Glutamate uptake in forebrain synaptosomes of control (CWT), control + ACEA (CWT + ACEA), stressed during 4 days (SWT), and stressed + ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. The data represents the mean ± SEM of six mice. *p<0.05 vs CWT; #p<0.05 vs SWT (one-way ANOVA followed by Newman–Keuls post hoc test). (b) Western blot and densitometric analysis of glial glutamate transporter EAAT-2 band in the prefrontal cortex of control (CWT), control + daily ACEA (CWT + ACEA), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. Data are normalized by β-actin (lower band), and are representative of three experiments. *p<0.05 vs CWT; #p<0.05 vs SWT (one-way ANOVA followed by Newman–Keuls post hoc test). AU, arbitrary units.
We also assessed the possibility that these effects might be due to expressional changes in the main brain glutamate transporter, EAAT-2 (astrocytic). Western blot analysis revealed that stress exposure reduced EAAT-2 expression (Figure 2b), and that treatment with ACEA prevented this stress-induced decrease. As in the case of synaptosomal glutamate transport, both stressed and nonstressed CB1 KO mice showed reduced EAAT-2 expression in brain cortical homogenates (Figure 2b).
Antiinflammatory Effects Elicited by CB1 Activation. Mechanisms Involved
In general, ECS has been proposed as an endogenous protective system against excessive inflammatory/immune responses in multiple CNS pathologies. Our following studies were aimed at clarifying the particular role of CB1 as a possible regulator of stress-induced inflammatory response.
Effects on HPA axis activity. Plasma corticosterone levels
Our first study evaluated CB1 effects on glucocorticoid synthesis, the main stress hormones, widely known as classical regulators of the inflammatory/immune response in brain and periphery.
The quantification by RIA of plasma corticosterone levels at the time of blood extraction (approximately 1500 h) in all groups studied revealed an expected increase in corticosterone in all groups of stressed animals compared with their respective controls (Figure 3); however, this increase is statistically higher in KO mice than in WT animals (Figure 3). Interestingly, daily ACEA administration did not change plasma corticosterone levels after stress at the time of blood extraction (Figure 3), suggesting that the mechanism(s) of this compound in modulating stress response could be independent of peripheral corticosterone levels.

CB1-related effects on plasma corticosterone levels. Corticosterone plasma levels (ng/ml) at the time of blood extraction (1500 h) of control (CWT), control + daily ACEA (CWT + ACEA), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. Data represents the mean ± SEM of six mice. *p<0.05 vs CWT; #p<0.05 vs SWT; &p<0.05 vs CKO (one-way ANOVA followed by Newman–Keuls post hoc test).
Brain proinflammatory cytokines and chemokines
Proinflammatory cytokines, such as TNF-α or MCP-1, are the first proinflammatory mediators activated in the brain by the massive release of glutamate after stress exposure. Q-PCR studies showed a consistent increase in TNF-α mRNA in WT mouse prefrontal cortex after stress, an effect that is blocked by ACEA (Figure 4a). Conversely, stressed CB1 KO animals presented a reliable increase in the mRNA of the proinflammatory cytokine, suggesting a potentially deleterious chronic inflammatory status in brain. Stress also increased MCP-1 levels in the prefrontal cortex of WT mice and pharmacological stimulation of CB1 decreased them. CB1 KO mice showed no differences from WT in nonstressed animals. However, stress increased MCP-1 levels in brain in SKO group compared with SWT group (Figure 4b).

CB1 antiinflammatory effects on brain I. Proinflammatory cytokines and chemokines. (a) Q-PCR analysis of the proinflammatory cytokine TNF-α mRNA in the prefrontal cortex of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. Data are normalized by tubulin, and are representative of three experiments. *p<0.05 vs CWT; #p<0.05 vs SWT (one-way ANOVA followed by Newman–Keuls post hoc test). AU, arbitrary units. (b) MCP-1 levels (pg/mg protein) in prefrontal cortex homogenates of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. Data represents the mean ± SEM of six mice. *p<0.05 vs CWT; #p<0.05 vs SWT; &p<0.05 vs CKO (one-way ANOVA followed by Newman–Keuls post hoc test).
In this set of neuroinflammation-related experiments (Figures 4, 5, 6 and 7) there were no differences between CWT and CWT + ACEA groups and the data are not shown to simplify figures.

CB1 antiinflammatory effects on brain II. NF-κB. (a) Western blot and densitometric analysis of the NF-κB inhibitory protein IκBα in prefrontal cortex cytosolic extracts of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. Data are normalized by β-actin (lower band), and are representative of three experiments. *p<0.05 vs CWT; #p<0.05 vs SWT (one-way ANOVA followed by Newman–Keuls post hoc test). AU, arbitrary units. (b) Western blot and densitometric analysis of the NF-κB proinflammatory subunit p65 in prefrontal cortex nuclear extracts of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. Data are normalized by β-actin (lower band), and are representative of three experiments. *p<0.05 vs CWT; #p<0.05 vs SWT (one-way ANOVA followed by Newman–Keuls post hoc test). AU, arbitrary units.

