
Abstract
India has the highest number of retinoblastoma (RB) patients among the developing countries owing to its increasing population. Of the patients with RB, about 40% have the heritable form of the disease, making genetic analysis of the RB1 gene an integral part of disease management. However, given the large size of the RB1 gene with its widely dispersed exons and no reported hotspots, genetic testing can be cumbersome. To overcome this problem, we have developed a rapid screening strategy by prioritizing the order of exons to be analyzed, based on the frequency of nonsense mutations, deletions and duplications reported in the RB1-Leiden Open Variation Database and published literature on Indian patients. Using this strategy for genetic analysis, mutations were identified in 76% of patients in half the actual time and one third of the cost. This reduction in time and cost will allow for better risk prediction for siblings and offspring, thereby facilitating genetic counseling for families, especially in developing countries.
Similar content being viewed by others


Introduction
Retinoblastoma (RB) is the most common intraocular malignancy in children under 5 years of age.1 RB is primarily caused by biallelic inactivation of RB1 gene. By identifying mutations in the proband, a risk assessment can be made for siblings and offspring, thereby improving the management of the disease. Only the individuals identified as having germline RB1 mutations will require further follow-up. This provides cost benefit to the family through elimination of unnecessary ophthalmic surveillance in those who do not have the mutations.2, 3
The RB1 gene spans more than 180 kb on chromosome 13q14, which consists of 27 exons with no reported hotspots.4, 5 The spectrum of RB1 mutations includes point mutations, indels, large deletions and duplications.6 Given the large size of the gene and multiple dispersed exons, genetic analysis is time intensive and costly, which is a major hindrance for molecular diagnosis of RB in developing countries.
Methods such as karyotyping and Southern blot were initially employed to detect loss of RB1 at the chromosomal level.7, 8, 9, 10 With the evolution of molecular methods such as single-strand conformation polymorphism11 and denaturing high-performance liquid chromatography,12 detection of point mutations was made possible. However, these methods have a low mutation detection rate. In order to get an enhanced mutation detection rate of both deletions and point mutation, multi-technique approaches were developed using methods such as quantitative multiplex PCR and exon-by-exon sequencing.3, 5, 13, 14 Nevertheless, these approaches remain time consuming, labor-intensive, and expensive. Allele-specific PCR was used to provide molecular diagnosis of RB at a faster rate, but could detect only known mutations.15, 16 A recent study of Tunisian families has suggested a low cost approach for the analysis of heritable RB.17
Therefore, it is essential to develop a sensitive method of screening for RB1 mutations that reduces time and cost. We developed a new strategy for analysis of germline and somatic RB with this in mind. Our strategy is based on using the frequency and location of mutations reported in the published literature and databases. We utilized Sanger sequencing and Multiplex Ligation-dependent Probe Amplification (MLPA) in a four-step manner with selected exons to identify point mutations, large deletions and duplications.
Materials and methods
Samples
The present study is approved by the Institutional Review Board of Aravind Eye Care System, Madurai and the samples were collected after obtaining written informed consent from parents of RB patients. The study included 21 patients (13 bilateral and 8 unilateral) who were randomly selected out of 42 patients seen at Aravind Eye Hospital, Madurai between March 2013 and February 2014 (Table 1). RB was diagnosed after a complete clinical examination including computed tomography/magnetic resonance imaging, Ultrasonography B-scan (USB) and retcam imaging. When possible, tumor samples of the proband were the first choice of analysis in both unilateral and bilateral cases. Blood samples were collected from all probands and family members for further analysis.
