
Abstract
X-linked dominant hypophosphatemic rickets (XLHR), is characterized mainly by renal phosphate wasting with hypophosphatemia, short stature and abnormal bone mineralization. PHEX, located at Xp22.1–p22.2, is the gene causing XLHR. We aim to characterize the pathogenesis of a Chinese boy who is apparently ‘heterozygous’ in PHEX gene. Direct sequencing showed two peaks: one was a wild-type ’G’ and the other was one base substitution to ‘A’, though the patient was a male. TA clone assay clearly showed each sequences and the ratios. The mutation effect was predicted via bioinformatics and validated by exon-trapping assay. Real-time PCR was applied to determine the copy number of PHEX. TA clone assay showed the frequency of normal (G) to mutant allele (A) as 19:13. Normal karyotype and real-time PCR results indicate the normal copy number of PHEX. This splice site mutation leads to 4 bp of exon 18 skipping out causing frame shift p.Gly590Glufs*28 that ends up with a loss of active site and Zn2+-binding site of PHEX, which probably interfere with renal phosphate reabsorption and bone mineralization. In conclusion, mutation at conserved splice acceptor site resulted in aberrant splicing, ending up with a damaged protein product. This novel mutation is de novo in mosaic pattern that may be induced during early postzygotic period. Taking mosaic somatic mutation of PHEX into consideration is strongly suggested in genetic counseling and etiology research for XLHR.
Similar content being viewed by others

Introduction
X-linked hypophosphatemic rickets or X-linked vitamin D-resistant rickets (XLHR, MIM#307800) is a disorder associated with low levels of phosphate in the blood (hypophosphatemia) caused by a decreased renal tubular reabsorption of inorganic phosphates. Additionally, the normal physiological response of an elevation of 1,25(OH)2 vitamin D toward hypophosphatemia is absent. The incidence of XLHR is approximately 3.9–5/100 000 live birth.1 Clinical manifestations include short stature, bone pain, dental anomalies, lower extremity deformities and radiographic evidence of rickets in children.2, 3 XLHR is caused by mutations in PHEX gene that is located at Xp22.1-p22.2. PHEX encodes proteins that has an important role in regulation of phosphate transport and bone mineralization.4 PHEX protein shows high homology to membrane-bound zinc metallopeptidases with the metal-binding site and active site close to C termination.5
In this study, we report an unusual case in that a Chinese boy with XLHR was identified with an apparently ‘heterozygous’ de novo splice site mutation in PHEX gene. We examined the mutant frequency and gene copy number and analyzed the mutation effects, which implies that the patient is mosaic with normal and mutated PHEX gene.
Materials and methods
Patient
The proband is a 4.5-year-old boy, presenting gait abnormalities and bone pain for 2 years. He does not have any siblings. No relevant family history was recorded. His parents showed normal appearance with no positive symptoms. They claimed that karyotyping was performed for the proband with a normal 46, XY reported.
We performed physical examination, X-ray and ultrasonic examinations. We measured biochemical parameters and bone mineral density. Informed consent was obtained and the study was approved by the ethics committee of the School of Medicine, Zhejiang University.
Molecular diagnosis
The proband’s DNA was extracted from peripheral white blood cells by RelaxGene Blood DNA system (TianGen, Beijing, China). We designed 22 primers (Table 1) that cover all exons and their exon–intron boundaries of PHEX based on the reference sequence (NC_000023.11) using standard PCR followed by Sanger sequencing.
TA cloning and real-time PCR (quantitative PCR (qPCR))
Using the pMD19-T Vector Cloning Kit (Takara, Shiga, Japan) we cloned PCR products of PHEX-exon18. After transformation and overnight culture, 32 colonies were selected randomly and directly sequenced. Quantization of the gene copies of PHEX of the proband was completed by qPCR assay with ABI 7900HT (ABI, Los Angeles, CA, USA) Real-time PCR System using the primer PHEX-qPCR (Table 1). Two normal males and two females were taken into the assay as controls. β-Globin gene was tested as internal reference gene. SYBR Green was applied as nucleic acid stain (Bestar qPCR mastermix, DBI, Shanghai, China).