CB1 antiinflammatory effects on brain III. Proinflammatory enzymes and lipid peroxidation. (a) Western blot and densitometric analysis of the proinflammatory enzyme NOS-2 in prefrontal cortex homogenates of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. Data are normalized by β-actin (lower band), and are representative of three experiments. *p<0.05 vs CWT; #p<0.05 vs SWT (one-way ANOVA followed by Newman–Keuls post hoc test). AU, arbitrary units. (b) COX-2 expression shown by western blot in all groups studied. (c) Proinflammatory PGE2 levels (index of COX-2 activity) in prefrontal cortex homogenates of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. Data represents the mean ± SEM of six mice. *p<0.05 vs CWT; #p<0.05 vs SWT; &p<0.05 vs CKO (one-way-ANOVA followed by Newman–Keuls post hoc test). (d) MDA levels in prefrontal cortex homogenates of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. Data represents the mean ± SEM of six mice. *p<0.05 vs CWT; #p<0.05 vs SWT; &p<0.05 vs CKO (one-way ANOVA followed by Newman–Keuls post hoc test).

CB1 interactions with the antiinflammatory pathway L-PGDS/15d-PGJ2/PPARγ. (a) Western blot and densitometric analysis of the enzyme L-PGDS in prefrontal cortex homogenates of control (CWT), stressed during 4 days (SWT) and stressed + daily ACEA (SWT + ACEA) wild-type mice and control (CKO) and stressed (SKO) CB1 KO mice. Data are normalized by β-actin (lower band) and are representative of three experiments. *p<0.05 vs CWT; #p<0.05 vs SWT (one-way ANOVA followed by Newman–Keuls post hoc test). AU, arbitrary units. (b) Antiinflammatory 15d-PGJ2 levels in prefrontal cortex homogenates of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. Data represents the mean ± SEM of six mice. *p<0.05 vs CWT; #p<0.05 vs SWT; &p<0.05 vs CKO (one-way ANOVA followed by Newman–Keuls post hoc test). (c) Western blot and densitometric analysis of the nuclear receptor PPARγ in prefrontal cortex homogenates of control (CWT), stressed during 4 days (SWT), and stressed + daily ACEA (SWT + ACEA) wild-type mice, and control (CKO) and stressed (SKO) CB1 KO mice. Data are normalized by β-actin (lower band), and are representative of three experiments. *p<0.05 vs CWT; #p<0.05 vs SWT (one-way ANOVA followed by Newman–Keuls post hoc test). AU, arbitrary units.
Nuclear factor-κB
The release of TNF-α after stress also accounts for NF-κB activation, and we also tested the possible involvement of CB1 in the level of NF-κB expression.
Western blot studies of the inhibitory protein of NF-κB (IκBα) in cytosolic extracts (Figure 5a) and proinflammatory NFκB subunit (p65) in nuclear extracts (Figure 5b) revealed that CB1 regulated NF-κB activation after stress. As it can be observed, stress exposure induced a decrease in the expression of IκBα in brain extracts from WT mice, which was reversed by ACEA (Figure 5a). Levels of nuclear p65 in the different groups studied (Figure 5b) mirrored the expression of its inhibitory factor. CB1 KO animals presented lower levels of IκBα in cytosolic extracts than their respective controls (CWT) (Figure 5a) and a consequent increase in p65 protein in nuclear extracts (Figure 5b), suggesting a state of chronic NF-κB activation in these animals. Interestingly, stress exposure did not affect IκBα/NF-κB expression in the prefrontal cortex of CB1 KO mice.
Proinflammatory enzymes (NOS-2 and COX-2)
NF-κB regulates the expression of genes involved in the accumulation of oxidative/nitrosative and inflammatory mediators after stress exposure. Among others, two main sources of these mediators dependent on NF-κB are inducible NOS-2 and COX-2. Their major products (NO and PGE2, respectively) are potent oxidant/proinflammatory molecules that have been widely connected with damage and even cellular death in multiple CNS pathologies. We decided to explore CB1 effects on the expression and/or activity of these enzymes.
NOS-2 expression increased in the cerebral cortex after stress exposure in WT mice (Figure 6a), although no changes in COX-2 expression were found after this stress protocol (Figure 6b). However, the presumed brain COX-2-derived product, PGE2, appeared in an increased quantity after the stress exposure Figure 6c).
CB1 receptor activation with ACEA was able to completely block NOS-2 upregulation produced by stress exposure (Figure 6a). As in the case of other proinflammatory mediators, CB1 KO mice, both in control and stress conditions, showed consistent NOS-2 upregulation compared with their WT control groups, and a higher increase after stress (Figure 6a).
The interaction between CB1 receptor and COX-2 is more complex, as suggested by our results. PGE2 cortical levels, a presumed COX-2 product in different tissues, including the brain, are affected by CB1 activation. As it can be observed in Figure 6c, ACEA reduced PGE2 tissue levels compared with those in the SWT animals, whereas CB1 SKO animals presented insignificant differences in PGE2 content compared with the SWT group.
