
Abstract
Comparing genetic and phenotypic similarity among unrelated individuals seems a promising way to quantify the genetic component of traits while avoiding the problematic assumptions plaguing twin- and other kin-based estimates of heritability. One approach uses a Genetic Relatedness Estimation through Maximum Likelihood (GREML) model for individuals who are related at less than 0.025 to predict their phenotypic similarity by their genetic similarity. Here we test the key underlying assumption of this approach: that genetic relatedness is orthogonal to environmental similarity. Using data from the Health and Retirement Study (and two other surveys), we show two unrelated individuals may be more likely to have been reared in a similar environment (urban versus nonurban setting) if they are genetically similar. This effect is not eliminated by controls for population structure. However, when we include this environmental confound in GREML models, heritabilities do not change substantially and thus potential bias in estimates of most biological phenotypes is probably minimal.
Similar content being viewed by others


Main
Ascertaining the proportion of variance in a quantitative trait—such as height or intelligence quotient—that is due to genetic variation has long been of interest to a wide range of scientists.1, 2, 3, 4, 5 For human populations, where experimentation is not possible, the workhorse of such analysis has been the twin or extended twin design, where the average relatedness of various kin pairs is correlated with their phenotypic similarity in order to ascertain the effect of shared genotype on a given outcome.6, 7 The reigning critique of this approach is that it is difficult to eliminate the possibility that increased similarity between, say, monozygotic twins as compared with, for example, dizygotic twins is due to more similar environments and not solely their greater genetic similarity.8, 9
Among the recent and novel approaches to overcome this potential environmental confounding are studies that correlate phenotypic similarity with genotypic similarity across the genome among pairs of individuals who are less than 2.5% related as computed by identity by state and are therefore considered non-kin.10, 11, 12 Simply described, a genetic relatedness matrix is constructed in which each cell is filled by a measure of genotypic correlation between pairs of individuals (the rows and columns) where the genotype is based on the summation of 2N gametic correlation at specific loci across the genome, after pruning single nucleotide polymorphisms for linkage disequilibrium. The genotypes are coded as the numbers of minor alleles for each single nucleotide polymorphism, standardized by a transformation that makes the sample variance independent of allele frequency. The genetic relatedness matrix may then be used in concert with a phenotypic distance matrix to estimate heritability without estimating the phenotypic effect of any individual single nucleotide polymorphism locus. This Genetic Relatedness Estimation through Maximum Likelihood (GREML) approach yields estimates of narrow-sense (additive) heritability (h2) that are lower than but approaching those obtained from traditional twin-based approaches and has been deployed for diverse phenotypes, including height,13 schizophrenia,10 asthma,14 smoking,15 body mass index,16 educational attainment17 and political and economic preferences.18
However, similar to twin-based models, the GREML approach relies on one key assumption about the relationship between genetic similarity and environmental similarity. Although those who share genetic variation may experience more similar environments owing to population structure, admixture and, of course, extended family ties, GREML assumes that those who are less related than second cousins share alleles in an essentially random manner that is itself uncorrelated with environmental similarity. The motivating notion is that at these low levels of relatedness, relative genetic similarity is driven by the randomness of recombination and allele segregation and not by underlying kinship structure. As such, parental relatedness and relevant environmental conditions should be orthogonal to respondent relatedness.
