
Abstract
Psoriasis is a chronic autoimmune disease with complex genetic architecture. Previous genome-wide association studies (GWAS) and a recent meta-analysis using Immunochip data have uncovered 36 susceptibility loci. Here, we extend our previous meta-analysis of European ancestry by refined genotype calling and imputation and by the addition of 5,033 cases and 5,707 controls. The combined analysis, consisting of over 15,000 cases and 27,000 controls, identifies five new psoriasis susceptibility loci at genome-wide significance (P<5 × 10−8). The newly identified signals include two that reside in intergenic regions (1q31.1 and 5p13.1) and three residing near PLCL2 (3p24.3), NFKBIZ (3q12.3) and CAMK2G (10q22.2). We further demonstrate that NFKBIZ is a TRAF3IP2-dependent target of IL-17 signalling in human skin keratinocytes, thereby functionally linking two strong candidate genes. These results further integrate the genetics and immunology of psoriasis, suggesting new avenues for functional analysis and improved therapies.
Similar content being viewed by others

Introduction
Psoriasis is a chronic immune-mediated skin disease with complex genetic architecture that affects ∼2% of the world population. It presents a significant economic burden for affected individuals and for society1, and can severely impact the patient’s quality of life2. Aided by advances in high-throughput genotyping technologies, genome-wide association studies have identified multiple genetic loci associated with psoriasis3,4,5,6. These studies have been accompanied by replication of the most promising signals to confirm the psoriasis-associated signals at genome-wide significance levels. More recently, large consortia have utilized meta-analyses to identify additional susceptibility loci with modest effect sizes7,8.
Previously, we performed a meta-analysis combining three existing GWAS [the Collaborative Association Study of Psoriasis (CASP)3, Kiel5, the Wellcome Trust Case Control Consortium 2 (WTCCC2)6 and two Immunochip data sets (the Genetic Analysis of Psoriasis Consortium (GAPC) and the Psoriasis Association Genetics Extension (PAGE))) and identified 15 novel psoriasis susceptibility loci8, increasing the number of confirmed loci to 36 for populations of European descent.
In this study, we enhanced our previous meta-analysis8 and added replication data sets to follow up the most promising signals. First, we applied the optiCall9 genotype calling algorithm to the GAPC and PAGE data sets, and performed genome-wide imputation to 1,000 Genomes haplotypes. In this way, we were able to examine association at over six times the number of variants examined in our previous meta-analysis8. Using these enhancements, we perform a discovery meta-analysis of the combined GAPC/PAGE Immunochip data set and the three psoriasis GWAS3,5,6,7, comprising 10,262 psoriasis cases and 21,871 controls. We then conduct replication analysis in 5,033 cases and 5,707 controls from four independent data sets, resulting in a combined analysis involving over 15,000 cases and 27,000 controls. We report five novel psoriasis susceptibility loci reaching genome-wide significance (P<5 × 10−8), which add to the collective understanding of the genetic basis of this common cutaneous disorder.
Results
Discovery meta-analysis
The discovery meta-analysis included the combined GAPC/PAGE Immunochip data set and three existing GWAS data sets (that is, CASP, Kiel and WTCCC2). We first performed genome-wide imputation of each data set using 1000 Genomes haplotypes as a reference panel10. After imputation, we tested association for 696,365 markers (52,444 of them are short insertion/deletion variations, INDELs) that had minor allele frequencies (MAF) ≥1% and imputation quality (r2) >0.7 in all the four data sets. This study analysed more than six times the 111,236 markers examined in the original meta-analysis8.
We have shown that linear mixed modelling could be used to correct population stratification for association analysis11 in the PAGE and GAPC data sets separately8. This is also the case in the current combined Immunochip data set consisting of 6,268 psoriasis cases and 14,172 controls, with the genomic inflation factor estimated to be 0.96 (Supplementary Fig. 1). The genomic inflation factors for the CASP, Kiel and WTCCC2 GWAS data were estimated to be 1.01, 1.04 and 1.04, respectively.
Among the 36 known psoriasis susceptibility loci, 35 yielded strong evidence of association (effective sample-size weighted z-score: Pdiscovery-meta≤5 × 10−7), of which 30 achieved genome-wide significance (Pdiscovery-meta≤5 × 10−8) in the discovery meta-analysis (Supplementary Information and Supplementary Fig. 2) involving 10,262 cases and 21,871 controls. The only previously described locus that did not yield association in this study maps near IL28RA, in which the strongest previously identified signals6 failed our quality filters in one of the GWAS data sets. However, the Immunochip data alone does show suggestive association in the IL28RA region (EMMAX: PImmunochip=3.5 × 10−7).