Lipid peroxidation
As a final index of stress-induced cellular damage that could be affected by CB1-selective modulation, we measured the accumulation of the lipid peroxidation marker MDA in the brain prefrontal cortex of the different groups of mice. Daily treatment with ACEA prevented MDA overaccumulation, observed after stress exposure in WT mice (Figure 6d). Conversely, CB1 KO mice presented higher levels of MDA after stress than the SWT group (Figure 6d).
CB1-Dependent Modulation of the Antiinflammatory Pathway L-PGDS/15d-PGJ2/PPARγ
So far, our results had shown that the daily administration of the selective CB1 receptor agonist ACEA prevented stress-induced excitotoxicity and proinflammatory mediator accumulation in the prefrontal cortex of WT mice, whereas CB1 KO mice presented an aggravated excitotoxic/neuroinflammatory process after stress exposure. Our final series of studies were aimed to elucidate possible alternative antiinflammatory mechanisms related to CB1 activation. One candidate of interest is the antiinflammatory pathway L-PGDS/15d-PGJ2/PPARγ, that is activated after inflammatory/immune stimuli (stress included) and whose activation (even by some cannabinoids) has been shown to confer neuroprotection after stress exposure, and in diverse CNS pathologies with an inflammatory component in their pathophysiology.
Western blot studies and subsequent densitometric analysis (Figure 7a) revealed a significant increase in the 15d-PGJ2 enzymatic source L-PGDS in the prefrontal cortex homogenates of mouse brains, daily treated with ACEA, compared with the SWT group. On the other hand, L-PGDS protein levels were reduced in the brain of CB1 KO mice in control conditions and after stress exposure (Figure 7a).
Consequently, 15d-PGJ2 levels, quantified by ELISA in brain prefrontal cortex samples, were increased by the administration of ACEA in WT mice compared with their controls (Figure 7b). Both in WT and CB1 KO mice, stress exposure produced a marked decrease in 15d-PGJ2 levels but there were no differences between WT and KO animals.
Western blot analysis of PPARγ protein revealed a significant increase of this 15d-PGJ2 nuclear receptor in prefrontal cortex homogenates of mouse brains, daily treated with ACEA, compared with their controls after stress exposure (SWT) (Figure 7c). CKO mice presented significant lower levels of PPARγ than the CWT group, but stress exposure did not affect PPARγ expression in the brains of CB1 KO animals (Figure 7c).
Validation of ACEA CB1-Dependent Antiexcitotoxic/Antiinflammatory Effects
Finally, to elucidate whether ACEA actions are CB1 mediated, we examined its effects in CB1 KO mice on some representative excitotoxic (EAAT-2 expression) and inflammatory parameters (NOS-2 expression and MDA levels) after sub-chronic stress exposure.
As has been previously shown (Figure 2), stress exposure downregulates EAAT-2 expression in mouse prefrontal cortex and this effect is completely prevented by ACEA pretreatment in WT mice, however this ACEA antiexcitotoxic effect is absent in CB1 KO mice (SKO + ACEA) (Figure 8a), suggesting the direct involvement of CB1 receptor. A similar pattern occurs with the proinflammatory parameter NOS-2 expression (Figure 8b) and MDA levels (Figure 8c), in which ACEA neuroprotective effects are lost in CB1 KO mice compared with WT. These results suggest that ACEA antiinflammatory effects are also dependent on CB1 in our model.
DISCUSSION
Our studies show for the first time to our knowledge, CB1 upregulation in mouse prefrontal cortex exposed to sub-chronic restraint/acoustic stress in a mechanism dependent on NMDA glutamate receptors. In addition, daily ACEA administration prevented stress-induced upregulation of CB1 mRNA and protein, and presented an antiexcitotoxic profile, regulating glutamate transport and uptake after stress exposure, at least in part by restoring protein expression of EAAT-2. CB1 activation also produces antiinflammatory effects, regulating stress-induced proinflammatory response at multiple levels. Notably, a possible related mechanism is the positive modulation of the antiinflammatory pathway L-PGDS/15d-PGJ2/PPARγ, although the precise mechanism/s responsible for this relationship needs to be further investigated (see Figure 9 for a schematic view of the regulatory role of CB1 in stress-induced excitotoxicity and neuroinflammatory balance).
Stress exposure is a major contributing factor to cell death and damage in neurological and neuropsychiatric diseases by three interrelated mechanisms: HPA axis dysregulation, excitotoxicity, and neuroinflammation. However, stress exposure also activates alternative mechanisms aimed at correctly resolving this response. In this way, the CB1 upregulation demonstrated here could be a mechanism related to this protective response of the brain. Similarly, endocannabinoids are released to negatively regulate HPA axis activation (Manzanares et al, 1999; Patel et al, 2004), and brain SEC activation has been considered as a protective mechanism aimed at reducing both neurodegenerative and inflammatory damage after an immune/inflammatory challenge (Centonze et al, 2008).