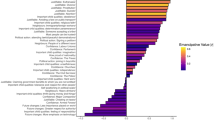
To support this claim that relatedness among these pairs of individuals is random (and thus uncorrelated with potential environmental confounders), Yang et al.11 show correlations in relatedness levels between chromosomes in a Supplementary Table.11 Their logic is that if the person-wide genetic relatedness measure between individuals (that is, gametic correlation) was reflecting population structure (and, thus, covaried with environment), pairwise genetic relatedness would be correlated across those individuals’ chromosomes. However, if the distribution of pairwise relatedness is really just the result of randomization during meiosis, then each chromosome should be independent, demonstrating no correlation. Yang et al. find no single pair of chromosomes for which the P-value of the correlation between the genetic relatedness of those two chromosomes is less than 0.00022, which corresponds to a 0.05 α-level with a Bonferroni correction for the 231 comparisons they make across the bivariate combinations of the autosomal chromosomes. However, this strikes us as the wrong statistical test; we are not concerned as to whether the relatedness of a specific pair of chromosomes co-varies below a strict type I error threshold. Rather, we are worried that there is an overall pattern of relatedness in the data and thus should apply a more sensitive test that minimizes type II error. Along these lines, in Figure 1, we present a histogram of their 231 reported P-values and show that there is indeed an excess of low P-values, particularly below the P<0.10 threshold as compared with a random distribution. Indeed, when we perform a Kolmogorov–Smirnov test on their reported distribution, we find it to deviate from the theoretically expected (uniform) distribution (D+=0.1892, P-value=7.037e−08). Although we do not know the signs of the associated coefficients (as they were not reported by Yang et al.), the overall non-random distribution of correlations suggests that the data fail the test for randomization of alleles across chromosomes.

Histogram of P-values from pairwise chromosome regressions of relatedness as presented in Supplementary Table 2 of Yang et al.11 ‘Common single nucleotide polymorphisms (SNPs) explain a large proportion of the heritability for human height.’ Note excess of low P-values, particularly less than 0.10. This suggests that there is a significant pattern of covariance between independently segregating genomic segments and thus potential non-randomness in overall relatedness (that is, potential covariance with population structure and thus environmental confounders): the Kolmogorov–Smirnov test: (D+=0.1892, P-value=7.037e−08).
With this in mind, we do not believe that this core assumption that the environmental similarity between pairs of unrelated persons is uncorrelated with their genetic similarity (below the 0.025 threshold) has not been adequately interrogated. In the present study, we test the key GREML assumption by asking whether the childhood environments of subjects are more similar if they are more related genetically. If pairs of individuals share the experience of urban environment during their respective childhoods—or, conversely, share a non-urban childhood experience—this is likely to have the effect of making their formative social and physical environments more similar than they would be by chance. Thus, if relatedness predicts environmental similarity in this way, it could confound the premise of GREML-based methods of estimating the genetic component of phenotypes. It makes no difference whether urbanicity is itself causal of the phenotype under consideration; it may be acting merely as a proxy for other, more relevant environmental factors—such as social class, nutritional status and so forth—that are themselves related, through environmental channels, to the offspring phenotype (such as height, body mass index or education). That said, a large literature shows that urbanicity is correlated with a range of outcomes studied by geneticists, ranging from mental health19, 20, 21 to immunological response21, 22 to education.23
Health and Retirement Study data allow us to estimate the heritability of urban childhood residence as well as how urban residence during childhood affects GREML estimates of other putatively heritable traits. We used the standard GREML analysis (using Genome-wide Complex Trait Analysis software12) to estimate heritability, with population stratification controlled by principal components (PCs; see Supplementary Materials: Methods). As shown in the first row of Table 1 below, in the Health and Retirement Study sample with two PCs controlled, urban childhood—putatively a childhood environmental variable based on circumstance and parental choices—is indeed highly heritable at 29%. As we suspected that the nonzero heritability might be a result of geographic population structure, we then reran the analysis with 10 and 25 PCs included as controls. These controls attenuated, but did not eliminate, the effect we discovered. Thus, it seems that controls for population structure through deployment of PCs do not adequately address this confounding. We replicated this finding with data from the National Longitudinal Survey of Adolescent Health as well as with another childhood phenotype—maternal education—in the National Longitudinal Survey of Adolescent Health and in the Framingham Heart Study. Both the National Longitudinal Survey of Adolescent Health and the Framingham Heart Study are underpowered to generate statistically precise GREML heritability estimates, but ordinary least square regressions show magnitudes of estimates in line with the Health and Retirement Study results (see Supplementary Materials).