Combined analysis identifies five new loci
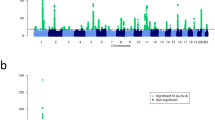
In the discovery meta-analysis, we identified six novel loci showing significant association with Pdiscovery-meta≤5 × 10−8 (Table 1, Fig. 1 and Supplementary Table 1). We evaluated the most strongly associated markers (that is, the ‘best markers’) identified in the new loci, and all of them have good imputation quality and none exhibited significant heterogeneity across data sets (all heterogeneity P-values>0.1; Supplementary Table 2). We then expanded our analysis utilizing genotyping data from four independent replication data sets, utilizing either the best markers or their best linkage disequilibrium (LD) proxies if LD-r2 ≥0.8. Notably, five of the six loci retained genome-wide significance in the combined meta-analysis (Table 2; Supplementary Table 3). Because one of the best markers (rs4685408) was genotyped separately in a substantial fraction (3,030 cases and 2,859 controls) of two of our replication data sets (that is, Exomechip 2 and PsA GWAS; Table 2), and the proxies for this marker in the two data sets were among the weakest of those listed in Table 2, we also treated this data as an additional independent data set (termed as ‘Michigan Genotyping’ in Supplementary Table 3; logistic regression: PMichigan=9 × 10−5; combined meta-analysis: Pcombined-meta=9 × 10−15). In all, the combined analysis consists of around 15,000 psoriasis cases and over 27,000 controls.

(a–f) This figure depicts LocusZoom-generated association plots for the six psoriasis susceptibility loci identified in the discovery meta-analysis (effective sample-size weighted approach). All but the 15q25.3 locus (f) showed genome-wide significant results in the combined meta-analysis.
The estimated odds ratios (ORs) for the five confirmed novel loci ranged from 1.12 to 1.17 (Table 1), similar to the 15 new loci identified in the original meta-analysis8. Among the five novel loci, 5q13.1 has the highest effect size (OR=1.17). Interestingly, this signal is situated in an intergenic region (Fig. 1), and was previously identified as a susceptibility locus for other autoimmune diseases including inflammatory bowel disease and multiple sclerosis (Table 3). While additional comparisons and more well-powered studies are needed, none of the five novel loci reported here have been identified as genome-wide psoriasis susceptibility loci in non-European samples4,12,13,14,15 (Supplementary Table 4).
As assessed using ANNOVAR16, the strongest signals from three of the confirmed loci map to intronic regions (Table 1), and the strongest signals from the other two loci map to intergenic regions. Using 1000 Genomes Project data, we did not identify any common (MAF>1%) protein-altering variants (that is, missense/nonsense mutations) in high LD (LD-r2 ≥0.8) with our strongest signals. We also performed conditional and interaction analyses using the five new loci identified in this study and did not identify any independent secondary signals within the five loci or evidence for epistasis effects among these loci or with previously described psoriasis loci.
Biological inferences for identified loci
Nearby genes within the three non-intergenic susceptibility loci (within ±200 kb boundary of the strongest signals) include: PLCL2 on 3p24.3; NFKBIZ, ZPLD1 and CEP97 on 3q12.3; and CAMK2G, FUT11, AGAP5, PLAU and MYOZ1 on 10q22.2 (Fig. 1). Among the above genes, NFKBIZ and MYOZ1 were differentially expressed when comparing psoriatic and normal skin samples17: NFKBIZ was 4-fold up-regulated (Wilcoxon rank-sum test: P=1.1 × 10−28) and MYOZ1 was down-regulated (fold change=0.5; P=3.5 × 10−14) in lesional psoriatic skin versus normal skin.
We searched the NHGRI catalogue 18 for previously identified genome-wide significant (P≤5 × 10−8) loci within ±200 kb of the best signals from the five novel loci identified in this study. Four of the five new loci are shared with other complex traits, most of which are immune-mediated inflammatory disorders (Table 3). For the most part, however, the risk variants from psoriasis and the other complex traits that map to the same loci are not in LD, meaning that the psoriasis risk haplotypes do not tend to be the same as those found for the other traits (Table 3). Although intergenic in nature, the 5p13.1 locus is shared with four other complex diseases, including allergy19, multiple sclerosis20, Crohn’s disease21,22 and ulcerative colitis23.