Screening strategy for RB1
All variants including point mutations and indels, reported in RB1-Leiden Open Variation Database (rb1-lovd.d-lohmann.de) and published literature were collected. Pathogenic variants that include nonsense, frameshift and canonical splice variants were segregated from all reported variants. Our strategy was derived from the observation that nonsense mutations alone contribute about 50% of these pathogenic variants (Table 2). Nonsense mutations often occur in fragile codons wherein a single base change leads to a stop codon.18 When we looked for the fragile codons that frequently get mutated in RB1, we found CGA (Arg) fragile codon has a higher frequency than other fragile codons. With this precursory look, we analyzed the number of fragile codons present in each exon of RB1 versus nonsense mutations as shown in Figure 1. Accordingly, we grouped them into three clusters. The cluster containing eight exons (8, 10, 11, 14, 15, 17, 18 and 23) corresponds to 39% of nonsense mutations among the pathogenic variants, thus forming the first step of our strategy. The remaining two clusters of exons with 7% and 2% of nonsense mutations form step III and IV, respectively. As deletions and duplications reported in RB1 gene contribute 15–25% of all variants in RB patients (Table 3), the MLPA analysis of all the exons (except 15) of RB1 was included in our strategy as step II.

Frequency of nonsense mutations reported in RB1 gene. Each of the exons was plotted based on the frequency of nonsense mutations reported in RB1-Leiden Open Variation Database (x axis) and the number of fragile codons that can become stop codon by a single base change (y axis). Percentage refers to the frequency of nonsense mutations in the above three cluster of exons, among pathogenic variants given in Table 2. There were no nonsense mutations reported in exons 26 and 27. RB, retinoblastoma.
DNA isolation
Genomic DNA was isolated from blood samples by the salting out method26 and tumor samples by QIAamp DNA Mini Kit (Qiagen, Germantown, MD, USA) following the manufacturer’s protocol. The quality and quantity of the DNA was determined by Nanodrop 1000 spectrophotometer (Thermo Scientific, Waltham, MA, USA).
Sanger sequencing and MLPA
Sanger sequencing was performed to identify all the variants including point mutations and indels using the primers and conditions as described elsewhere.27 Briefly, each 20 μl reaction contained 40 ng of genomic DNA, 10 × PCR Buffer, 100 mm deoxy nucleotide tri phosphates (dNTPS), 10 μm of each forward and reverse primer and 2 U of Taq DNA polymerase (Sigma-Aldrich, St Louis, MO, USA). Cycle sequencing was performed using the BigDye Terminator kit version 3.1 (Thermo scientific) and purified products were analyzed on a 3130 Genetic Analyzer (Life Technologies, Carlsbad, CA, USA). Variants were then identified by aligning the sequences with the reference (NCBI accession no. L11910). Missense variants were selected if SIFT (Sorting Intolerant from Tolerant),28 PolyPhen229 and MutationTaster,30 all suggested pathogenic. Variants in intronic regions causing splicing alterations were chosen, if they are predicted by Human Splice Site Finder (http://www.umd.be/HSF/) and MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html).
MLPA was performed with SALSA MLPA kit P047-RB1 kit v.C1 (MRC-Holland, Amsterdam, The Netherlands) according to manufacturer’s instructions. Fragment analysis was performed with GeneMapper software (Life Technologies) and data were analyzed using Coffalyser software (MRC-Holland), where DNA copy number ratios of samples were computed using the matched normal controls. The threshold for recording duplications was a ratio of >1.3 and for deletion was a ratio of <0.7. Parents and siblings were screened for the presence of mutations identified in probands using the same methods described above.
Results
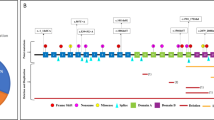
Using our four-step sequential screening strategy, we analyzed 21 (13 bilateral and 8 unilateral) RB patients and identified mutations in 16 within 5 weeks (Table 4). The mutation details of the 16 patients are presented in Figure 2.

Mutations identified in 16 retinoblastoma patients in four steps as described in Table 4. del, deletion; dup, duplication; RB, retinoblastoma.