In particular, our data are in agreement with several studies that propose a role for CB1 as a main regulator of glutamate neurotransmission and metabolism in multiple pathological scenarios (Parmentier-Batteur et al, 2002; Marsicano et al, 2003; Veldhuis et al, 2003; Fernández-López et al, 2006; Kim et al, 2006; Gilbert et al, 2007; Loría et al, 2010). In relation to our findings, stress-induced CB1 upregulation could represent a compensatory response aimed at controlling the massive release of glutamate into the synaptic cleft that might produce cellular damage and even death by excitotoxicity after stress exposure (Lowy et al, 1993; Stein-Behrens et al, 1994; García-Bueno et al, 2007). As reported here, CB1 receptor activation with ACEA enhances the main glutamate uptake mechanism, by restoring protein expression of the glutamate transporter EAAT-2. Astrocyte uptake of glutamate is a high-affinity process regulated by EAAT-2 (Hertz et al, 1983); the fact that stress decreases EAAT-2 expression in the cerebral cortex (García-Bueno et al, 2007), indicates a specific damaging effect at this level, and makes the effect of ACEA more interesting. CB1 effects on EAAT-2 have also been demonstrated in other experimental settings, such as an in vitro model of multiple sclerosis (MS) (Loría et al, 2010), or after prenatal exposure to WIN 55,212-2 (Castaldo et al, 2007). It has been widely demonstrated that presynaptic CB1 inhibits glutamate release (Freund et al, 2003; Chevaleyre et al, 2006; Steiner and Wotjak, 2008), but our data suggests that glutamatergic neurotransmission is also regulated by CB1 at the level of postsynaptic reuptake clearance mechanisms.
How neuronal CB1 activation enhances glutamate glial uptake may be explained by neuron–actrocyte communication that regulates PPARγ activity in astrocytes. Notably, EAAT-2 glutamate transporter is a target gene of PPARγ in excitotoxic-related models, such as stress exposure (García-Bueno et al, 2007) or brain ischemia (Romera et al, 2007). As well, a recent study identified endocannabinoids as PPARγ agonists affording neuroprotection targeting EAAT-2 in an in vitro model of MS (Loría et al, 2010).
Regarding stress-induced neuroinflammation, although cannabinoid-dependent antiinflammatory and antioxidant actions have been commonly related with CB2 receptor activation in brain immune cells (Cabral and Griffin-Thomas, 2009; Ashton and Glass, 2007; Arévalo-Martín et al, 2008), or even with receptor-independent mechanisms (Hampson et al, 2000; Marsicano et al, 2002)), our and other studies revealed a significant role for CB1 as well in inflammatory/immune responses in animal models of MS (Arévalo-Martín et al, 2003; Pryce et al, 2003; Maresz et al, 2007) and brain ischemia (Fernández-López et al, 2006).
Here, we demonstrated that CB1 activation interacted with the proinflammatory response elicited by stress at multiple levels. In the case of cytokines, CB1 activation has been shown to reduce TNFα and IL-1β synthesis in neurons, astrocytes, and microglia (Fernández-López et al, 2006; Maresz et al, 2007; Marchalant et al, 2009). Notably, the opposite relationship, in which cytokines presumably upregulate cannabinoid receptor expression, has also been suggested in some immune/inflammatory scenarios (Jean-Gilles et al, 2010), stress included (Lee et al, 2006).
Regarding other stress-induced proinflammatory mediators, like the key transcription factor NF-κB, our results coincide with others showing a role for CB1 in NF-κB regulation in injured mouse brains (Panikashvili et al, 2005); however regulation of NF-κB by cannabinoids has been commonly related with CB2 in some immune cells such as macrophages or monocytes (Jeon et al, 1996; Rajesh et al, 2008); and even with CB1/CB2 receptor-independent mechanisms, as is the case of cannabidiol in LPS-activated microglia (Kozela et al, 2010) or 2-arachidonyl glycerol through PPARγ (Rockwell et al, 2006).
Two main NF-κB-dependent enzymatic sources of oxidative/nitrosative mediators after stress in the brain are NOS-2 and COX-2. NOS-2 expression is downregulated by selective CB1 activation in neurons and astrocytes both in in vitro and in vivo inflammatory-related models (Fernández-López et al, 2006; Esposito et al, 2006; Sheng et al, 2005), as it is also reported in our study. However, cannabinoid–cyclooxygenase interactions may be more intricate. In our stress model, CB1 regulates the levels of the presumed COX-2 product PGE2 without altering enzyme expression. These results suggest possible effects of ACEA on the catalytic activity or protein stability of COX-2, or a possible effect on the activity of tissue-specific prostaglandin E2 synthases, which is yet to be explored. Some authors have reported opposite COX-2/CB1 interactions: protection afforded by 2-arachidonoylglycerol and THC by downregulation of COX-2 expression and activity in cultured neurons, and LPS stimulated J774 macrophages (Zhang and Chen, 2008; Chang et al, 2001), or THC neurotoxicity in hippocampal neurons in a mechanism attributable to COX-2 activity and the concomitant generation of free radicals (Chan et al, 1998).