Computational analysis
After genotyping, the impact of this splicing mutation was evaluated by bioinformatic tools online: Human-Splicing-Finder (http://www.umd.be/HSF/), MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html), and NNSPLICE (http://www.fruitfly.org/seq_tools/splice.html).
Exon-trapping assay
To examine the predicted results, a mini-gene assay was carried out. We designed a pair of primers (Table 1, PHEX-et_18) covering exon 18 and exon–intron boundaries (943 bp). PCR products and pSPL3 vector (Invitrogen Corporation, Carlsbad, CA, USA) were double digested by EcoRI and XhoI followed by a ligation reaction. After transformation into competent cell DH5α, plasmid DNA was prepared by the NucleoBond Xtra Midi Kit (MN, Neumann-Neander-Straße, Germany) for transfection. African green monkey cell line COS7 was cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum in 5% CO2 at 37 °C. We transfected COS7 with empty pSPL3 vector as well as normal/mutant recombinant plasmids using PolyJet reagent (SignaGen Laboratories, Gaithersburg, MD, USA), respectively. After 24 h, cells were harvested and total RNA was extracted. Then reverse transcriptase–PCR was performed using primers SD6 and SA2 (Table 1) followed by agarose gel electrophoresis and sequencing.
Results
The patient was 93 cm (below 0.4th percentile), weighing 14 kg (2nd percentile), with a stature shorter than peers. The shape of his head became box-like (caput quadratum) owing to thickening of the parietal and frontal bones. The patient had a typical hypophosphatemia and elevated serum alkaline phosphatase for his age (Table 2). In addition, his chest and extremities X-ray indicated an enlargement of head of rib, decreased bone density and rough metaphysis of both bilateral scapula and upper segment of humerus. Bilateral left radius, distal end of femur, ulna, tibia and fibula had cup-shaped change, rough margin of metaphysis, uneven bone density and sparse bony trabeculae. Bilateral distal femoral ossification centers had uneven density and rough margin; bilateral femora slightly curved outward and medial tibial cortex thickened. Anteroposterior pelvis X-ray showed coarse bone trabecula, increased bone density and slightly wider space between bone trabecula that arrayed in a grid-like arrangement. The upper outer margins of both femoral heads were vague, while the central part had focal reduced bone density and patchy increased bone density. These physical examination and radiographic findings implied an active rickets. Biochemical testing showed a normal serum calcium level and typical hypophosphatemia. Therefore, this case was highly suspected to be a category of hereditary hypophosphatemic rickets.
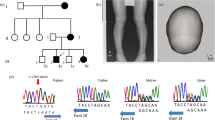
We first focused on PHEX gene as XLHR is the most common form of hereditary hypophosphatemic rickets. Through direct sequencing, a novel splice site mutation was found at c.1769-1 (IVS 17) on the PHEX gene of the proband, changing from G to A (Figure 1b), which has not been documented by 1000 Genomes Projects yet. This result indicated that the patient is likely affected with XLHR. Interestingly, rather than a single peak at the mutation site, the sequencing result showed two peaks (Figure 1b). This is not common considering that PHEX is located on X chromosome of the proband who is a male. In order to confirm the result and distinguish the different alleles, we carried out a TA clone assay and picked out 32 colonies. It is shown that both c.1769-1G and c.1769-1A indeed exist (Figures 1c and d) and the frequency of G to A is 19:13. Karyotype is claimed normal 46, XY (data not shown). qPCR was applied to further confirm this karyotype result and investigate the copy number of PHEX gene. The average 2−ΔΔct values of proband/control male and proband/control female are 1.002 and 0.541, respectively, which indicates that the patient has the same amount of gene copies of PHEX as normal males do (Figure 1e).

Sanger and TA clone sequencing and real-time PCR of proband. (a) Pedigree of PHEX family. Black symbol shows an affected individual for hypophosphatemic rickets. The letter ‘P’ and an arrow show the proband. (b) Sanger sequencing of the proband. The arrow indicates two peaks: one is a wild-type ‘G’ and the other is one base substitution to ‘A’. (c, d) show TA clone sequencing; the arrow shows the different alleles that are isolated (A/G). (e) Real-time PCR of PHEX gene including c.1769-1G>A mutation demonstrates that the proband has a normal copy number of PHEX as male controls do. Ctrl (M) and Ctr2 (M): male control; Ctr3 (F) and Ctr4 (F): female control. A full color version of this figure is available at the Journal of Human Genetics journal online.