As the novel signals do not tend to be in LD with any amino-acid altering variants, we then investigated their potential regulatory roles. Of the five novel loci, four of them overlapped with histone marks or transcription factor binding sites in lymphoblastoid cells or keratinocytes according to data from ENCODE24 (Supplementary Table 5). The transcription factors (for example, NF-κB, IRF4, BATF, JUN-D) for the binding sites overlapped by these loci tend to be involved in immune responses: NF-κB and IRF4 are important transcription factors that regulate both innate and adaptive immune response; and the BATF–JUN family protein complexes are essential for IRF4-mediated transcription in T cells25. We then asked whether our best markers from the novel loci are eQTLs and could alter the expression of nearby genes. Interestingly, we found that rs7637230 (P=2.22 × 10−8) and rs2675662 (P=5.78 × 10−10) are both cis-eQTLs in lymphoblastoid cells26, and affect the expression levels of NFKBIZ and FUT11, respectively.
The IL-17 pathway has been shown to play an important role in psoriasis27. TRAF3IP2, a susceptibility locus for both psoriasis and psoriatic arthritis5,8,28, encodes adaptor protein Act1, one of the key components in the IL-17 signalling pathway29. NFKBIZ, a gene in one of the newly discovered loci of this study, has been shown to be an Act1-dependent IL-17 target gene in mice30,31. Because psoriasis is a uniquely human disease, and because keratinocytes appear to be a key target of IL-17 action in psoriasis27, we set out to determine whether NFKBIZ is also an Act1-dependent target of IL-17 signalling in human keratinocytes. To do this, we investigated the time course of NFKBIZ expression in human keratinocytes engineered to silence TRAF3IP2 expression under the control of tetracycline. As shown in Fig. 2, expression of NFKBIZ mRNA and protein could be significantly induced by IL-17 (P<0.01), and these inductions were significantly (P<0.01) blocked by TRAF3IP2 silencing. As illustrated above, variant rs7637230 is in strong LD (r2≥0.8) with markers that overlap with chromatin marks in lymphoblastoid cells and keratinocytes (Supplementary Table 5) and it is an eQTL for the expression of NFKBIZ in lymphoblastoid cells26. Together with the results from our NFKBIZ experiments, these findings nominate NFKBIZ as a strong candidate gene in the 3q12 locus and suggest a potential disease mechanism of the IL-17 pathway in psoriasis.

(a) Time courses of TRAF3IP2 (top) and NFKBIZ (bottom) mRNA expression following IL-17 induction. Black bars represent mRNA relative expression levels measured by qPCR in the absence of tetracyline (Tet) while white bars represent levels after silencing TRAF3IP2 expression via Tet-inducible expression of shTRAF3IP2. Bars are mean+s.e.m. of three independent experiments and **P<0.01; Student’s t-test. (b) Western blot analysis of time courses of IκB-zeta and Act1 expression (upper panel) and quantifications of IκB-zeta band intensity relative to β-actin. Bars are mean+s.d. of two independent experiments (lower panel).
Discussion
In this study, we identified five novel psoriasis susceptibility loci reaching genome-wide significance, increasing the number of known psoriasis susceptibility loci to 41 in Caucasians and to 49 worldwide (Supplementary Table 4).
We performed three main procedures to ensure the validity of the identified novel loci in this study: First, to ensure that the imputed dosage of each marker is accurate and to avoid batch effects, we required a stringent imputation quality threshold (r2≥0.7). Furthermore, we only considered markers that passed the quality threshold in all the four data sets when performing the associations (Supplementary Table 2). Second, we calculated the heterogeneity P-value from the meta-analysis to be sure that the associations were not heterogeneous (P>0.05) among the data sets (Supplementary Table 2). Finally, we performed a replication analysis to further validate that the 5 loci still achieve genome-wide significance in the combined meta-analysis (Table 2).
Although estimates of allele frequencies based on imputed dosages will have added uncertainty compared with those based on experimentally determined genotypes, the allele frequencies reported in our study do show consistent differences between cases and controls for each of the associated markers across different data sets (Supplementary Table 2). We also compared risk allele frequencies for 41 psoriasis susceptibility loci across the four discovery data sets, and we observed very high concordance (r2≥0.99) of the estimated frequencies in controls for all six possible pairs of data sets (Supplementary Fig. 3).