Our results show expression changes in the antiinflammatory pathway L-PGDS/15d-PGJ2/PPARγ after CB1 pharmacological and genetic manipulations; although still speculative, the involvement of this antiinflammatory pathway in CB1 neuroprotective effects is an interesting idea that deserves further investigation. This potential relationship is especially relevant as some pharmacological and genetic studies using CB1/CB2 KO mice indicate possible CB1/CB2 alternative mechanisms of action. PPARs could be excellent candidates, taking into account that diverse cannabinoids are potent ligands of PPARs (O’Sullivan, 2007). Thus, the main endocannabinoids, anandamide, and 2-arachidonoylglycerol, and also ajulemic acid, a THC structural analog, have antiinflammatory properties mediated by PPARγ (Sun and Bennett, 2007). In addition, the endogenous acylethanolamines, oleoylethanolamide, and palmitoylethanolamide have neuroprotective effects mediated through PPARα. The possibility of these related acylethanolamines modulating the stress response through interaction with PPARα remains unexplored.
Our results show a significant upregulation of L-PGDS/15d-PGJ2 after ACEA administration. This upregulation has been also demonstrated after the administration of ajulemic acid to human synovial cells (Stebulis et al, 2008), inducing an antiinflammatory profile of eicosanoids. PPARγ regulation is not yet completely understood, but one of the main mechanisms may be ligand availability (Daynes and Jones, 2002). This kind of regulation could be important in our stress model because we also found increased 15d-PGJ2 levels after ACEA administration. Other authors have proposed alternative mechanisms by which cannabinoids can potentially lead to PPARs activation. For example, CB1 binding could stimulate MAPK pathways, as it occurs in other scenarios (Lépicier et al, 2006), which regulates PPARγ transcriptional activity by direct phosphorylation on serine residues (Burns and Vanden Heuvel, 2007).
In contrast to ACEA neuroprotective effects in stress conditions, we and others have showed a dysregulated HPA axis activity in basal and after stress exposure in CB1 KO mice (Barna et al, 2004; Cota, 2008; Aso et al, 2008). In addition, a status of excessive excitotoxicity and neuroinflammation is also presented, increasing their susceptibility to experience damage and even cellular death. Similarly, in an animal model of Parkinson's disease, other authors have also found an increased vulnerability to 6-hydroxydopamine lesion in CB1 KO mice, presenting a higher index of lipid peroxidation and NOS-2 expression in selected brain areas (Pérez-Rial et al, 2009).
In addition to HPA axis dysregulation, chronic excitotoxicity, and neuroinflammation, CB1 KO mice present defective adult neurogenesis (Jin et al, 2004), increased anxiety (Urigüen et al, 2004; Mikics et al, 2009), and deficiency in neuronal plasticity by decreasing BDNF levels in the hippocampus (Aso et al, 2008), suggesting a possible role of CB1 in the pathophysiology of depression and its pharmacological manipulation as a potential therapeutic strategy.
In summary, the multifaceted neuroprotective effects described here suggest that CB1 activation is an attractive therapeutic strategy against diverse neuropsychiatric pathologies with HPA axis dysregulation and an excitotoxic/neuroinflammatory component in their pathophysiology.
References
Arévalo-Martín A, García-Ovejero D, Gómez O, Rubio-Araiz A, Navarro-Galve B, Guaza C et al (2008). CB2 cannabinoid receptors as an emerging target for demyelinating diseases: from neuroimmune interactions to cell replacement strategies. Br J Pharmacol 153: 216–225.
Arévalo-Martín A, Vela JM, Molina-Holgado E, Borrell J, Guaza C (2003). Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J Neurosci 23: 2511–2516.
Ashton JC, Glass M (2007). The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr Neuropharmacol 5: 73–80.
Aso E, Ozaita A, Valdizán EM, Ledent C, Pazos A, Maldonado R et al (2008). BDNF impairment in the hippocampus is related to enhanced despair behavior in CB1 knockout mice. J Neurochem 105: 565–572.
Bambico FR, Duranti A, Tontini A, Tarzia G, Gobbi G (2009). Endocannabinoids in the treatment of mood disorders: evidence from animal models. Curr Pharm Des 15: 1623–1646.
Barna I, Zelena D, Arszovszki AC, Ledent C (2004). The role of endogenous cannabinoids in the hypothalamo-pituitary-adrenal axis regulation: in vivo and in vitro studies in CB1 receptor knockout mice. Life Sci 75: 2959–2970.
Baum A, Posluszny DM (1999). Health psychology: mapping biobehavioral contributions to health and illness. Annu Rev Psychol 50: 137–163.
Begley JG, Butterfield DA, Keller JN, Koppal T, Drake J, Mattson MP (1998). Cryopreservation of rat cortical synaptosomes and analysis of glucose and glutamate transporter activities, and mitochondrial function. Brain Res Brain Res Protoc 3: 76–82.
Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D et al (2003). A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci USA 100: 1920–1925.
Bradford MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254.
Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, Southwick SM et al (1995). MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 152: 973–981.
Burns KA, Vanden Heuvel JP (2007). Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta 1771: 952–960.
Cabral GA, Griffin-Thomas L (2009). Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev Mol Med 11: e3.
Castaldo P, Magi S, Gaetani S, Cassano T, Ferraro L, Antonelli T et al (2007). Prenatal exposure to the cannabinoid receptor agonist WIN 55,212-2 increases glutamate uptake through overexpression of GLT1 and EAAC1 glutamate transporter subtypes in rat frontal cerebral cortex. Neuropharmacology 53: 369–378.
