
Abstract
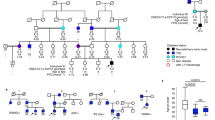
Hirschsprung disease (HSCR) is characterized by a congenital absence of enteric ganglia along a variable length of the intestine. Although long considered to be a multifactorial disease, we have identified linkage in a subset of five HSCR families to the pericentromeric region of chromosome 10, thereby proving monogenic inheritance in some families. A maximum two–point lod score of 3.37 (θ̂ = 0.045) was observed between HSCR and D10S176, under an incompletely penetrant dominant model. Multipoint, affecteds–only and non–parametric analyses supported this finding and localize this gene to a region of ≈7 centiMorgans, in close proximity to the locus for multiple endocrine neoplasia type 2 (MEN2). The co–occurrence of these two entities in some families might be attributable to shared pathogenetic origins.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others


References
Badner, J.A., Sieber, W.K., Garver, K.L. & Chakravarti, A. A genetic study of Hirschsprung disease. Am. J. hum. Genet. 46, 568–580 (1990).
Swenson, O. Historical review. In Hirschsprung's Disease (ed. Holschneider, A.M.) 3–6 (Thieme-Stratton, New York, 1982).
Swenson, O. My early experience with Hirschsprung's disease. J. ped. Surg. 24, 839–845 (1989).
Bodian, M. & Carter, C.O. A family study of Hirschsprung's disease. Ann. hum. Genet. 26, 261–277 (1963).
Passarge, E. The genetics of Hirschsprung's disease: evidence for heterogeneous etiology and a study of sixty-three families. New Engl. J. Med. 276, 138–143 (1967).
Passarge, E. Genetics of common gastrointestinal malformations and the heterogeneity of Hirschsprung's disease. In The Genetics and Heterogeneity of Common Gastrointestinal Disorders (eds Rotter, J.I., Samloff, I.M. & Rimoin, D.L) 441–449 (Academic Press, New York, 1980).
Garver, K.L., Law, J.C. & Garver, B. Hirschsprung disease: a genetic study. Clin. Genet. 28, 503–508 (1985).
Lipson, A.H. & Harvey, J. Three-generation transmission of Hirschsprung's disease. Clin. Genet. 32, 175–178 (1987).
Lipson, A.H., Harvey, J. & Oley, C.A. Three-generation transmission of Hirschsprung's disease. Clin. Genet. 37, 235 (1990).
Slaugenhaupt, S.A. A genetic linkage study of Hirschsprung disease. Ph.D. thesis, (University of Pittsburgh, Pittsburgh, Pennsylvania, 1991).
Lane, P.W. Association of megacolon with two recessive spotting genes in the mouse. J. Hered. 57, 181–183 (1966).
Lane, P.W. & Liu, H.M. Association of megacolon with a new dominant spotting gene (Dom) in the mouse. J. Hered. 75, 435–439 (1984).
Ikadai, H. et al. Observation of congenital aganglionosis rat (Hirschsprung's disease rat) and its genetic analysis. Congen. Anom. 19, 31–36 (1979).
Angrist, M. et al. Hirschsprung disease: identification and analysis of candidate regions. (Abstract) Am. J. hum. Genet. 51, A359 (1992).
Puffenberger, E.G. et al. Pedigree analysis of a large Mennonite kindred segregating Hirschsprung disease. (Abstract) Am. J. hum. Genet. 51, A106 (1992).
Khan, A.H., Desjardins, J.G., Youssef, S., Grégoire, H. & Seidman, E. Gastrointestinal manifestations of Sipple syndrome in children. J. ped. Surg. 22, 719–723 (1987).
Mahaffey, S.M., Martin, L.W., McAdams, A.J., Ryckman, F.C. & Torres, M. Multiple endocrine neoplasia type II B with symptoms suggesting Hirschsprung's disease: a case report. J. ped. Surg. 25, 101–103 (1990).
Verdy, M. et al. Hirschsprung's disease in a family with multiple endocrine neoplasia type 2. J. ped. Gastroenterol. Nutr. 1, 603–607 (1982).