In step I, we identified mutations in eight (six bilateral and two unilateral) patients, of whom five had a CGA to TGA (stop) codon mutations causing premature protein truncation. Among these five patients, a homozygous mutation in exon 15 (c.1399C>T p.R467X) was identified in the tumor sample of patient 5. We then screened the blood samples of the proband and family members and found the mutation to be de novo germline (Supplementary Figure 1A). In patient 12, a C>T change was observed in exon 8 (c.763C>T p.R255X) in a heterozygous state both in the proband tumor and blood, thus indicating that the mutation identified was the first hit and also a de novo germline event. In patient 4 (bilateral RB without family history), a homozygous somatic mutation in exon 10 (c.958C>T p.R320X) was found in the tumor but not in the blood. In patients 9 and 15, a C>T change was found in a heterozygous state in both the tumor and blood. De novo germline indels causing frameshift and premature protein termination were identified in exon 8 (c.792delAp.A264AfsX6) and exon 23 (c.2327insCp.S794RfsX19) in patient 11 and 14, respectively. A novel C>T change in intron 17 (c.1696-14) causing an aberrant splicing was seen in the blood sample of patient 6. Familial analysis showed the presence of this mutation in the unaffected father but not in the other family members.
In step II, analysis by MLPA was performed in the remaining 13 samples that did not show mutations in step I. We detected deletions/duplications in five (three unilateral and two bilateral) patients. Among the unilateral patients, deletion of the whole RB1 gene and exons 2–27 were detected in patient 8 and 10, respectively (Figure 2). In patient 3, duplication of the whole RB1 gene was observed in a heterozygous pattern. All three unilateral patients had mutations only in the tumor but not in the blood samples. Among the bilateral patients, patient 1 showed a heterozygous deletion of exons 18 and 19, which was found to be a de novo germline event. In patient 7, a heterozygous deletion of exon 10 was detected in the proband blood. This was found to have been inherited from the father who was diagnosed with regressed RB during clinical examination (Supplementary Figure 1B).
After step I and II, the remaining eight patients were analyzed for nine exons having moderate frequency of mutations in fragile codons in step III. An insertion of 'G' (c.2209insG p.Q735EfsX8) in exon 21 causing a frameshift mutation was observed in the blood sample of patient 13, who had bilateral disease. A missense mutation (c.2117G>A p.C706Y) in exon 21, predicted as pathogenic by SIFT, PolyPhen2 and MutationTaster, was identified in the tumor sample of patient 2, who had unilateral disease. This was also found to be present in the blood sample (Figure 2). In both cases, family members did not harbor these mutations.
The remaining six cases underwent step IV of our analysis. Here, the exons containing low-frequency mutations were examined. A change in a canonical splice site in intron 16 (c.1498+1G>A) was identified in the tumor of patient 16 (Figure 2).
Using our strategy we could identify oncogenic mutations in 76% of RB patients in our cohort of 21 patients within a short period of 5 weeks. Five patients (four bilateral and one unilateral), who did not show any mutations in our sequential analysis, were also found to be negative for the essential promoter region, as well as exons 26 and 27.
Discussion
Of all developing countries, India has the highest number of RB patients.31, 32 Therefore, efficient and rapid screening at a low cost is essential. To achieve this objective, we employed the Sanger sequencing and MLPA in a sequential four-step manner instead of analyzing all 27 exons at a time. The strategy we devised was based on the selection of exons reported to have a higher frequency of nonsense mutations as well as the deletions and duplications. As predicted, we found the highest number of mutations (50%) in the selected exons in step I, followed by 31% deletions and duplications in step II, 12.5% in step III and 6.25% in step IV (Figure 2).
For the detection of the nonsense mutations, allele-specific PCR was used in earlier studies, which could detect only known mutations.15, 16 However, we made use of Sanger sequencing of the eight exons in step I of our strategy and were thus able to detect novel variants including two indels and one splice variant as well. In step II of our strategy using MLPA, we detected five variants (four deletions and one duplication), the frequency of which was substantially higher than the predicted frequency of deletions and duplications in other ethnic groups. This increased ratio of deletions and duplications may be unique to the Indian population (Table 3). We have observed a low frequency of mutations (3 out of 16) in the exons of step III and IV. Thus, the increased number of mutations identified in step I and II compared with step III and IV justified our strategy developed for rapid genetic testing.