In order to evaluate the impact of this mutation, Human Splicing Finder was applied to analyze the adjacent DNA sequence.6 It suggested that the native splicing acceptor (c.1769-2_c.1769-1) is destroyed, and there are two loci (c.1769-1_c.1769 and c.1771_c.1772), conforming to the criteria of a typical alternative splicing site, which would probably be adopted instead (Table 3).
The results of exon trapping, a cell-based assay, demonstrated that c.1771_c.1772 is actually the cryptic site. The first 4 bp (GTAG) of exon 18 were skipped out abnormally in the mutational group (Figure 2). A deletion of 4 bp in the transcripts will lead to a frame shift during translation (p.Gly590Glufs*28). A premature stop codon occurred at p.617 that truncated 133 amino acids.

pSPL3 exon-trapping analysis and sequencing. The illustrations of partial structure of the recombinant plasmids are shown at the bottom, with the wild type on the left and the mutant on the right. Agarose gel electrophoresis and sequencing of RT-PCR products are shown above. Lane M: DNA marker; Lane 1: normal splicing RT-PCR product (394 bp); Lane 2: aberrant splicing RT-PCR products losing 4 bp due to the splice site mutation (390 bp); Lane 3: empty pSPL3 vector (263 bp). A full color version of this figure is available at the Journal of Human Genetics journal online.
Discussion
Hereditary hypophosphatemic rickets are a group of genetically heterogeneous disorders. Mutations in PHEX gene cause the most common X-linked dominant form. Mutations in FGF23 gene causes autosomal-dominant form (ADHR), DMP1 and ENPP1 gene for autosomal-recessive form (ARHR) and CLCN5 gene for X-linked recessive form. Given an isolated patient, all forms are possible and thus molecular diagnosis is critical. ADHR and ARHR are much rarer than XLHR.7, 8 X-linked recessive form is associated with increased 1,25(OH)2 vitamin D and hypercalciuria that was not observed in this case.9 Accordingly, we focus on PHEX gene in the first place. This gene, which comprises 22 exons coding for a 749 amino-acid protein (NP_000435.3), is the cause of the most common XLHR. In this study, we identified a novel splice site acceptor mutation in PHEX gene c.1769-1G>A in intron 17. Splicing acceptor site is highly conserved and crucial for accurate splicing process. Predictions of the impact of this mutation through different algorithms as well as the mini-gene assay come into the same conclusion that the splicing acceptor would be damaged and a cryptic acceptor is probably generated. The truncated PHEX proteins lost its active site, Zn2+-binding site and four conserved Cytosines, which probably causes loss of function and structural instability (Figure 3). PHEX protein is essential in bone and dentin mineralization and renal phosphate reabsorption, and thus the loss of function of PHEX is probably the cause of the low phosphate concentration and the abnormal bone mineral density. Moreover, it is known that splice site mutations of PHEX always lead to typical phenotype, such as lower tubular resorption of phosphate and lower calcitriol levels.10, 11 Therefore, we consider that this novel splice site mutation is probably pathogenic.

Prediction of the functional influence of PHEX mutation c.1769-1G>A. (a) A predicted amino-acid sequence of normal and mutant transcript. The aberrant splicing leads to p.G590E fs*28. (b) The position of potential premature stop codon and associated functional domain. The numbers indicate residues' number. Two important functional domains are damaged owing to premature stop codon.
Although DNA sample of patient’s mother was unavailable, we ruled out the possibility of his phenotypic normal mother as a carrier because the penetrance of XLHR is often said to be 100% by age 1 year.12 The father was validated negative as expected by sequencing analysis. Thereby, this novel mutation is deduced to be de novo rather than by inheritance.13, 14
The proband is a boy who carried two alleles on X chromosome, which is absolutely uncommon. The chromosome analysis displayed a normal 46, XY, which ruled out the possibility of heterozygous XXY. Also, there is no duplication of PHEX that occurred on X chromosome, because the copy number of PHEX is identical to normal males based on qPCR. As those possibilities ruled out, the proband is probably a somatic mosaicism, which was rarely reported before.