Three of the five newly identified loci contained protein-coding genes; however, we found no evidence suggesting that our signals are missense or nonsense mutations, nor are they in LD with such variations. Our results are in agreement with data for other known psoriasis susceptibility loci, in that fewer than 25% of the psoriasis-associated signals are in LD with codon-changing variations8. Although no deleterious functional variants were identified in the three protein-coding loci (PLCL2 at 3p24.3; NFKBIZ, ZPLD1 and CEP97 at 3q12.3; and CAMK2G, FUT11, AGAP5, PLAU and MYOZ1 at 10q22.2), variation in PLCL2 has previously been shown to be associated at genome-wide significance (P=2.3 × 10−8) with primary biliary cirrhosis32 and nominally (P=1.7 × 10−3) in a cohort of 982 Caucasian cases of psoriatic arthritis and 8,676 Caucasian controls 33. In mice, NFKBIZ deletion has been functionally associated with inflammatory skin eruptions34 and CAMK2G has been functionally implicated in thymic development35. In addition, PLAU, encoding urokinase-type plasminogen activator, has been reported to be overexpressed in psoriatic skin36 and was up-regulated 1.49-fold (P=3.7 × 10−13) in our psoriasis RNA-seq transcriptome data, albeit short of the 2-fold change threshold we used to declare significance17. Of the remaining protein-coding genes in these three loci, MYOZ1 encodes myozenin, a muscle protein of no obvious relevance to psoriasis, and very little information is available for ZPLD1, CEP97 and AGAP5 in Online Mendelian Inheritance in Man37. The identification of the disease-associated single-nucleotide polymorphisms (SNPs) rs7637230 (P=2.22 × 10−8) and rs2675662 (P=5.78 × 10−10) as cis-eQTLs in lymphoblastoid cells26 prioritizes NFKBIZ and FUT11 as strong candidate genes in their respective loci.
Several recent clinical studies have shown benefit of blockade of IL-17 and its receptors in psoriasis38. In this context, the connection between NFKBIZ and TRAF3IP2 is of biological and therapeutic interest. TRAF3IP2 encodes Act1 (also known as CIKS), an ubiquitin ligase that binds to IL-17 receptors39,40. NFKBIZ encodes IκB-zeta, a transcriptional regulator that binds to the p50 subunit of NF-kB41. Act1 and IκB-zeta are required for IL-17-dependent signalling associated with autoimmune and inflammatory diseases42,43. Nfkbiz-deficient mice manifest defective Th17 development, and IκB-zeta regulates this process by cooperating with ROR nuclear receptors43. While Act1 has previously been shown to regulate Nfkbiz expression in mouse embryonic fibroblasts30 and mouse skin keratinocytes31, this is the first demonstration of Act1-dependent NFKBIZ/IκB-zeta expression in human keratinocytes. Interestingly, TNFAIP3 encoding A20 is also a strong candidate gene in psoriasis and several other inflammatory diseases, which appears to act as a brake on cytokine-medicated signalling44. Interestingly TNFAIP3 is also an Act1-dependent target of IL-17 signalling, which, in turn, functions as a negative regulator of IL-17 receptor function29. These results highlight NFKBIZ as an IL17 target and identify an immunoregulatory network downstream of the IL-17 receptor involving at least three psoriasis candidate genes: TRAF3IP2, NFKBIZ and TNFAIP3.
Evidence is accumulating to indicate that genetic variants in regulatory regions may play important biological roles in complex genetic disorders45,46, and large scale projects such as ENCODE24 have been using sequencing technologies and integrative approaches to illuminate the functional elements of the human genome. Our results illustrate the potential regulatory roles of the psoriasis susceptibility loci, and will facilitate the design of future functional analyses.
Similar to other complex traits and diseases47,48, large inter-continental consortia have been forming to gather hundreds of thousands of samples to identify susceptibility loci with very mild effect sizes for psoriasis. Moreover, the study of psoriasis genetics is entering a new era, with more efforts on investigating the missing heritability explained by secondary independent signals (fine-mapping analysis49,50) and rare variants (by using exome-array or target-/exome-sequencing15) in known loci. Not only will these studies provide a higher explained variance by the genetic components, more importantly, they will also shed light on the pathogenesis and disease mechanisms, and ultimately provide new approaches and targets for effective drug discovery.