The comprehensive analysis of RB1 has been carried out by several groups.3, 5, 12, 13, 14, 33, 34 Recently, a stepwise protocol was developed for the genetic analysis of Tunisian families.17 Our strategy had notable differences, including the order of analysis and the method of selection of exons for various steps. For example, MLPA was included as the last step in the Tunisian study, whereas we employed it as the second step and could detect more number of deletions/duplications in our cohort of patients. Further, the number of exons analyzed in the first step in the Tunisian study was 12 compared with eight exons in our strategy with a comparable mutation detection rate. Increased sensitivity of genetic analysis in Tunisian families (95%) compared with our method (76%) may be attributed to the small set of samples in both the studies.
We have looked for the possible involvement of other exons of RB1 and other genes in the five patients who did not show RB1 mutations. Genetic analysis of exons 26 and 27 in these patients didnot show mutation, confirming the earlier report.17 Although there is no probe for exon 15 designed for MLPA, we did not see any additional bands denoting deletion in the gel electrophoresis of exons 15–16 amplicon (data not shown). We further analyzed the status of MYCN amplification in these five samples and found one (patient 18) tumor sample having MYCN amplification (>10 copies; data not shown). Other possible reasons may be RB1 inactivation by promoter methylation or mosaicism, which could possibly be detected by deep sequencing.35
Biallelic inactivation of RB1 gene is vital for tumor development.36 Of the nine tumor samples analyzed, we could not identify a second mutational event in seven samples. This could be attributed to methylation status that has not been studied by us. Another possibility is that the second allele presumably harbors an unidentified splicing mutation, which may give rise to an aberrant transcript.13 Alternative mechanisms of RB1 gene inactivation like chromothripsis, aberrant phosphorylation of pRB, degradation of the pRB protein by proapoptotic molecules may be involved.37, 38
Molecular genetic studies on RB have been carried out in India for the past 15 years and mutation detection rates have improved with time by adoption of newer methodologies. In a study by Kiran et al.,39 mutations were found in 22 out of 47 patients, a 47% detection rate. Parsam et al.14 showed a sensitivity of about 66% (49 out of 74 patients). In the present study, using the described strategy and taking into consideration one case with a high copy number of MYCN, the detection rate was as much as 81%. Since the first step could identify the mutations in about 50% of patients within 2 weeks (Table 4), the screening results could be delivered to clinic faster than the conventional exon by exon screening of RB1. In our strategy, the number of samples for further analysis is sequentially reduced. With the reduction in consumption of reagents and hands on time, genetic screening can be done with 1/3 of the cost compared with the analysis of all exons, confirming the earlier report.17 In addition, the time taken for the analysis of all 27 exons would be about 10 weeks, in contrast to 5 weeks following our strategy for the same number of samples. This reduction in cost and time will be beneficial for genetic counseling of patients’ families, especially in developing countries like India.
References
Chintagumpala, M., Chevez-Barrios, P., Paysse, E. A., Plon, S. E. & Hurwitz, R. Retinoblastoma: review of current management. Oncologist 12, 1237–1246 (2007).
Noorani, H. Z., Khan, H. N., Gallie, B. L. & Detsky, A. S. Cost comparison of molecular versus conventional screening of relatives at risk for retinoblastoma. Am. J. Hum. Genet. 59, 301–307 (1996).
Richter, S., Vandezande, K., Chen, N., Zhang, K., Sutherland, J., Anderson, J. et al. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am. J. Hum. Genet. 72, 253–269 (2003).
Friend, S.H., Bernards, R., Rogelj, S., Weinberg, R.A., Rapaport, J.M., Albert, D.M. et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323, 643–646 (1986).
Price, E. A., Price, K., Kolkiewicz, K., Hack, S., Reddy, M. A., Hungerford, J. L. et al. Spectrum of RB1 mutations identified in 403 retinoblastoma patients. J. Med. Genet. 51, 208–214 (2014).
Sellner, L. N., Edkins, E. & Smith, N. Screening for RB1 mutations in tumor tissue using denaturing high performance liquid chromatography, multiplex ligation dependent probe amplification, and loss of heterozygosity analysis. Pediatr. Dev. Pathol. 9, 31–37 (2006).