Generally, de novo mutations are present in the sperm or egg of one parent or in a fertilized egg. Once inherited to daughter cells, they would be present in all tissues of an individual.13, 14, 15 On the other hand, it is reported that de novo mutations in PHEX gene are mostly found in females; obviously,13, 14 it is, however, not the case here. Given the observation of the mosaic mutations in the peripheral blood of this patient, we think that this mutation could be generated at an early postzygotic period so that mutation presented in most tissues, including leukocytes, and in a mosaic pattern according to similar studies in neurological disease.16, 17 In terms of the phenotype of this patient, he displayed typical hypophosphatemia and skeleton deformity but without severe lower extremity bowing. Considering that the frequency of normal (G) to mutant allele (A) is 19:13, we suppose this, to some extent, supports the point of view that the dosage effect (mutant copy number) have influence on skeleton phenotype while mineral ion homeostasis remain unaffected.18
To date, >200 mutations in PHEX have been identified, yet the somatic mutation is rarely reported with the only case documented in Japan in 2006.17 Goji et al.17 reported a girl with XLHR in Japan whose father turned out to have mosaic mutation, indicating that germline cells are involved in mutant type as well. Additionally, Saito et al.19 reported three kinds of PHEX alleles in a female through single-nucleotide polymorphism analysis, suggesting a somatic mosaicism. Both studies are similar to our observation of the de novo mosaic mutation, which may provide an exciting opportunity for further study through cell lineage tracing in order to understand the etiology of XLHR at the cellular level. On the other hand, we realized that it is not easy to detect mosaic mutations for PHEX, especially in females, because they could probably be regarded as common heterozygous. As a result, we suspect that some of the mosaic mutations might have been missed out in previous study.13, 14 Moreover, it is noteworthy that somatic mosaic mutation could change the mode of inheritance, which may confuse the genetic diagnosis and genetic counseling.
In summary, we identified a novel splice site mutation that caused an aberrant splicing, ending up with a loss of active site and Zn2+-binding site of PHEX protein. This apparently ‘heterozygous’ mutation in an affected male had a normal karyotype and PHEX copy number and his parents show completely normal phenotype. Therefore, we suppose that it is a de novo mutation generated at early postzygotic period as a mosaic pattern was even observed in peripheral blood. This study emphasizes the significance to consider the possibility of de novo mutation in mosaic pattern during molecular diagnosis.
References
Beck-Nielsen, S. S., Brock-Jacobsen, B., Gram, J., Brixen, K. & Jensen, T. K. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. Eur. J. Endocrinol. 160, 491–497 (2009).
Gaucher, C., Walrant-Debray, O., Nguyen, T.-M., Esterle, L., Garabedian, M. & Jehan, F. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum. Genet. 125, 401–411 (2009).
Pereira, C. M., de Andrade, C. R., Vargas, P. A., Della Coletta, R., de Almeida, O. P. & Lopes, M. A. Dental alterations associated with X-linked hypophosphatemic rickets. J. Endod. 30, 241–245 (2004).
Shih, N. R., Jo, O. D. & Yanagawa, N. Effects of PHEX antisense in human osteoblast cells. J. Am. Soc. Nephrol. 13, 394–399 (2002).
Grieff, M., Mumm, S., Waeltz, P., Mazzarella, R., Whyte, M. P., Thakker, R. V. et al. Expression and cloning of the human X-linked hypophosphatemia gene cDNA. Biochem. Biophys. Res. Commun. 231, 635–639 (1997).
Desmet, F.-O., Hamroun, D., Lalande, M., Collod-Béroud, G., Claustres, M. & Béroud, C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, 67 (2009).
Econs, M. & McEnery, P. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J. Clin. Endocrinol. Metab. 82, 674–681 (1997).
Lorenz-Depiereux, B, Bastepe, M, Benet-Pagès, A, Amyere, M, Wagenstaller, J, Müller-Barth, U et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat. Genet. 38, 1248–1250 (2006).
Akuta, N., Lloyd, S. E., Igarashi, T., Shiraga, H., Matsuyama, T., Yokoro, S. et al. Mutations of CLCN5 in Japanese children with idiopathic low molecular weight proteinuria, hypercalciuria and nephrocalcinosis. Kidney Int. 52, 911–916 (1997).