Centonze D, Battistini L, Maccarrone M (2008). The endocannabinoid system in peripheral lymphocytes as a mirror of neuroinflammatory diseases. Curr Pharm Des 14: 2370–2442.
Chan GC, Hinds TR, Impey S, Storm DR (1998). Hippocampal neurotoxicity of delta 9-tetrahydrocannabinol. J Neurosci 18: 5322–5332.
Chang YH, Lee ST, Lin WW (2001). Effects of cannabinoids on LPS-stimulated inflammatory mediator release from macrophages: involvement of eicosanoids. J Cell Biochem 81: 715–723.
Chevaleyre V, Takahashi KA, Castillo PE (2006). Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci 29: 37–76.
Cota D (2008). The role of the endocannabinoid system in the regulation of hypothalamic pituitary-adrenal axis activity. J Neuroendocrinol 20 (Suppl 1): 35–38.
Das NP, Ratty AK (1987). Studies on the effects of the narcotic alkaloids, cocaine, morphine and codeine on nonenzymatic lipid peroxidation in rat brain mitochondria. Biochem Med Metab Biol 37: 256–264.
Daynes RA, Jones DC (2002). Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol 2: 748–759.
Depke M, Fusch G, Domanska G, Geffers R, Völker U, Schuett C et al (2008). Hypermetabolic syndrome as a consequence of repeated psychological stress in mice. Endocrinology 149: 2714–2723.
Esposito G, De Filippis D, Steardo L, Scuderi C, Savani C, Cuomo V et al (2006). CB1 receptor selective activation inhibits beta-amyloid-induced iNOS protein expression in C6 cells and subsequently blunts tau protein hyperphosphorylation in co-cultured neurons. Neurosci Lett 404: 342–346.
Fernández-López D, Martínez-Orgado J, Nuñez E, Romero J, Lorenzo P, Moro MA et al (2006). Characterization of the neuroprotective effect of the cannabinoid agonist WIN-55212 in an in vitro model of hypoxic-ischemic brain damage in newborn rats. Pediatr Res 60: 169–173.
Freund TF, Katona I, Piomelli D (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83: 1017–1066.
Galve-Roperh I, Aguado T, Palazuelos J, Guzmán M (2008). Mechanisms of control of neuron survival by the endocannabinoid system. Curr Pharm Des 14: 2279–2288.
García-Bueno B, Caso JR, Leza JC (2008a). Stress as a neuroinflammatory condition in brain: damaging and protective mechanisms. Neurosci Biobehav Rev 32: 1136–1151.
García-Bueno B, Caso JR, Pérez-Nievas BG, Lorenzo P, Leza JC (2007). Effects of peroxisome proliferator-activated receptor gamma agonists on brain glucose and glutamate transporters after stress in rats. Neuropsychopharmacology 32: 1251–1260.
García-Bueno B, Madrigal JL, Pérez-Nievas BG, Leza JC (2008b). Stress mediators regulate brain prostaglandin synthesis and peroxisome proliferator-activated receptor gamma activation after stress in rats. Endocrinology 149: 1969–1978.
Gilbert GL, Kim HJ, Waataja JJ, Thayer SA (2007). Delta 9-tetrahydrocannabinol protects hippocampal neurons from excitotoxicity. Brain Res 1128: 61–69.
Hampson AJ, Grimaldi M, Lolic M, Wink D, Rosenthal R, Axelrod J (2000). Neuroprotective antioxidants from marijuana. Ann N Y Acad Sci 899: 274–282.
Hertz L, Bender AS, Richardson JS (1983). Benzodiazepines and beta-adrenergic binding to primary cultures of astrocytes and neurons. Prog Neuropsychopharmacol Biol Psychiatry 7: 681–686.
Jean-Gilles L, Gran B, Constantinescu CS (2010). Interaction between cytollines cannabinoids and the nervous system. Immunobiology 215: 606–610.
Jeon YJ, Yang KH, Pulaski JT, Kaminski NE (1996). Attenuation of inducible nitric oxide synthase gene expression by delta 9-tetrahydrocannabinol is mediated through the inhibition of nuclear factor- kappa B/Rel activation. Mol Pharmacol 50: 334–341.
Jezova D, Tokarev D, Rusnak M (1995). Endogenous excitatory amino acids are involved in stress-induced adrenocorticotropin and catecholamine release. Neuroendocrinology 62: 326–333.
Jin K, Xie L, Kim SH, Parmentier-Batteur S, Sun Y, Mao XO et al (2004). Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol Pharmacol 66: 204–208.
Kiank C, Holtfreter B, Starke A, Mundt A, Wilke C, Schütt C (2006). Stress susceptibility predicts the severity of immune depression and the failure to combat bacterial infections in chronically stressed mice. Brain Behav Immun 20: 359–368.
Kim JJ, Yoon KS (1998). Stress: metaplastic effects in the hippocampus. Trends Neurosci 21: 505–509.
Kim SH, Won SJ, Mao XO, Jin K, Greenberg DA (2006). Molecular mechanisms of cannabinoid protection from neuronal excitotoxicity. Mol Pharmacol 69: 691–696.