Toguchida, J., McGee, T. L., Paterson, J. C., Eagle, J. R., Tucker, S., Yandell, D. W. et al. Complete genomic sequence of the human retinoblastoma susceptibility gene. Genomics 17, 535–543 (1993).
Szijan, I., Lohmann, D. R., Parma, D. L., Brandt, B. & Horsthemke, B. Identification of RB1 germline mutations in Argentinian families with sporadic bilateral retinoblastoma. J. Med. Genet. 32, 475–479 (1995).
Harbour, J. W. Overview of RB gene mutations in patients with retinoblastoma. Implications for clinical genetic screening. Ophthalmology 105, 1442–1447 (1998).
Harini, R., Ata-ur-Rasheed, M., Shanmugam., M. P., Amali, J., Das, D. & Kumaramanickavel, G. Genetic profile of 81 retinoblastoma patients from a referral hospital in southern India. Indian J. Ophthalmol. 49, 37–42 (2001).
Sugano, K., Yoshida, T., Izumi, H., Umezawa, S., Ushiama, M., Ichikawa, A. et al. Outpatient clinic for genetic counseling and gene testing of retinoblastoma. Int. J. Clin. Oncol. 9, 25–30 (2004).
Houdayer, C., Gauthier-Villars, M., Laugé, A., Pagès-Berhouet, S., Dehainault, C., Caux-Moncoutier, V. et al. Comprehensive screening for constitutional RB1 mutations by DHPLC and QMPSF. Hum. Mutat. 23, 193–202 (2004).
Nichols, K. E., Houseknecht, M. D., Godmilow, L., Bunin, G., Shields, C., Meadows, A. et al. Sensitive multistep clinical molecular screening of 180 unrelated individuals with retinoblastoma detects 36 novel mutations in the RB1 gene. Hum. Mutat. 25, 566–574 (2005).
Parsam, V. L., Kannabiran, C., Honavar, S., Vemuganti, G. K. & Ali, M. J. A comprehensive, sensitive and economical approach for the detection of mutations in the RB1 gene in retinoblastoma. J. Genet. 88, 517–527 (2009).
Mamatha, G., Joseph, B., Shanmugam, M. P. & Kumaramanickavel, G. CGA codons multiplex PCR in rapid diagnosis of retinoblastoma. Indian J. Hum. Genet. 12, 34–38 (2006).
Saliminejad, K., Behnam, B., Akbari, M. T., Khorshid, H. R. K., Ghassemi, F., Amoli, F. A. et al. Rapid detection of RB1 recurrent mutations in retinoblastoma by ARMS-PCR. J. Genet. 92, e36–e40 (2013).
Ayari Jeridi, H., Bouguila, H., Ansperger-Rescher, B., Baroudi, O., Mdimegh, I., Omran, I. et al. Genetic testing in Tunisian families with heritable retinoblastoma using a low cost approach permits accurate risk prediction in relatives and reveals incomplete penetrance in adults. Exp. Eye Res. 124, 48–55 (2014).
Cusack, B. P., Amdt, P. F., Duret, L. & Crollius, H. Preventing dangerous nonsense: selection for robustness to transcriptional error in human genes. PLoS Genet. 7, e1002276 (2007).
Bamne, M. E., Ghule, P. N., Jose, J., Banavali, S. D., Kurkure, P. A. & Amare Kadam, P. S. Constitutional and somatic RB1 mutation spectrum in nonfamilial unilateral and bilateral retinoblastoma in India. Genet Test 9, 200–211 (2005).
Joseph, B., Raman, R., Uthra, S., Jagadeesan, M., Ganesh, A., Paul, P. G. et al. Genotype-phenotype correlation analysis in retinoblastoma patients from India. Asian Pac. J. Cancer Prev. 7, 619–622 (2006).