Despite the apparent heritability of childhood residence, when we control for this possible confounder in analysis of common human phenotypes of interest—height, body mass index and years of schooling—we find that the differences between the ‘naive’ models and the ones that hold childhood urbanicity constant are negligible and not statistically significant. In fact, the only phenotype for which the heritability changes to any noticeable degree is respondent education, which drops by a statistically insignificant two percentage points (P=0.8203) in the model with only two PCs. This makes sense: of the three phenotypes, we would expect height to be the least influenced by childhood environment, body mass index in the middle and education to be the most affected by potential environmental confounds. As controlling for more PCs did not appear to eliminate the heritability of a putatively environmental confound—urban childhood—we then tried to see whether using a more restrictive relatedness cutoff (0.01) would address the ‘problem.’ However, when we used this more restrictive cutoff, sample sizes dropped too drastically to yield adequate power. (Results are shown in Supplementary Table 1.)
Our findings have implications not only for GREML analysis of heritability but also for genome-wide analysis more broadly. Namely, some scholars have claimed that PCs adequately control for population stratification, especially when data show no evidence of ‘early take-off’ (that is, across the vast majority of the distribution of P-values, they match what one would expect from chance).24, 25 Our results suggest that directly modeling error terms as a linear function of relatedness in a sample may also be necessary to adjust for stratification.26, 27 Finally, and most importantly, while the key assumption of GREML analysis that the genotype–environment correlation is zero is violated, the consequences of that violation appear to be trivial. We cautiously conclude that GREML is a valid estimation technique for heritability but recommend that going forward, researchers test for the violation of this assumption (and robustness to violations) in their own data sets as a standard sensitivity analysis.
Change history
25 June 2014
This article has been corrected since Advance Online Publication, and a corrigendum is also printed in this issue.
References
Breen, F., Plomin, R. & Wardle, J. Heritability of food preferences in young children. Physiol. Behav. 88, 443–447 (2006).
Rodgers, J. L., Kohler, H. P., Kyvik, K. O. & Christensen, K. Behavior genetic modeling of human fertility: findings from a contemporary Danish twin study. Demography 38, 29–42 (2001).
Van den Oord, E. J. A study of genetic and environmental effects on the co-occurrence of problem behaviors in three-year-old-twins. J. Abnorm. Psychol. 109, 360 (2000).
Rodgers, J., Rowe, D. & Buster, M. Nature, nurture and first sexual intercourse in the USA: fitting behavioural genetic models to NLSY kinship data. J. Biosoc. Sci. 31, 29–41 (1999).
Allison, D. B., Kaprio, J., Korkeila, M., Koskenvuo, M., Neale, M. C. & Hayakawa, K. The heritability of body mass index among an international sample of monozygotic twins reared apart. Int. J. Obes. 20, 501–506 (1996).
Plomin, R., Owen, M. & McGuffin, P. The genetic basis of complex human behaviors. Science 264, 1733–1739 (1994).
Purcell, S. Variance components models for geneenvironment interaction in quantitative trait locus linkage analysis. Twin Res. 5, 572–576 (2002).
Goldberger, A. Heritability. Economica 46, 327–347 (1979).
Scarr, S. & Carter-Saltzman, L. Twin method: defense of a critical assumption. Behav. Genet. 9, 527–542 (1979).
Purcell, S., Wray, N. R., Stone, J. L., Visscher, P. M., O’Donovan, M. C., Sullivan, P. F. et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (2009).
Yang, J., Benyamin, B. & McEvoy, B. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Yang, J., Lee, S., Goddard, M. & Visscher, P. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Davies, G., Tenesa, A., Payton, A., Yang, J., Harris, S. E., Liewald, D. et al. Genome-wide association studies establish that human intelligence is highly heritable and polygenic. Mol. Psychiatry 16, 996–1005 (2011).
Belsky, D. W., Sears, M. R., Hancox, R. J., Harrington, H., Houts, R., Moffitt, T. E. et al. Polygenic risk and the development and course of asthma: an analysis of data from a four-decade longitudinal study. Lancet Respir. Med. 1, 453–461 (2013).
Belsky, D. W., Moffitt, T. E., Baker, T. B., Biddle, A. K., Evans, J. P., Harrington, H. et al. Polygenic risk and the developmental progression to heavy, persistent smoking and nicotine dependence: evidence from a 4-decade longitudinal study. JAMA Psychiatry 70, 534–542 (2013).