Methods
Discovery meta-analysis data sets
Samples were collected at the participating institutions after obtaining informed consent, and enrolment of human subjects for this study was approved by the following ethics boards: UK samples—St Thomas' Hospital Research Ethics Committee; Estonian Biobank—Research Ethics Committee of the University of Tartu; Michigan samples—Institutional Review Board of the University of Michigan Medical School; German samples—Ethical review board of the Medical Faculty of the CAU (Christian-Albrechts-University of Kiel, Germany); Toronto samples—the ethics board is the University Health Network; Swedish samples—the ethics board is the Local Ethical Review Board at Linkóping University; Newfoundland samples—the ethics board is the Health Research Ethics Authority; Utah samples—the ethics board is the Institutional Review Board of the University of Utah. We first performed the discovery meta-analysis using four data sets: (i) combined GAPC/PAGE Immunochip data set; (ii) CASP GWAS; (iii) Kiel GWAS; and (iv) WTCCC2 GWAS. Since the algorithm implemented in optiCall, which uses both within- and across-sample signal intensities, can outperform standard methods (for example, GenCall or GenoSNP)9 when applied to Immunochip, we used optiCall9 to recall the PAGE and GAPC data sets. We removed samples with outlying intensity values based on optiCall default settings by using the ‘–meanintfilter’ flag. Samples with >2% missing genotypes and markers with >5% missing values were rejected. We merged the PAGE and GAPC data sets to form a unified Immunochip data set, re-applying the above call rate criteria to the combined data. These and subsequent genotype-dependent quality control procedures resulted in the removal of 1,222 (5.6%) samples from this analysis, compared with the previous study8. The quality-controlled Immunochip data set consisted of 2,858 cases and 8,636 controls in GAPC, and 3,410 cases and 5,536 controls in PAGE. For each of the three GWAS data sets, we first obtained the quality-controlled genotyped data used by the previous studies3,5,6. We then applied additional filters (sample/marker call rate ≥0.95; HWE P≥1 × 10−6) to the genotyped data before phasing/imputation (see below). Supplementary Table 6 shows the quality measurements for each data set.
Imputation and association
For each of the GWAS/Immunochip data sets, we performed haplotype phasing of the genotypes using ShapeIT51, we then performed imputation using minimac52 using haplotypes from the EUR subset of version 3 of the 1000 Genomes Project Phase I release as the reference panel10. After imputation, we analysed markers with minor allele frequency (MAF) ≥1% and with imputation quality r2≥0.7 in all the four data sets.
For the Immunochip data, a linear mixed model was used to perform association tests as implemented in EMMAX11. The kinship matrix was computed using common (MAF>1%) independent markers located outside the 36 known psoriasis loci, having PGWA≥0.5 (where PGWA represents the association P-values obtained from the meta-analysis using only the three independent GWAS data sets; this choice is common for analyses using ImmunoChip markers which are enriched for variants associated with psoriasis in our original GWAS). Using the same procedures described in the previous study for quality control8 (that is, SNPs with call rate <95% or with a Hardy–Weinberg equilibrium P-value <1 × 10−6 were excluded; samples with <95% call rate were excluded), we performed logistic regression on the three GWAS data sets (that is, CASP, Kiel and WTCCC2), using principal components as covariates to correct for population stratification3,5,6.
Discovery meta-analysis
We used METAL to perform meta-analysis, using the sample-size weighted approach53, adjusting the genomic control inflation factor separately for each data set. To estimate the odds ratios (ORs) for each marker, we first computed the ORs from the Immunochip data using logistic regression, and then used a sample-size weighted approach to compute the ORs from the full data sets8. The regional association plots were created by using the software LocusZoom54. We used ANNOVAR to perform variant annotations16 using Gencode v19 (ref. 55) as gene references.
Replication data sets and combined meta-analysis
For each of the most strongly associated genome-wide significant (P<5 × 10−8) markers (that is, the ‘best markers’) from the novel signals identified in the discovery meta-analysis, we used genotyping data obtained from four different collaborative data sets (here referred to as Exomechip 1, Exomechip 2, PsA GWAS and Genizon GWAS) for replication. If the best associated markers were not genotyped in the replication data sets, we identified the best proxies based on the LD (LD-r2) in the European ancestry subset of version 3 of the 1000 Genomes Project Phase I release10. Only proxies with LD-r2≥0.8 against our best signals were considered. Using only genetically independent samples, the 5,033 additional cases and 5,707 controls were combined with the discovery meta-analysis using the sample-size weighted approach53.