Morey, M., Castro-Feijóo, L., Barreiro, J., Cabanas, P., Pombo, M., Gil, M. et al. Genetic diagnosis of X-linked dominant hypophosphatemic rickets in a cohort study: Tubular reabsorption of phosphate and 1, 25 (OH) 2D serum levels are associated with PHEX mutation type. BMC Med. Genet. 12, 116 (2011).
Holm, I. A., Nelson, A. E., Robinson, B. G., Mason, R. S., Marsh, D. J., Cowell, C. T. et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J. Clin. Endocrinol. Metab. 86, 3889–3899 (2001).
Sabbagh, Y., Tenenhouse, H., Econs, M. & Auricchio, A. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID) (eds Valle, D., Beaudet, A., Vogelstein, B., Kinzler, K., Antonarakis, S., Ballabio, A., emeritus eds Scriver, C.R., Childs, B., Sly, W.S.) (Available online, 2014).
Durmaz, E., Zou, M., Al-Rijjal, R. A., Baitei, E. Y., Hammami, S., Bircan, I. et al. Novel and de novo PHEX mutations in patients with hypophosphatemic rickets. Bone 52, 286–291 (2013).
Zhu, X., Li, M., Pan, H., Bao, X., Zhang, J. & Wu, X. Analysis of the parental origin of de novo MECP2 mutations and X chromosome inactivation in 24 sporadic patients with Rett syndrome in China. J. Child Neurol. 25, 842–848 (2010).
Poduri, A., Evrony, G. D., Cai, X. & Walsh, C. A. Somatic mutation, genomic variation, and neurological disease. Science 341, 1237758 (2013).
Sicca, F., Kelemen, A., Genton, P., Das, S., Mei, D., Moro, F. et al. Mosaic mutations of the LIS1 gene cause subcortical band heterotopia. Neurology 61, 1042–1046 (2003).
Goji, K., Ozaki, K., Sadewa, A. H., Nishio, H. & Matsuo, M. Somatic and germline mosaicism for a mutation of the PHEX gene can lead to genetic transmission of x-linked hypophosphatemic rickets that mimics an autosomal dominant trait. J. Clin. Endocrinol. Metab. 91, 365–370 (2006).
Ichikawa, S., Gray, A. K., Bikorimana, E. & Econs, M. J. Dosage effect of a Phex mutation in a murine model of X-linked hypophosphatemia. Calcif. Tissue Int. 93, 155–162 (2013).
Saito, T., Nishii, Y., Yasuda, T., Ito, N., Suzuki, H., Igarashi, T. et al. Familial hypophosphatemic rickets caused by a large deletion in PHEX gene. Eur. J. Endocrinol. 161, 647–651 (2009).
Acknowledgements
We thank the families of the patient for their cooperation in the study. The study was partially supported by the grant National Natural Science Foundation of China (31371271), grant Natural Science Foundation of Zhejiang province, China (LZ14C06000) and Zhejiang province medical and health science and technology project (201493975). We sincerely thank Professor Jörg Gromoll for providing pSPL3 vector.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Weng, C., Chen, J., Sun, L. et al. A de novo mosaic mutation of PHEX in a boy with hypophosphatemic rickets. J Hum Genet 61, 223–227 (2016). https://doi.org/10.1038/jhg.2015.133
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.133
This article is cited by
-
Identification and functional characterization of a novel homozygous intronic variant in the fumarylacetoacetate hydrolase gene in a Chinese patient with tyrosinemia type 1
BMC Medical Genomics (2022)
-
Skeletal and extraskeletal disorders of biomineralization
Nature Reviews Endocrinology (2022)
-
Two De Novo Mosaic Variants Within the Same Site of PHEX Gene in a Girl with X-Linked Hypophosphatemic Rickets
Calcified Tissue International (2022)
-
Identification and functional characterization of a hemizygous novel intronic variant in OCRL gene causes Lowe syndrome
Clinical and Experimental Nephrology (2020)
-
Novel variants and uncommon cases among southern Chinese children with X-linked hypophosphatemia
Journal of Endocrinological Investigation (2020)