Kozela E, Pietr M, Juknat A, Rimmerman N, Levy R, Vogel Z (2010). Cannabinoids delta (9)-tetrahydrocannabinol and cannabidiol differentially inhibit the lipopolysaccharide-activated NF-kappaB and interferon-beta/STAT proinflammatory pathways in BV-2 microglial cells. J Biol Chem 285: 1616–1626.
Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F et al (1999). Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 283: 401–404.
Lee YL, Choi B, Lee EH, Choi KS, Sohn S (2006). Immobilization stress induces cell death through production of reactive oxygen species in the mouse cerebral cortex. Neurosci Lett 392: 27–31.
Lépicier P, Bibeau-Poirier A, Lagneux C, Servant MJ, Lamontagne D (2006). Signaling pathways involved in the cardioprotective effects of cannabinoids. J Pharmacol Sci 102: 155–166.
Loría F, Petrosino S, Hernangómez M, Mestre L, Spagnolo A, Correa F et al (2010). An endocannabinoid tone limits excitotoxicity in vitro and in a model of multiple sclerosis. Neurobiol Dis 37: 166–176.
Lowy MT, Gault L, Yamamoto BK (1993). Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J Neurochem 61: 1957–1960.
Madrigal JL, García-Bueno B, Caso JR, Pérez-Nievas BG, Leza JC (2006). Stress-induced oxidative changes in brain. CNS Neurol Disord Drug Targets 5: 561–568.
Madrigal JL, Hurtado O, Moro MA, Lizasoain I, Lorenzo P, Castrillo A et al (2002). The increase in TNF-alpha levels is involved in NF-kappaB activation and inducible nitric oxide synthase expression in brain cortex after immobilization stress. Neuropsychopharmacology 26: 155–163.
Madrigal JL, Moro MA, Lizasoain I, Lorenzo P, Castrillo A, Boscá L et al (2001a). Inducible nitric oxide synthase expression in brain cortex after acute restraint stress is regulated by nuclear factor kappaB-mediated mechanisms. J Neurochem 76: 532–538.
Madrigal JL, Moro MA, Lizasoain I, Lorenzo P, Fernández AP, Rodrigo J et al (2003). Induction of cyclooxygenase-2 accounts for restraint stress-induced oxidative status in rat brain. Neuropsychopharmacology 28: 1579–1588.
Madrigal JL, Olivenza R, Moro MA, Lizasoain I, Lorenzo P, Rodrigo J et al (2001b). Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology 24: 420–429.
Manzanares J, Corchero J, Fuentes JA (1999). Opioid and cannabinoid receptormediated regulation of the increase in adrenocorticotropin hormone and corticosterone plasma concentrations induced by central administration of delta (9)-tetrahydrocannabinol in rats. Brain Res 839: 173–179.
Marchalant Y, Brothers HM, Norman GJ, Karelina K, DeVries AC, Wenk GL (2009). Cannabinoids attenuate the effects of aging upon neuroinflammation and neurogenesis. Neurobiol Dis 34: 300–307.
Maresz K, Pryce G, Ponomarev ED, Marsicano G, Croxford JL, Shriver LP et al (2007). Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat Med 13: 492–497.
Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A et al (2003). CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302: 84–88.
Marsicano G, Moosmann B, Hermann H, Lutz B, Behl C (2002). Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1. J Neurochem 80: 448–456.
McEwen BS (1998). Protective and damaging effects of stress mediators. N Engl J Med 338: 171–179.
McLeod TM, Lopez-Figueroa AL, Lopez-Figueroa MO (2001). Nitric oxide, stress, and depression. Psychopharmacol Bull 35: 24–41.
Mechoulam R, Shohami E (2007). Endocannabinoids and traumatic brain injury. Mol Neurobiol 36: 68–74.
Mikics E, Vas J, Aliczki M, Halasz J, Haller J (2009). Interactions between the anxiogenic effects of CB1 gene disruption and 5-HT3 neurotransmission. Behav Pharmacol 20: 265–272.
Moghaddam B (1993). Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J Neurochem 60: 1650–1657.
Olfe J, Domanska G, Schuett C, Kiank C (2010). Different stress-related phenotypes of BALB/c mice from in-house or vendor: alterations of the sympathetic and HPA axis responsiveness. BMC Physiol 10: 2.
O’Sullivan SE (2007). Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol 152: 576–582.
Panikashvili D, Mechoulam R, Beni SM, Alexandrovich A, Shohami E (2005). CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J Cereb Blood Flow Metab 25: 477–484.
Parmentier-Batteur S, Jin K, Mao XO, Xie L, Greenberg DA (2002). Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci 22: 9771–9775.
Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ (2004). Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitaryadrenal axis. Endocrinology 145: 5431–5438.
Pérez-Rial S, García-Gutiérrez MS, Molina JA, Pérez-Nievas BG, Ledent C, Leiva C et al (2009). Increased vulnerability to 6-hydroxydopamine lesion and reduced development of dyskinesias in mice lacking CB1 cannabinoid receptors. Neurobiol Aging. (Epub ahead of print 4 May 2009).
Pryce G, Ahmed Z, Hankey DJ, Jackson SJ, Croxford JL, Pocock JM et al (2003). Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain 126 (Part 10): 2191–2202.