NFKBIZ expression
N/TERT-TR keratinocytes were engineered to express a tetracycline (Tet)-inducible shRNA as described56. These cells (N/TERT-TR-shTRAF3IP2) were seeded at 8,000 cells cm−2 in the presence or absence of 1 μg ml−1 Tet. When confluent (after 7 days), cells were stimulated for 1 to 4 h with 200 ng ml−1 recombinant human (rh) IL-17A (R&D Systems). RNA was isolated using RNAeasy columns (Qiagen) and reverse-transcribed using the High Capacity cDNA Reverse Transcription Kit (Life Technologies). Quantitative real-time polymerase chain reaction (qPCR) was performed using Taqman primers (Life Technologies) specific for nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, zeta (NFKBIZ, Hs00230071-m1), TNF receptor associated factor 3 interacting protein 2 (TRAF3IP2, Hs00974570-m1) and the control gene RPLP0 (Hs99999902_m1). From the same experiments, NFKBIZ and TRAF3IP2/Act1 proteins were assessed by western blotting, utilizing rabbit polyclonal IgG (respectively from Cell Signal Transduction #9244 and Santa Cruz Biotechnology sc-11444, both diluted 1:1,000) to detect protein, with β-actin (Cell Signal Transduction #4967 dilution 1:5,000) as a loading control. Statistical analysis was performed using GraphPad software (Prizm).The uncropped scans of the blots are shown in Supplementary Fig. 4.
Overlap with ENCODE genomic features
We investigated whether the best signals identified from the novel loci are in LD with markers within chromatin marks or transcription factor binding sites. For each best associated marker, we first identified a set of high LD-markers based on the European population of the 1000 Genomes. We then examined human keratinocytes and lymphoblastoid cell lines (relevant cell types for psoriasis) and identified any chromatin marks or transcription factor binding sites overlapping with the LD-marks.
Additional information
How to cite this article: Tsoi, L. C. et al. Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci. Nat. Commun. 6:7001 doi: 10.1038/ncomms8001 (2015).
References
Mustonen, A., Mattila, K., Leino, M., Koulu, L. & Tuominen, R. The costs of psoriasis medications. Dermatol. Ther. (Heidelb) 3, 169–177 (2013).
Hawro, T. et al. Impact of psoriasis severity on family income and quality of life. J. Eur. Acad. Dermatol. Venereol. 29, 438–443 (2014).
Nair, R. P. et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 41, 199–204 (2009).
Sun, L. D. et al. Association analyses identify six new psoriasis susceptibility loci in the Chinese population. Nat. Genet. 42, 1005–1009 (2010).
Ellinghaus, E. et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat. Genet. 42, 991–995 (2010).
Genetic Analysis of Psoriasis, C.. et al. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 42, 985–990 (2010).
Stuart, P. E. et al. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat. Genet. 42, 1000–1004 (2010).
Tsoi, L. C. et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 44, 1341–1348 (2012).
Shah, T. S. et al. optiCall: a robust genotype-calling algorithm for rare, low-frequency and common variants. Bioinformatics 28, 1598–1603 (2012).
1000 Genomes Project Consortium. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Kang, H. M. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010).
Li, Y. et al. Association analyses identifying two common susceptibility loci shared by psoriasis and systemic lupus erythematosus in the Chinese Han population. J. Med. Genet. 50, 812–818 (2013).
Zhang, X. J. et al. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat. Genet. 41, 205–210 (2009).
Sheng, Y. et al. Sequencing-based approach identified three new susceptibility loci for psoriasis. Nat. Commun. 5, 4331 (2014).
Tang, H. et al. A large-scale screen for coding variants predisposing to psoriasis. Nat. Genet. 46, 45–50 (2014).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Li, B. et al. Transcriptome analysis of psoriasis in A large case-control sample: RNA-Seq provides insights into disease mechanisms. J. Invest. Dermatol. 134, 1828–1838 (2014).
Welter, D. et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–D1006 (2014).
Hinds, D. A. et al. A genome-wide association meta-analysis of self-reported allergy identifies shared and allergy-specific susceptibility loci. Nat. Genet. 45, 907–911 (2013).
Sawcer, S. et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 (2011).
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Julia, A. et al. A genome-wide association study on a southern European population identifies a new Crohn's disease susceptibility locus at RBX1-EP300. Gut 62, 1440–1445 (2013).
Anderson, C. A. et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 43, 246–252 (2011).