Mathew, C.G.P. et al. A linked genetic marker for multiple endocrine neoplasia type 2A on chromosome 10. Nature 328, 527–528 (1987).
Simpson, N.E. et al. Assignment of multiple endocrine neoplasia type 2A to chromosome 10 by linkage. Nature 328, 528–530 (1987).
Martucciello, G. et al. Total colonic aganglionosis with interstitial deletion of the long arm of chromosome 10. Ped. Surg. Int. 7, 308–310 (1992).
Puliti, A. et al. Deleted and normal chromosome 10 homologs from a patient with Hirschsprung disease isolated in two cell hybrids through enrichment by immunomagnetic selection. Cytogenet Cell. Genet. 63, 102–106 (1993).
Decker, R.A., Moore, J., Ponder, B. & Weber, J.L. Linkage mapping of human chromosome 10 microsatellite polymorphisms. Genomics 12, 604–606 (1992).
Papi, L., Tunnacliffe, A. & Ponder, B.A.J. Dinucleotide repeat polymorphism at the RBP3 locus in chromosome band 10q11.2. Hum. molec. Genet. 1, 450 (1992).
Weissenbach, J. et al. A second-generation linkage map of the human genome. Nature 359, 794–801 (1992).
Cox, T.K., Perlin, M. & Chakravarti, A. MultiMap: automatic construction of linkage maps. Am. J. hum. Genet. 51, A33 (1992).
Weeks, D.E. & Lange, K. The affected-pedigree-member method of linkage analysis. Am. J. hum. Genet. 42, 315–326 (1988).
Weeks, D.E. & Lange, K. A multilocus extension of the affected-pedigree-member method of linkage analysis. Am. J. hum. Genet. 50, 859–868 (1992).
Sherman, J.O. et al. A 40-year multinational retrospective study of 880 Swenson procedures. J. ped. Surg. 24, 833–838 (1989).
Bickler, S.W., Harrison, M.W., Campbell, T.J. & Campbell, J.R., Long-segment Hirschsprung's disease. Arch. Surg. 127, 1047–1051 (1992).
Smith, B. Pre- and postnatal development of the ganglion cells of the rectum and its surgical implications. J. ped. Surg. 3, 386–391 (1968).
Whitehead, R. Mucosal Biopsy of the Gastrointestinal Tract 4th edn 391–398 (Saunders, Philadelphia, 1990).
Bolande, R.P. The neurocristopathies: a unifying concept of disease arising in neural crest maldevelopment. Hum. Path. 5, 409–429 (1974).
Gutmann, D.H., Wood, D.L. & Collins, F.S. Identification of the neurofibromatosis type 1 gene product. Proc. natn. Acad. Sci. U.S.A. 88, 9658–9662 (1991).
Xu, G. et al. The neurofibromatosis type 1 gene product encodes a protein related to GAP. Cell 62, 599–608 (1990).
Baldwin, C.T., Hoth, C.F., Amos, J.A., da-Silva, E.O. & Milunsky, A. Anexonic mutation in the HuP2 paired domain gene causes Waardenburg's syndrome. Nature 355, 637–638 (1992).
Tassabehji, M. et al. Waardenburg's syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature 355, 635–636 (1992).
Chakravarti, A. & Lander, E.S. Genetic approaches to the dissection of complex diseases. Banbury Report 33, 307–315 (1990).
Cass, D.T. Aganglionosis: associated anomalies. J. Paediatr. Child Health 26, 351–354 (1990).
Le Marec, B. et al. Cancer de la thyroide a stroma amyloide, syndrome de Sipple, megacolon congenital avec hyperplasie des plexus: une seule et meme affection autosomique dominante a penetrance complete. J. Genet. Hum. 28, 169–174 (1980).
McKusick, V.A. Mendelian Inheritance in Man. 10th edn 848–850 (Johns Hopkins University Press, Baltimore, 1992).
Lairmore, T.C. et al. Familial medullary thyroid carcinoma and multiple endocrine neoplasia 2B map to the same region of chromosome 10 as multiple endocrine neoplasia type 2A. Genomics 9, 181–192 (1991).