Belsky, D. W., Moffitt, T. E., Houts, R., Bennett, G. G., Biddle, A. K., Blumenthal, J. A. et al. Polygenic risk, rapid childhood growth, and the development of obesity: evidence from a 4-decade longitudinal study. Arch. Pediatr. Adolesc. Med. 166, 515–521 (2012).
Rietveld, C., Medland, S., Derringer, J. & Yang, J. GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science 340, 1467–1471 (2013).
Benjamin, D. J., Cesarini, D., van der Loos, M. J., Dawes, C. T., Koellinger, P. D., Magnusson, P. K. et al. The genetic architecture of economic and political preferences. Proc. Natl Acad. Sci. USA 109, 8026–8031 (2012).
Krabbendam, L. & Van Os, J. Schizophrenia and urbanicity: a major environmental influence—conditional on genetic risk. Schizophr. Bull. 31, 795–799 (2005).
Stefanis, N., Delespaul, P., Smyrnis, N., Lembesi, A., Avramopoulos, D. A., Evdokimidis, I. K. et al. Is the excess risk of psychosis-like experiences in urban areas attributable to altered cognitive development? Soc. Psychiatry 39, 364–368 (2004).
Spauwen, J., Krabbendam, L., Lieb, R., Wittchen, H. U. & Van Os, J. Evidence that the outcome of developmental expression of psychosis is worse for adolescents growing up in an urban environment. Psychol. Med. 36, 407–415 (2006).
Priftis, K., Anthracopoulos, M. B., Nikolaou-Papanagiotou, A., Matziou, V., Paliatsos, A. G., Tzavelas, G. et al. Increased sensitization in urban vs. rural environment–rural protection or an urban living effect? Pediatr. Allergy Immunol. 18, 209–216 (2007).
Jencks, C. & Mayer, S. in Inn. poverty United States (1990). http://www.books.google.com/books?hl=en&lr=&id=P7IV4eaGcxwC&oi=fnd&pg=PA111&dq=The+social+consequences+of+growing+up+in+poor+city+neighborhood&ots=Zn2Q7Z3SD4&sig=CxdXMCb0dKjkt1zrCr-6Av_UnwA.
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A. & Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Price, A., Zaitlen, N., Reich, D. & Patterson, N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 11, 459–463 (2010).
Kang, H. M., Sul, J. H., Service, S. K., Zaitlen, N. A., Kong, S. Y., Freimer, N. B. et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 42, 348–354 (2010).
Manichaikul, A., Mychaleckyj, J. C., Rich, S. S., Daly, K., Sale, M. & Chen, W. M. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Acknowledgements
This research uses data from The National Longitudinal Study of Adolescent Health (Add Health), a program project directed by Kathleen Mullan Harris and designed by J Richard Udry, Peter S Bearman, and Kathleen Mullan Harris at the University of North Carolina at Chapel Hill, and funded by grant P01-HD31921 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, with cooperative funding from 23 other federal agencies and foundations. Information on how to obtain the Add Health data files is available on the Add Health website (http://www.cpc.unc.edu/addhealth). The Framingham Heart Study (FHS; accession #7909-7) was supported by the National Heart Lung and Blood Institute of the National Institutes of Health and Boston University School of Medicine, and the National Heart, Lung and Blood Institute's Framingham Heart Study. The Health and Retirement Study (HRS; accession number 0925-0670) is sponsored by the National Institute on Aging (grant numbers NIA U01AG009740, RC2AG036495, and RC4AG039029) and is conducted by the University of Michigan. Additional funding support for genotyping and analysis were provided by NIH/NICHD R01 HD060726.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Conley, D., Siegal, M., W Domingue, B. et al. Testing the key assumption of heritability estimates based on genome-wide genetic relatedness. J Hum Genet 59, 342–345 (2014). https://doi.org/10.1038/jhg.2014.14
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.14
Keywords
This article is cited by
-
Genetic variation, brain, and intelligence differences
Molecular Psychiatry (2022)
-
Familial Influences on Neuroticism and Education in the UK Biobank
Behavior Genetics (2020)
-
Genomic analysis of family data reveals additional genetic effects on intelligence and personality
Molecular Psychiatry (2018)
-
Cohort Effects in the Genetic Influence on Smoking
Behavior Genetics (2016)