Bernstein, B. E. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Li, P. et al. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 490, 543–546 (2012).
Liang, L. et al. A cross-platform analysis of 14,177 expression quantitative trait loci derived from lymphoblastoid cell lines. Genome Res. 23, 716–726 (2013).
Lowes, M. A., Suarez-Farinas, M. & Krueger, J. G. Immunology of psoriasis. Annu. Rev. Immunol. 32, 227–255 (2014).
Huffmeier, U. et al. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat. Genet. 42, 996–999 (2010).
Gaffen, S. L., Jain, R., Garg, A. V. & Cua, D. J. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol. 14, 585–600 (2014).
Chang, S. H., Park, H. & Dong, C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J. Biol. Chem. 281, 35603–35607 (2006).
Sonder, S. U., Paun, A., Ha, H. L., Johnson, P. F. & Siebenlist, U. CIKS/Act1-mediated signaling by IL-17 cytokines in context: implications for how a CIKS gene variant may predispose to psoriasis. J. Immunol. 188, 5906–5914 (2012).
Mells, G. F. et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 43, 329–332 (2011).
Bowes, J. et al. Comprehensive assessment of rheumatoid arthritis susceptibility loci in a large psoriatic arthritis cohort. Ann. Rheum. Dis. 71, 1350–1354 (2012).
Yamamoto, M. et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature 430, 218–222 (2004).
Bui, J. D. et al. A role for CaMKII in T cell memory. Cell 100, 457–467 (2000).
Spiers, E. M., Lazarus, G. S. & Lyons-Giordano, B. Expression of plasminogen activator enzymes in psoriatic epidermis. J. Invest. Dermatol. 102, 333–338 (1994).
Hamosh, A., Scott, A. F., Amberger, J., Valle, D. & McKusick, V. A. Online Mendelian Inheritance in Man (OMIM). Hum. Mutat. 15, 57–61 (2000).
Martin, D. A. et al. The emerging role of IL-17 in the pathogenesis of psoriasis: preclinical and clinical findings. J. Invest. Dermatol. 133, 17–26 (2013).
Liu, C. et al. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci. Signal. 2, ra63 (2009).
Sonder, S. U. et al. IL-17-induced NF-kappaB activation via CIKS/Act1: physiologic significance and signaling mechanisms. J. Biol. Chem. 286, 12881–12890 (2011).
Yamazaki, S., Muta, T. & Takeshige, K. A novel IkappaB protein, IkappaB-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappaB in the nuclei. J. Biol. Chem. 276, 27657–27662 (2001).
Qian, Y. et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 8, 247–256 (2007).
Okamoto, K. et al. IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 464, 1381–1385 (2010).
Martin, F. & Dixit, V. M. A20 edits ubiquitin and autoimmune paradigms. Nat. Genet. 43, 822–823 (2011).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Sakabe, N. J., Savic, D. & Nobrega, M. A. Transcriptional enhancers in development and disease. Genome Biol. 13, 238 (2012).
Okada, Y. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014).
Willer, C. J. et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 45, 1274–1283 (2013).
Knight, J. et al. Conditional analysis identifies three novel major histocompatibility complex loci associated with psoriasis. Hum. Mol. Genet. 21, 5185–5192 (2012).
Fan, X. et al. Fine mapping of the psoriasis susceptibility locus PSORS1 supports HLA-C as the susceptibility gene in the Han Chinese population. PLoS Genet. 4, e1000038 (2008).
Delaneau, O., Marchini, J. & Zagury, J. F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2012).
Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G. R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 (2012).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Stoll, S. W., Johnson, J. L., Li, Y., Rittie, L. & Elder, J. T. Amphiregulin carboxy-terminal domain is required for autocrine keratinocyte growth. J. Invest. Dermatol. 130, 2031–2040 (2010).
