
Abstract
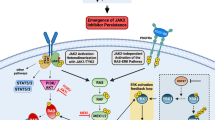
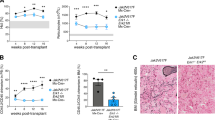
The identification of somatic activating mutations in JAK2 (refs 1–4) and in the thrombopoietin receptor gene (MPL)5 in most patients with myeloproliferative neoplasm (MPN) led to the clinical development of JAK2 kinase inhibitors6,7. JAK2 inhibitor therapy improves MPN-associated splenomegaly and systemic symptoms but does not significantly decrease or eliminate the MPN clone in most patients with MPN. We therefore sought to characterize mechanisms by which MPN cells persist despite chronic inhibition of JAK2. Here we show that JAK2 inhibitor persistence is associated with reactivation of JAK–STAT signalling and with heterodimerization between activated JAK2 and JAK1 or TYK2, consistent with activation of JAK2 in trans by other JAK kinases. Further, this phenomenon is reversible: JAK2 inhibitor withdrawal is associated with resensitization to JAK2 kinase inhibitors and with reversible changes in JAK2 expression. We saw increased JAK2 heterodimerization and sustained JAK2 activation in cell lines, in murine models and in patients treated with JAK2 inhibitors. RNA interference and pharmacological studies show that JAK2-inhibitor-persistent cells remain dependent on JAK2 protein expression. Consequently, therapies that result in JAK2 degradation retain efficacy in persistent cells and may provide additional benefit to patients with JAK2-dependent malignancies treated with JAK2 inhibitors.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
James, C. et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434, 1144–1148 (2005)
Kralovics, R. et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790 (2005)
Baxter, E. J. et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365, 1054–1061 (2005)
Zhao, R. et al. Identification of an acquired JAK2 mutation in polycythemia vera. J. Biol. Chem. 280, 22788–22792 (2005)
Pikman, Y. et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 3, e270 (2006)
Verstovsek, S. et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 363, 1117–1127 (2010)
Pardanani, A. et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J. Clin. Oncol. 29, 789–796 (2011)
Druker, B. J. et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 344, 1031–1037 (2001)
Flaherty, K. T. et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819 (2010)
Rosell, R. et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 361, 958–967 (2009)
Mok, T. S. et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957 (2009)
Kobayashi, S. et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792 (2005)
Pao, W. et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2, e73 (2005)
Gorre, M. E. et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 293, 876–880 (2001)
Engelman, J. A. et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043 (2007)
Pao, W. et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2, e17 (2005)
Johannessen, C. M. et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968–972 (2010)
Nazarian, R. et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977 (2010)
Azam, M., Latek, R. R. & Daley, G. Q. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 112, 831–843 (2003)
Shah, N. P. et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117–125 (2002)
Sharma, S. V. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010)
Parganas, E. et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 93, 385–395 (1998)
Ihle, J. N. & Gilliland, D. G. Jak2: normal function and role in hematopoietic disorders. Curr. Opin. Genet. Dev. 17, 8–14 (2007)
Mullighan, C. G. et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc. Natl Acad. Sci. USA 106, 9414–9418 (2009)
Marubayashi, S. et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Invest. 120, 3578–3593 (2010)
Koppikar, P. et al. Efficacy of the JAK2 inhibitor INCB16562 in a murine model of MPLW515L-induced thrombocytosis and myelofibrosis. Blood 115, 2919–2927 (2010)
Rider, L., Shatrova, A., Feener, E. P., Webb, L. & Diakonova, M. JAK2 tyrosine kinase phosphorylates PAK1 and regulates PAK1 activity and functions. J. Biol. Chem. 282, 30985–30996 (2007)
Andraos, R. et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2, 512–523 (2012)
Wang, Y. et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 114, 5024–5033 (2009)
Guerini, V. et al. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2V617F. Leukemia 22, 740–747 (2007)
He, H. et al. Identification of potent water-soluble purine-scaffold inhibitors of the heat shock protein 90. J. Med. Chem. 49, 381–390 (2006)
Mikkelsen, T. S. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 (2007)
Acknowledgements
We thank C. Sawyers, J. Licht, P. Poulikakos and N. Rosen for advice and suggestions; P. Bhatt for his help in the saturation mutagenesis screen; T. Taldone for synthesis of PU-H71; L. Staudt and T. Look for shRNA constructs against JAK2 and TYK2, respectively; and T. Radimerski and P. Manley for providing BBT-594. We are grateful to the Genomics Core Laboratories at Memorial Sloan-Kettering Cancer Center and the Geoffrey Beene Core for their assistance with 454 sequencing. This work was supported in part by National Cancer Institute grant 1R01CA151949-01 to R.L.L., by a grant from the Leukemia and Lymphoma Society to R.L.L. and by a grant from the Myeloproliferative Neoplasms Foundation and the Starr Cancer Consortium to R.L.L., B.E.B. and B.L.E. B.E.B. is a Howard Hughes Medical Institute Early Career Scientist.
Author information
Authors and Affiliations
Contributions
P.K. and R.L.L. conceived the project. P.K., N.B., O.K. and R.L.L. designed experiments. P.K., N.B., O.K., T.M., M.A., F.L., O.A.W., L.L., A.W., S.M. and A.G. performed experiments. P.K., N.B., T.H., M.G. and M.A. analysed data. L.M.S., A.M., B.L.E. and G.C. provided reagents. Z.E. and S.V. provided patient samples. P.K., N.B. and R.L.L. wrote the paper with input from S.V., Z.E., O.K., B.L.E., B.E.B. and S.D.N.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1-17 and Supplementary Tables 1-3. (PDF 2799 kb)
Rights and permissions
About this article
Cite this article
Koppikar, P., Bhagwat, N., Kilpivaara, O. et al. Heterodimeric JAK–STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 489, 155–159 (2012). https://doi.org/10.1038/nature11303
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature11303
This article is cited by
-
Role of allogeneic hematopoietic stem cell transplantation in patients with high-risk T-cell lymphoblastic leukaemia/lymphoma: an analysis of clinical outcomes
Bone Marrow Transplantation (2024)
-
AKT2S128/CCTαS315/319/323-positive cancer-associated fibroblasts (CAFs) mediate focal adhesion kinase (FAK) inhibitors resistance via secreting phosphatidylcholines (PCs)
Signal Transduction and Targeted Therapy (2024)
-
Targeting PP2A-dependent autophagy enhances sensitivity to ruxolitinib in JAK2V617F myeloproliferative neoplasms
Blood Cancer Journal (2023)
-
Research progress of additional pathogenic mutations in chronic neutrophilic leukemia
Annals of Hematology (2023)
-
Philadelphia-like acute lymphoblastic leukemia: the journey from molecular background to the role of bone marrow transplant—review article
Annals of Hematology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