Albrecht, P., Ansperger-Rescher, B., Schüler, A., Zeschnigk, M., Gallie, B. & Lohmann, D. R. Spectrum of gross deletions and insertions in the RB1 gene in patients with retinoblastoma and association with phenotypic expression. Hum. Mutat. 26, 437–445 (2005).
Ahani, A., Behnam, B., Khorshid, H. R. & Akbari, M. T. RB1 gene mutations in Iranian patients with retinoblastoma: report of four novel mutations. Cancer Genet. 204, 316–322 (2011).
Ahani, A., Akbari, M. T., Saliminejad, K., Behnam, B., Akhondi, M. M. & Vosoogh, P. et al. Screening for large rearrangements of the RB1 gene in Iranian patients with retinoblastoma using multiplex ligation-dependent probe amplification. Mol. Vis. 19, 454–462 (2013).
Rushlow, D. E., Mol, B. M., Kennett, J. Y., Yee, S., Pajovic, S., Thériault, B. L. et al. Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol. 14, 327–334 (2013).
He, M-. Y., An, Y., Gao, Y-. J., Qian, X-. W., Li, G. & Qian, J. Screening of RB1 gene mutations in Chinese patients with retinoblastoma and preliminary exploration of genotype–phenotype correlations. Mol. Vis. 20, 545–552 (2014).
Miller, S. A., Dykes, D. D. & Polesky, H. F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 11, 1215 (1988).
Ganguly, A. & Shields, C. L. Differential gene expression profile of retinoblastoma compared to normal retina. Mol. Vis. 16, 1292–1303 (2010).
Ng, P. C. & Henikoff, S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Gallie, B. L. Key note lecture. World internet survey of classification of retinoblastoma. XI International Congress of Ocular Oncology, Hyderabad, India (2004).
Arora, R. S., Eden, T. O. B. & Kapoor, G. Epidemiology of childhood cancer in India. Indian J. Cancer 46, 264–273 (2009).
Lohmann, D. R., Brandt, B., Höpping, W., Passarge, E. & Horsthemke, B. The spectrum of RB1 germ-line mutations in hereditary retinoblastoma. Am. J. Hum. Genet. 58, 940–949 (1996).
Dommering, C. J., Mol, B. M., Moll, A. C., Burton, M., Cloos, J., Dorsman, J. C. et al. RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. J. Med. Genet. 51, 366–374 (2014).
Chen, Z., Moran, K., Richards-Yutz, J., Toorens, E., Gerhart, D., Ganguly, T. et al. Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum. Mutat. 35, 384–391 (2014).
Aerts, I., Lumbroso-Le Rouic, L., Gauthier-Villars, M., Brisse, H., Doz, F. & Desjardins, L. Retinoblastoma. Orphanet. J. Rare Dis. 1, 31 (2006).
Chau, B. N. & Wang, J. Y. Coordinated regulation of life and death by RB. Nat. Rev. Cancer 3, 130–138 (2003).
McEvoy, J., Nagahawatte, P., Finkelstein, D., Richards-Yutz, J., Valentine, M., Ma, J. et al. RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget 5, 438–450 (2014).
Kiran, V. S., Kannabiran, C., Chakravarthi, K., Vemuganti, G. K. & Honavar, S. G. Mutational screening of the RB1 gene in Indian patients with retinoblastoma reveals eight novel and several recurrent mutations. Hum. Mutat. 22, 339 (2003).
Acknowledgements
We acknowledge Aravind Eye Foundation, USA, for the funding. We specially thank Professor Arupa Ganguly, Department of Genetics, University of Pennsylvania, Philadelphia for providing the PCR conditions for exons 1 and 15 and also for the training of AVR in the molecular analysis of RB at her lab during his Indo-US Research Fellowship. We also thank Dr Ashley Campbell, Massachusetts Eye and Ear, Boston for language editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Thirumalairaj, K., Abraham, A., Devarajan, B. et al. A stepwise strategy for rapid and cost-effective RB1 screening in Indian retinoblastoma patients. J Hum Genet 60, 547–552 (2015). https://doi.org/10.1038/jhg.2015.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.62