Acknowledgements
Major support for this study was provided by the National Institutes of Health (R01AR042742, R01AR050511, R01AR054966, R01AR062382, R01AR065183 to JTE), the Wellcome Trust and the German Research Foundation. We acknowledge use of the British 1958 Birth Cohort DNA collection, funded by the Medical Research Council (G0000934) and the Wellcome Trust (068545/Z/02), and of the UK National Blood Service controls funded by the Wellcome Trust. F.C., R.C.T. and J.N.W.N.B. were supported by the Medical Research Council Stratified Medicine award (MR/L011808/1). A.F. and E.E. received infrastructure support through the DFG Clusters of Excellence 306 ‘Inflammation at Interfaces’ and is supported by the German Ministry of Education and Research (BMBF) through the e:Med sysINFLAME grant. JEG was supported by Doris Duke Foundation. We acknowledge support from the Department of Health via the NIHR comprehensive Biomedical Research Center award to GSTT NHS Foundation Trust in partnership with King’s College London and KCH NHS Foundation Trust. We acknowledge the Collaborative Association Study of Psoriasis (CASP) and the Wellcome Trust Case Control Consortium 2 (WTCCC2) for the contribution of GWAS data, as well as the provision of control DNA samples by the Cooperative Research in the Region of Augsburg (KORA) and genotyping data generated by the Dietary, Lifestyle and Genetic determinants of Obesity and Metabolic syndrome (DILGOM) consortium. We also thank the Genetic Analysis of Psoriasis Consortium (GAPC) and Psoriasis Association Genetics Extension (PAGE) for the contribution of Immunochip data. We thank the Barbara and Neal Henschel Charitable Foundation for their support of the National Psoriasis Victor Henschel BioBank. The Heinz Nixdorf Recall (HNR) cohort was established with the support of the Heinz Nixdorf Foundation. The Estonian Psoriasis cohort was supported by institutional research funding IUT20-46 of the Estonian Ministry of Education and Research, by the Centre of Translational Genomics of University of Tartu (SP1GVARENG) and by the European Regional Development Fund (Centre of Translational Medicine, University of Tartu). The genotyping of control individuals from the HNR cohort was financed by the German Federal Ministry of Education and Research (BMBF), within the context of the NGFNplus Integrated Genome Research Network MooDS (Systematic Investigation of the Molecular Causes of Major Mood Disorders and Schizophrenia). J.T.E. is supported by the Ann Arbor Veterans Affairs Hospital. Detailed consortium contributor lists for the Collaborative Association Study of Psoriasis, Genetic Analysis of Psoriasis Consortium, Psoriasis Association Genetics Extension, Cooperative Research in the Region of Augsburg and Heinz Nixdorf Recall (Risk Factors, Evaluation of Coronary Calcification, and Lifestyle) are listed in the Supplementary Note 1.
Author information
Authors and Affiliations
Contributions
J.T.E., R.C.T. and G.R.A. designed and directed the study. R.P.N., M.W., J.J.V., J.T.E., F.C., J.N.W.N.B., T.T., V.C., C.F.R., M.H.A., A.R., R.C.T., A.F., S.W., S.K., K.K., T.E., G.G.K., C.E., A.M., D.D.G., P.R. and S.L.S. contributed to sample collection and phenotyping. J.K. coordinated the GAPC samples and data sets. J.T.E. coordinated the PAGE samples and data sets. J.K., P.E.S, G.R.A. and H.M.K. advised on the statistical analysis. E.E. and R.P.N. performed the genotyping. S.L.S., L.C.T., and E.E. performed the genotype calling. L.C.T., S.L.S. and S.D. performed genotype imputation. L.C.T. performed statistical analysis. S.L. and S.W. performed the NFKBIZ expression experiments. F.C., T.T., S.L., S.W.S., J.E.G., J.N.W.N.B., R.C.T., J.T.E. and L.C.T. contributed to interpretation and biological inference for the results. L.C.T. and J.T.E. drafted the manuscript. L.C.T. and S.L. prepared the figures and tables. All the authors approved the final draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-4, Supplementary Tables 1-6 and Supplementary Note 1 (PDF 411 kb)
Rights and permissions
About this article

Cite this article
Tsoi, L., Spain, S., Ellinghaus, E. et al. Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci. Nat Commun 6, 7001 (2015). https://doi.org/10.1038/ncomms8001
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms8001
This article is cited by
-
Identification immune response genes in psoriasis after treatment with secukinumab
BMC Medical Genomics (2023)
-
Inflammatory and infectious upper respiratory diseases associate with 41 genomic loci and type 2 inflammation
Nature Communications (2023)
-
Shared genetic risk factors and causal association between psoriasis and coronary artery disease
Nature Communications (2022)
-
IκBζ controls IL-17-triggered gene expression program in intestinal epithelial cells that restricts colonization of SFB and prevents Th17-associated pathologies
Mucosal Immunology (2022)
-
Ischemic heart injury leads to HIF1-dependent differential splicing of CaMK2γ
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