Qiu BS, Vallance BA, Blennerhassett PA, Collins SM (1999). The role of CD4+ lymphocytes in the susceptibility of mice to stress-induced reactivation of experimental colitis. Nat Med 5: 1178–1182.
Radley JJ, Arias CM, Sawchenko PE (2006). Regional differentiation of the medial prefrontal cortex in regulating adaptive responses to acute emotional stress. J Neurosci 26: 12967–12976.
Robinson MB, Hunter-Ensor M, Sinor J (1991). Pharmacologically distinct sodium-dependent L-[3H]glutamate. Brain Res 544: 196–202.
Rajesh M, Mukhopadhyay P, Haskó G, Huffman JW, Mackie K, Pacher P (2008). CB2 cannabinoid receptor agonists attenuate TNF-alpha-induced human vascular smooth muscle cell proliferation and migration. Br J Pharmacol 153: 347–357.
Rockwell CE, Snider NT, Thompson JT, Vanden Heuvel JP, Kaminski NE (2006). Interleukin-2 suppression by 2-arachidonyl glycerol is mediated through peroxisome proliferator-activated receptor gamma independently of cannabinoid receptors 1 and 2. Mol Pharmacol 70: 101–111.
Romera C, Hurtado O, Mallolas J, Pereira MP, Morales JR, Romera A et al (2007). Ischemic preconditioning reveals that GLT1/EAAT2 glutamate transporter is a novel PPARgamma target gene involved in neuroprotection. J Cereb Blood Flow Metab 27: 1327–1338.
Schreiber E, Matthias P, Müller MM, Schaffner W (1989). Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acid Res 17: 6419.
Sheng WS, Hu S, Min X, Cabral GA, Lokensgard JR, Peterson PK (2005). Synthetic cannabinoid WIN55,212-2 inhibits generation of inflammatory mediators by IL-1beta stimulated human astrocytes. Glia 49: 211–219.
Slimmer LM, Lyness JM, Caine ED (2001). Stress, medical illness, and depression. Semin Clin Neuropsychiatry 6: 12–26.
Stebulis JA, Johnson DR, Rossetti RG, Burstein SH, Zurier RB (2008). Ajulemic acid, a synthetic cannabinoid acid, induces an antiinflammatory profile of eicosanoids in human synovial cells. Life Sci 83: 666–670.
Stein-Behrens BA, Lin WJ, Sapolsky RM (1994). Physiological elevations of glucocorticoids potentiate glutamate accumulation in the hippocampus. J Neurochem 63: 596–602.
Steiner MA, Wotjak CT (2008). Role of the endocannabinoid system in regulation of the hypothalamic-pituitary-adrenocortical axis. Prog Brain Res 170: 397–432.
Sun Y, Bennett A (2007). Cannabinoids: a new group of Agonists of PPARs. PPAR Res 2007: 23513.
Urigüen L, Pérez-Rial S, Ledent C, Palomo T, Manzanares J (2004). Impaired action of anxiolytic drugs in mice deficient in cannabinoid CB1 receptors. Neuropharmacology 46: 966–973.
Veldhuis WB, van der Stelt M, Wadman MW, van Zadelhoff G, Maccarrone M, Fezza F et al (2003). Neuroprotection by the endogenous cannabinoid anandamide and arvanil against in vivo excitotoxicity in the rat: role of vanilloid receptors and lipoxygenases. J Neurosci 23: 4127–4133.
Wolf SA, Tauber S, Ullrich O (2008). CNS immune surveillance and neuroinflammation: endocannabinoids keep control. Curr Pharm Des 14: 2266–2278.
Zhang J, Chen CJ (2008). Endocannabinoid 2-arachidonoylglycerol protects neurons by limiting COX-2 elevation. Biol Chem 283: 22601–22611.
Acknowledgements
This research was supported by the Regional Government of Madrid (S-SAL/0261/2006), the Spanish Ministeries of Science and Innovation (SAF07-63138 and CIBERSAM), and Universidad Complutense-Santander (2878-920140). The research was also funded by Red Temática de Investigación Cooperativa en Salud (RETICS, Instituto de Salud Carlos III, MICINN/FEDER): Red de Trastornos Adictivos, RD06/0001/1004 (JM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Zoppi, S., Pérez Nievas, B., Madrigal, J. et al. Regulatory Role of Cannabinoid Receptor 1 in Stress-Induced Excitotoxicity and Neuroinflammation. Neuropsychopharmacol 36, 805–818 (2011). https://doi.org/10.1038/npp.2010.214
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2010.214
Keywords
This article is cited by
-
How depression and antidepressant drugs affect endocannabinoid system?—review of clinical and preclinical studies
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
-
The semantics of microglia activation: neuroinflammation, homeostasis, and stress
Journal of Neuroinflammation (2021)
-
Cannabinoid receptor 1 signalling modulates stress susceptibility and microglial responses to chronic social defeat stress
Translational Psychiatry (2021)
-
Cognitive behavioral therapy program for cannabis use cessation in first-episode psychosis patients: study protocol for a randomized controlled trial
Trials (2016)
-
Neurobiological Interactions Between Stress and the Endocannabinoid System
Neuropsychopharmacology (2016)