Carnahan, J.F., Anderson, D.J. & Patterson, P.H. Evidence that enteric neurons may derive from the sympathoadrenal lineage. Dev. Biol. 148, 552–561 (1991).
Anderson, D.J. Cell fate determination in the peripheral nervous system: the sympathoadrenal progenitor. J. Neurobiol. 24, 185–198 (1993).
Lo, L.C., Johnson, J.E., Wuenschell, C.W., Saito, T. & Anderson, D.J., Mammalian achaete-scute homolog 1 is transiently expressed by spatially restricted subsets of early neuroepithelial and neural crest cells. Genes Dev. 5, 1524–1537 (1991).
Patterson, P.H. New blossoms in the neural crest field: trophic and transcription factors. Curr. Opin. cell Biol. 4, 962–966 (1992).
Wu, J.S. et al. The beta subunit locus of the human fibronectin receptor: DNA restriction fragment length polymorphism and linkage mapping studies. Hum. Genet. 83, 383–390 (1989).
Troelstra, C. et al. Localization of the nucleotide excision repair gene ERCC6 to human chromosome 10q11-q21. Genomics 12, 745–749 (1992).
McDonald, H. et al. Identification and characterization of a gene at D10S94 in the MEN2A region. Genomics 13, 344–348 (1992).
Rousseau-Merck, M.F., Berger, R., Ponder, B.A.J. & Thiesen, H.J. A cluster of expressed zinc finger protein genes in the pericentromeric region of human chromosome 10. Genomics 13, 845–848 (1992).
Hirschsprung, H. Stuhlträgheit neugeborener infolge dilatation und hypertrophie des colons. Jahresb. Kinderh. 27, 1–7 (1888).
Sorant, A.J.M., Bonney, G.E. & Elston, R.C. Segregation analysis of a discrete trait under a class A regressive logistic model (REGD, version 2. 0). In Statistical Analysis for Genetic Epidemiology (S.A.G.E.) Users Manual (ed Elston, R.C.) (Louisiana State University Medical Center, New Orleans, Louisiana, 1989).
Howe, J.R. et al. Presymptomatic identification of carriers of the multiple endocrine neoplasia type 2A gene using flanking DNA markers. Surgery 112, 219–226 (1992).
Howe, J.R. et al. Improved predictive testfor MEN2, using flanking dinucleotide repeats and RFLPs. Am. J. hum. Genet. 51, 1430–1442 (1992).
Lander, E. & Green, P. Construction of multi-locus genetic linkage maps in humans. Proc. natn. Acad. Sci. U.S.A. 84, 2363–2367 (1987).
Miller, S.A., Dykes, D.D. & Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16, 1215 (1988).
Lathrop, G.M., Lalouel, J.M., Julier, C. & Ott, J. Strategies for multilocus linkage analysis in humans. Proc. natn. Acad. Sci. U.S.A. 81, 3443–3446 (1984).
Ott, J. Analysis of Human Genetic Linkage. 2nd edn (Johns Hopkins University Press, Baltimore, 1991).
Chi, D. et al. A highly informative dinucleotide repeat polymorphism at the ret proto-oncogene locus within human chromosome 10q11.2. Am. J. hum. Genet. 51, A184 (1992).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Angrist, M., Kauffman, E., Slaugenhaupt, S. et al. A gene for Hirschsprung disease (megacolon) in the pericentromeric region of human chromosome 10. Nat Genet 4, 351–356 (1993). https://doi.org/10.1038/ng0893-351
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/ng0893-351
This article is cited by
-
Genetics of Hirschsprung’s disease
Pediatric Surgery International (2023)
-
Comprehensive characterization of the genetic landscape of familial Hirschsprung’s disease
World Journal of Pediatrics (2023)
-
mRNA sequencing provides new insights into the pathogenesis of Hirschsprung’s disease in mice
Pediatric Surgery International (2023)
-
Genetic variants in RET and risk of Hirschsprung's disease in Southeastern Chinese: a haplotype-based analysis
BMC Medical Genetics (2011)
-
Genetic basis of Hirschsprung’s disease
Pediatric Surgery International (2009)
